

1 Introduction
Hydrogen is considered as a promising fuel with regards to environment compatibility, high energy content, versatility, efficiency and cost effectiveness when compared to oil, gasoline, methane, methanol and liquefied petroleum gas [1,2]. Hydrogen can be produced via water electrolysis with high yield and purity. Although the hydrogen evolution reaction (her) has been extensively investigated since the last century [3,4], the interest in finding lower cost electro-catalysts with higher efficiency and stability to her has been growing tremendously in recent years.
The cost of hydrogen production by water electrolysis is directly proportional to the operating cell voltage and can be reduced by minimizing the overpotentials of the electrode reactions. The most efficient electrocatalysts for her in acid and alkaline media are the Pt group of metals [5,6], with platinum exhibiting the highest activity [3]. Novel electrode materials have been investigated, aiming at the reduction of the material cost and energy consumption by using bi-metallic Pt-based alloys [7,8]. Nickel and its alloys with Co [9–11], Mo [12] and Fe [13], as well as Raney-type Ni-Zn-P [14] are promising candidates for her in alkaline solutions, and show satisfactory electrocatalytic activity and stability [4].
Recent progress in nanotechnology and surface science techniques opens up new opportunities in understanding and rational design of nanostructured electro-catalysts with low metal content and increased activity to her. Reactivity of nanosized electro-catalysts is associated with the high ratio of surface to bulk atoms. Several parameters control the electrocatalytic activity of particles; among them are particle size, shape, size distribution, surface and bulk composition, structure, metal-support interaction, etc. Nanosized nickel materials that exhibit high electrochemical surface area are attractive for her [15].
In the present work, Ni particles were synthesized using the modified polyol method and tested for her in 1 M KOH. In order to improve their catalytic activity a simple electrochemical treatment was used. The treatment consisted of applying a sinusoidal wave with the impulse amplitudes of −0.8 V — 0.1 V vs. MSE in 0.1 M Na2SO4 + 30 mM ascorbic acid solution. The proposed treatment is based on the work of Tian et al. [16,17] in which a square-wave potential treatment was applied to Pt microspheres (750 nm average size) in order to obtain high Miller's index tetrahexahedral particles. The authors reported that resulting particles had enhanced the catalytic activity towards ethanol and formic acid electro-oxidation.
2 Experimental
2.1 Synthesis of Ni particles
Nickel particles were prepared by a modified polyol technique [18,19]. First, 0.4 g of Ni(OH)2 (Acros Organics) was mixed with 150 mL of ethylene glycol (Sigma Aldrich) for one hour in a three-neck flask and refluxed for 10 min. Second, 8.7 mg of PdCl2 (Alfa Aesar) were dissolved in ethylene glycol (EG) containing 0.5 mL of HCl (Sigma Aldrich) for one hour. When Ni(OH)2 in EG solution reached 196 °C, the Pd-containing solution was injected into the former, resulting in 1 at. % of Pd in the final Ni content. Formed particles had an Ni to Pd ratio of 99 to 1 at. % (Ni99Pd1); however, these are denoted as Ni particles hereafter. Once the PdCl2-containing EG solution was injected, the colour of the reaction mixture turned from bright green to black, indicating the formation of metallic Ni. The reflux was kept at 196 °C for two hours to ensure the completion of the reaction, and then cooled down to room temperature. After extensive washing with ethanol (Fisher Scientific) and deionized water, the resulting Ni particles were separated by centrifugation and stored in ethanol.
2.2 Electrochemistry
Electrochemical experiments were carried out in a conventional three-electrode cell using BioLogic VSP Potentiostat/Galvanostat/EIS in conjunction with EC Lab software. Highly oriented pyrolytic graphite (HOPG) electrode (S = 14.96 mm2) was used as a current collector for Ni particles. First, 5 μL of Ni particles-in-ethanol solution was deposited onto HOPG and dried in air for 10 min, then 1.5 μL of Nafion solution (Sigma Aldrich, 5 wt. %) was placed on the top of the particles. Large surface area Pt gauze was used as a counter electrode, and all electrochemical experiments are performed with respect to a mercury sulphate electrode Hg/Hg2SO4/K2SO4 (MSE). Her on as-prepared and electrochemically treated Ni particles was studied using cyclic voltammetry (CV) and chronoamperometry (CA) in a de-aerated 1 M KOH (EMD, ACS grade).
