

1 Introduction
La coupure oxydante de la liaison C-C des cycloalcanones et des α-cétols est largement utilisée dans la chimie organique soit au niveau de laboratoire ou à l’échelle industrielle [1–4]. L’une des applications très importante de ces réactions est la synthèse de l’acide adipique (> 2 Mt/an), produit primaire destiné aux fibres et matières synthétiques à base de polyamide et au polyester [5]. L’industrie automobile et des matières plastiques est le principal acheteur de ce produit. La production de l’acide adipique à l’échelle industrielle est réalisée par l’oxydation catalytique du cyclohexanone ou mélange cyclohexanone/cyclohexanol, ‘olone’, en présence de l’acide nitrique comme oxydant et les sels de vanadium (V) et de cuivre (II) qui catalysent certaines étapes de la réaction (Schéma 1).
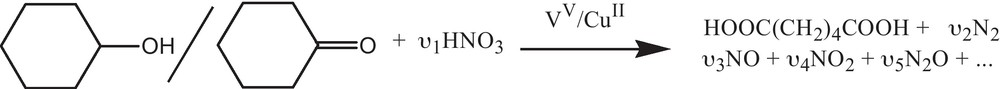
Le problème majeur de ce procédé est la formation de l’oxyde nitreux N2O (400 000 t/an) qui a un effet de serre 200 fois plus dangereux que le dioxyde de carbone [6]. Dans la dernière décennie, sous la peine de pénalité, certains industriels ont ajouté de système de décomposition catalytique ou thermique de l’oxyde nitreux à leurs unités de production comme le cas de Lanxess en 2009. De même, l’oxyde nitreux a été réutilisé dans l’oxydation catalytique du benzène en phénol par Solutia aux États-Unis en 2003 [7,8]. Il reste que ces remèdes, à moindre coût, relève d’un simple ‘replâtrage’ du procédé existant auparavant plutôt que d’une véritable refonte. La chimie verte ne se conçoit que dans le bilan global d’un procédé, d’où la nécessité d’utiliser le dioxygène comme oxydant terminal ou autre oxydant propre.
Antérieurement, il a été montré qu’en présence de dioxygène, les complexes à base de vanadium tels que les hétéropolyacides, H3+n[PMo12−nVnO40], aq, [noté HPA-n (n = 1–6)] ou bien les sels de cuivre, réagissent avec les cycloalcanones pour donner des acides carboxyliques [2,9]. Ainsi, le traitement d’un substrat modèle, comme la 2-méthylcyclohexanone, conduit à l’acide 6-oxoheptanoïque avec de très bons rendements (90 %), la réaction d’oxydation de la cyclohexanone au sein du système ‘HPA-n/AcOH-H2O/O2’ fournit bien l’acide adipique, mais aussi les acides glutarique, succinique… et le dioxyde de carbone. Ces sous-produits proviennent principalement de la dégradation de l’acide 6-oxoheptanoïque et d’autres réactions homolytiques parasites. Il apparaît aussi que les catalyseurs hétérogènes sont activement recherchés car le recyclage du catalyseur est souvent l’étape qui augmente le coût total d’un procédé. Récemment, il a été développé un système catalytique ‘homogène supporté’ de résines de type Nafion® échangées par l’ion vanadyle pour l’ouverture oxydante des cycloalcanones et des α-cétols en présence de dioxygène [10]. Cependant, l’oxydation de certains substrats a conduit à une déperdition importante de vanadium ; l’effet de la lixiviation.
De nombreux travaux menés visent à trouver des transpositions des systèmes homogènes en procédés hétérogènes. En chimie organique, l’hétérogénéisation des systèmes connaît d’importants développements. Ainsi, nous nous sommes intéressés à reconstituer la réactivité des systèmes homogènes pour développer des applications en catalyse solide/liquide. Pour ce faire le phosphate naturel (PN) sera dopé avec des précurseurs simples du vanadium. Le PN est un matériau naturel très abondant. Il est insoluble dans l’eau et dans les solvants organiques. Son activité catalytique a été démontrée dans plusieurs réactions organiques [11]. Les matériaux préparés seront éprouvés dans la coupure oxydante de cycloalcanones α-substituées et des α-cétols en présence du dioxygène.
2 Partie expérimentale
2.1 Produits utilisés
Tous les réactifs utilisés dans ce travail proviennent d’Aldrich, Merck, Prolabo ou Janssen Chimica. Ils ont été utilisés sans purification. Les solvants organiques, très purs, ont été obtenus à partir de la SDS et Carlo Erba.
2.2 Préparation du catalyseur phosphaté V/PN
D’abord, nous préparons le PN tout seul par le protocole déjà décrit par S. Sebti et al. [12].
Une fois le PN préparé, nous prenons 10 g de ce dernier qu’on ajoute à 1 mmol de vanadate d’ammonium (NH4VO3) dissous dans 10 mL d’une solution aqueuse d’acide oxalique (0,1 M). Ensuite, le mélange est agité pendant 30 minutes à température ambiante. L’excès de solvant a été évaporé sous la pression réduite jusqu’à le séchage. Ensuite, le solide obtenu est chauffé à 150 °C pendant toute la nuit et calciné à différentes températures : 400, 600 et 800 °C pour deux heures. La notation V/PN (1 mmol) signifie que le catalyseur phosphaté est préparé à partir de 10 g de PN et 1 mmol de NH4VO3. Pour les catalyseurs phosphatés V/PN (0,5, 1,5, 2 et 2,5 mmol), le protocole de préparation est identique, nous changeons seulement le nombre de mole de NH4VO3.
2.3 Techniques de caractérisation du V/PN
Diffraction des rayons X (DRX) sur poudre : les diffractogrammes ont été obtenus à partir d’échantillons pulvérulents à température ambiante à l’aide d’un diffractomètre SIMENS D 500 automatisé et équipé d’une anticathode de cuivre. La radiation Kα du cuivre a été utilisée avec une tension d’accélération de 30 kV. Le support est une plaque de verre. Spectrométrie infrarouge à transformé de Fourier (IRFT) : les spectres infrarouge ont été enregistrés à l’aide d’un spectromètre Brucker Vector-22 à transformée de Fourier équipé d’un détecteur de type MCT. Les analyses ont été effectuées par la technique de pastillage dans la gamme spectrale 400–4000 cm−1. Mesure de la surface spécifique et porosité : la mesure de la surface spécifique et de la porosité de V/PN (1 mmol) a été réalisée avec un appareil Micrometrics Instrument Corporation ASAP 2000 V2.02 qui utilise le procédé d’adsorption en multicouches de gaz d’azote à 77 K selon la théorie de Brunauer