Electrochemical treatment of Ni particles consisted of applying a sinusoidal-wave signal with upper potential 0.1 V and lower potential −0.8 V vs. MSE at 10 Hz in a solution of 0.1 M Na2SO4 + 30 mM ascorbic acid (Sigma Aldrich). In addition, solutions of 0.5 M H2SO4, 1 M KOH and 0.1 M Na2SO4 without ascorbic acid were tested for the treatment of Ni particles. Potential amplitudes between −0.61 and 0.1 V at 10 Hz were studied as well. All experiments were conducted at room temperature.
2.3 Particles characterization
The X-ray diffraction (XRD) patterns were measured with a Rigaku ultima IV diffractometer equipped with a Cu tube between 20 to 60° 2θ at a scanning rate of 0.030° 2θ/s. Scanning electron microscopy (SEM) analysis of Ni particles was carried out using a JEOL JSM-7500F FESEM. Samples were prepared by depositing a droplet from the Ni-in-ethanol solution onto a freshly cleaned silicon wafer followed by ethanol evaporation at room temperature. Surface composition of Ni catalysts was analyzed by X-ray photoelectron spectroscopy (XPS) using a KRATOS Axis Ultra DLD with a Hybrid lens mode. XPS measurements were conducted at 140 W and pass energy 20 eV using a monochromatic AlKα. In order to compensate the accumulation of positive charges, the XPS measurements were performed under the presence of an electron flood gun. The XPS O 1 s core level spectra were analyzed using the XPS Peak program with a fitting routine which decomposes each spectrum into individual mixed Gaussian-Lorentzian peaks using a Shirley background subtraction over the energy rate of the fit. The Lorentzian contribution to the peak shape was kept in all cases under 40. The Binding Energy (BE) scale was assigned by adjusting the C 1 s peak at 284.6 eV.
3 Results and discussion
The XRD patterns of as-synthesized Ni particles are shown in Fig. 1. The structure of the resulting Ni is face-centred cubic (fcc) with well-defined peaks at 44.46 and 51.84° 2θ corresponding to Ni (111) and (200) crystalline planes, respectively. Small broad peaks at 40 and 46° 2θ are attributed to fcc Pd (111) and (200) indicating that separate crystalline phase of Pd is formed. Using the full-width at half maximum of Ni (111) reflection and Scherrer formula [20], the average crystallite size of Ni was estimated to be 30.1 nm.
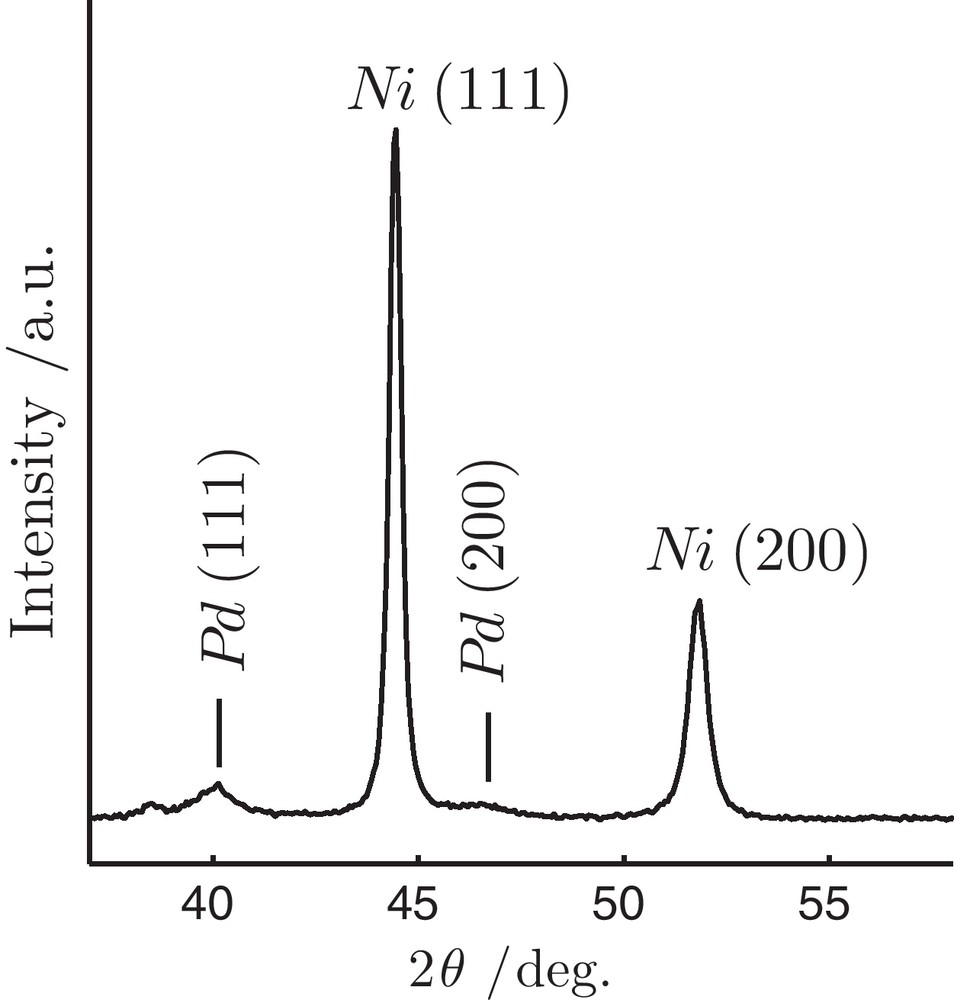
X-ray diffraction patterns of the as-synthesized Ni particles by modified polyol technique.
SEM micrographs of as-prepared Ni particles are shown in Fig. 2 (a). The resulting particles are spherical in shape with wide size distribution in the range of 100 to 500 nm. Compared to the crystallite size calculated from the XRD data, the micrometer-sized particles are constituted of several crystallites of 30.1 nm average size.

Electron micrographs of Ni particles before (a) and after (b) electrochemical treatment (sine-wave −0.8 — 0.1 V at 10 Hz for 50 min in 0.1 M Na2SO4 + 30 mM ascorbic acid).
As-prepared Ni particles were tested by cyclic voltammetry (CV) in 1 M KOH solution in the anodic (Fig. 3 (a), solid line) and cathodic regions (Fig. 3 (b), solid line). Observed peaks are typical for the bulk Ni metal. The surface redox couple centred at ∼ −0.1 V, can be attributed to Ni(II)/Ni(III) redox reaction [21,22]:
| (1) |
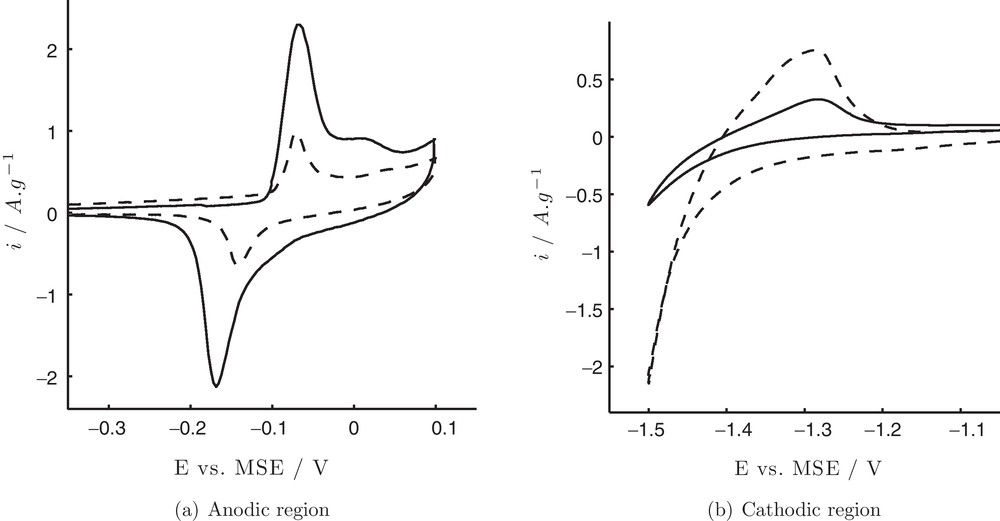
Cyclic voltammograms of Ni particles before (—) and after (- -) electrochemical treatment (parameters as in Fig. 2) in the anodic (a) and cathodic (b) region.
Fig. 3 (b) shows an anodic peak at −1.3 V due to the formation of α-phase of Ni(OH)2 according to Eq. (2) [12,23,24]:
| (2) |
Hydrogen desorption from metallic Ni also can take place in this potential region:
| (3) |
At potentials more negative than −1.4 V, the her starts to take place.
As-prepared Ni particles were subject to sinusoidal-wave treatment. The treatment was applied to the particles so as to improve their catalytic response towards her. The procedure was based on the recent work by Tian et al. [17], where square-wave treatment resulted in the structural modifications of Pt nanospheres electrodeposited on glassy carbon substrate. In the present study, several supporting electrolytes and two potential limits of the waveform were tested to find the most noticeable changes in the current density of her. The following supporting electrolytes were tested: 0.1 M KOH and 0.5 M H2SO4 (results are not shown here), as well as 0.1 M Na2SO4 with and without 30 mM of ascorbic acid. Treatment in the two former solutions at either −0.61 — 0.1 V (anodic) or −0.8 — 0.1 V (anodic/cathodic) vs. MSE at 10 Hz did not lead to the improvement Ni performance in the her region. On the contrary, treatment in 0.1 M Na2SO4 + 30 mM ascorbic acid at −0.8 — 0.1 V at 10 Hz gave the most noticeable improvement, i.e. increase in the current density of her in 1 M KOH. It is worth noting that in the absence of ascorbic acid the resulting electrochemical response was much lower, nevertheless, additional experiments are necessary to clarify its role in the modification of Ni.
CVs of Ni particles in 0.1 M KOH after treatment (sine-wave −0.8 — 0.1 V at 10 Hz for 50 min in 0.1 M Na2SO4 + 30 mM ascorbic acid) are shown in Fig. 3 (dashed lines). It can be seen that a pair of peaks Ni(II)/Ni(III) in the anodic region decreases significantly (Fig. 3 (a) dashed line) indicating that after the treatment the amount of β-Ni(OH)2, which participates in the reaction depicted by Eq. (1), decreases. Potential difference between anodic and cathodic peak maxima decreases to 0.06 V when compared to 0.1 V before the treatment suggesting higher reversibility of the surface redox reaction. At the same time, the peak corresponding to α-Ni(OH)2 formation and Hads oxidation process increases considerably (Fig. 3 (b) dashed line). This is accompanied by an increase of the current density for H2 evolution along with a shift of the onset potential to more positive values: −1.3 vs. −1.35 V for treated and as-prepared Ni particles, respectively.
To evaluate the changes that occur in Ni particles after the treatment, the electrochemical surface area (ECSA) was evaluated for as-prepared and treated materials. ECSA was estimated using the peak at around −1.3 V following the method proposed in [12,24]:
| (4) |
The ECSA in m2·g−1 is calculated by dividing the charge Q in Coulomb corresponding to reaction of α-Ni(OH)2 formation (Eq. (2)) at the Ni surface [25] by the mass mNi of nickel deposited on the electrode in g and the theoretical charge 0.514 mC·m−2 [12,21,24]. The theoretical value is associated with α-Ni(OH)2 monolayer formation and takes in account that two electrons are involved in the reaction (Eq. (2)) [12]. The charge Q was calculated by peak integration between −1.18 and −1.41 V. ECSA before and after the treatment is 0.84 and 7.75 cm2·mg−1, respectively, which corresponds to 9.2 times increase, whereas current density for her is more than four times higher after the treatment (Fig. 3 (b)).
Chronoamperograms of Ni particles before and after treatment at −1.4 V for one hour are shown in Fig. 4. As it can be seen, cathodic current densities obtained at the treated Ni particles are over two times higher than for the untreated sample. Satisfactory stability of the electrode is observed, and cathodic current densities, for both samples, continue to increase without reaching a steady-state after one hour.

Chronoamperograms of Ni particles before (—) and after (- -) treatment (parameters as in Fig. 2) at −1.4 V vs. MSE.
Effect of the treatment time on the resulting catalytic activity of Ni was investigated and ECSA before and after treatment served as a point of quantitative reference. Fig. 5 shows the ratio of ECSA after to ECSA before the treatment (
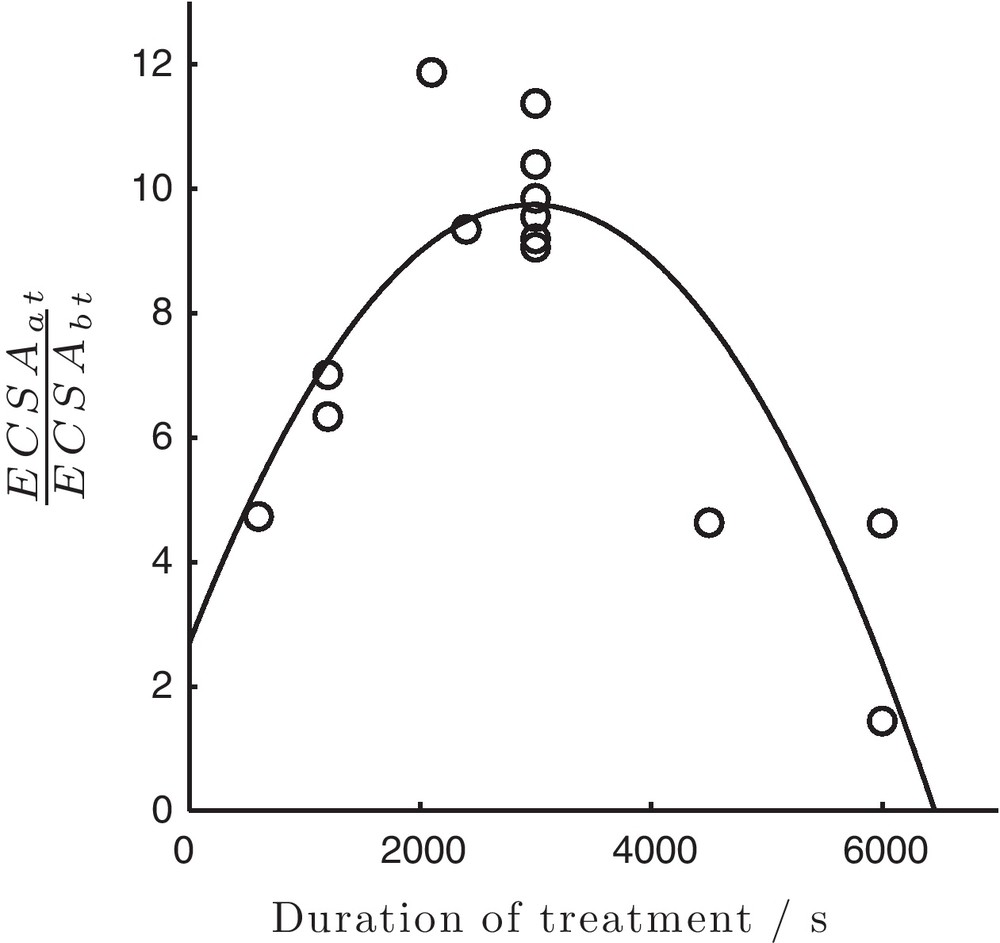
Ratio of the electrochemical active surface areas (ECSA) of Ni particles after and before treatment (parameters as in Fig. 2) with respect to the duration of the electrochemical treatment.
SEM micrographs of Ni particles after treatment are shown in Fig. 2 (b). No visible changes in particle shape, size or structure were found after the treatment indicating that the observed increase in the catalytic activity of the treated particles might be mainly attributed to the changes in the surface chemistry and not to the material morphology.
Detailed surface analysis of the catalysts surface before and after the treatment was carried out by XPS. Before treatment, beyond the existence of carbon, oxygen and nickel, XPS analysis showed the existence of Pd as well, in the form of PdO. The characteristic peaks of Pd are of very low intensity implying its very low concentration in the sample. After treatment no Pd was detected on the surface possibly due to its dissolution in the bulk of Ni. The XPS spectra of Ni 2p3/2 and O 1 s peaks before (upper spectra) and after treatment (lower spectra) are presented in Fig. 6 (a) and (b), respectively. The Ni 2p3/2 spectrum shows a complex structure with intense and wide satellite peaks at higher BEs adjacent to the primary photolines. These peaks that are usually observed in the case of transition metal ions are called shake-up; they are due to a charge transfer ligand-metal process by which an excitation of a valence ligand electron to a previously unoccupied metal orbital takes place simultaneously with the primary core photoelectron process. The satellite band is placed at higher binding energy and its position depends on the chemical environment that surrounds the nickel atom. Thus, for metallic Ni it is reported to be 6 eV [26], while it is 5.8 eV in the case of NiOOH and Ni(OH)2 [27,28] and about 7 eV for NiO respectively [29]. Before treatment four different peaks are detected at 852.2, 854.1, 855.4 and 857.4 eV that according to their BEs are assigned to Ni metal, NiO, Ni(OH)2 and NiOOH, respectively [26,30–32].
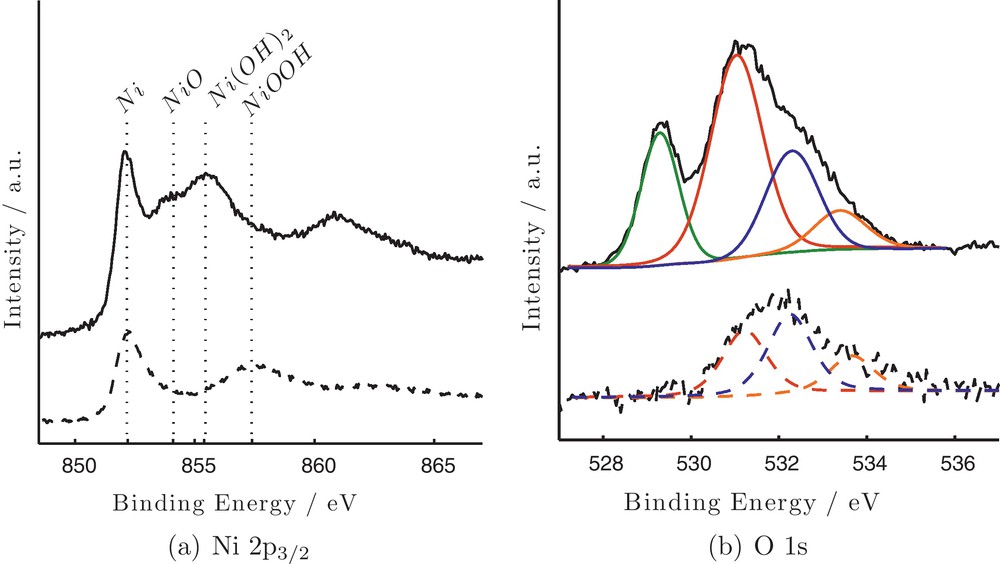
X-ray photoelectron spectra of Ni 2p3/2 (a) and O 1 s (b) before (—) and after (- -) electrochemical treatment (parameters as in Fig. 2) of Ni particles.
The O 1 s peak before treatment is deconvoluted into four components at 529.2, 531.1, 532.3 and 533.4 eV. It must be noted that O 1 s peak should be masked by the Pd 3p3/2 (∼532 eV), but in this case due to the very low concentration of Pd in the sample, its contribution is rather low. Further studies are necessary so as to use the approach of Titkov for the substraction of the Pd 3p peak from the O 1 s peak [33]. The O 1 s peak at the lower BE is characteristic of oxygen atoms in NiO [34] and PdO [35] while the one at 533.4 eV is assigned to adsorbed water [31] with a small contribution also of the Pd 3p peak. The peak at 531.1 eV, that represents about 50% of the total peak area is assigned to oxygen atoms in CO contaminants [36,37]; in the literature it is also attributed to oxygen atoms of the defect oxide Ni2O3 [38]. Although Ni2O3 is referred as not a stable bulk compound [39], its existence cannot be excluded, at this point.
After treatment the shape of the Ni 2p3/2 XPS peak has changed significantly. At the same time the signal of oxygen is much lower compared to the one before treatment, while no Pd is detected on the surface. This decrease of the O 1 s signal suggests that the treatment caused an extended reduction of the as-synthesized Ni atoms. As it is seen, the Ni 2p3/2 peak now consists of two components. The first at about 852.3 eV is attributed to Ni atoms in the metallic form. The second peak appears at about 857.4 eV and its signal has significantly increased. The peak is at ∼0.7 eV lower BE from the energy position of Ni0 satellite peak (∼858.3 eV) and is attributed to NiOOH surface species, which significantly increases after treatment [32]. The corresponding satellite peak is detected at about 863 eV. The above analysis involving the formation of nickel oxyhydroxide is also justified by the analysis of O 1 s peak (Fig. 6 (b) lower spectrum). The most characteristic change is that no component at 529.2 eV is detected after treatment. This, in combination with the lack of the line at about 854.3 eV in Ni 2p3/2 spectrum strongly denote the extended reduction of NiO species due to the treatment. At the same time the component at ∼532.5 eV that is characteristic of OH species demonstrates now a higher relative intensity, implying that OH− species are dominating the surface of nickel particles mainly in the oxyhydroxide form, because the line at 855.5 eV that is characteristic of Ni(OH)2 and present before treatment is not clearly distinguished on the spectrum after treatment. The components attributed to CO contamination and adsorbed H2O are now at 531.3 and 533.7 eV, respectively.
4 Conclusion
Ni particles were synthesized via modified polyol method using 1 at. % of Pd for homogeneous nucleation. The particles had face-centred cubic structure with crystallite size of 30.1 nm. SEM showed wide particle size distribution in the range of 100–500 nm. Sinusoidal-wave treatment of Ni particles in 0.1 M Na2SO4 + 30 mM ascorbic acid at the amplitudes of −0.8 — 0.1 V, for 50 min lead to a significant decrease of the pair of peaks (Ni(II)/Ni(III)) in the anodic region and increase of the peak corresponding to formation of α-Ni(OH)2 and hydrogen desorption. ECSA was found to be almost nine times higher after the treatment. Moreover, catalytic activity of Ni towards her increased significantly and around two times higher current densities were found during chronoamperometric measurements at −1.45 V vs. MSE. According to SEM, no visible changes in particle shape and size were observed after the treatment. XPS analysis revealed significant changes in the surface chemistry of Ni particles. After treatment the dominant surface species were metallic Ni and NiOOH. It is believed that treatment with impulse current lead to the dissolution of Ni oxide and hydroxide, as well as α- and β-Ni(OH)2 and formation of catalytically active Ni metal. Nevertheless, further studies are necessary to clarify the role of various treatment parameters on the performance of Ni particles and their stability in her.
Acknowledgement
Authors would like to thank the financial support from Fonds québécois de la recherche sur la nature et les technologies (FQRNT) “Recherche partenariat contribuant réduction et séquestration gaz effet de serre” and the Center for Catalysis Research and Innovation (CCRI) at the University of Ottawa for microscopy services.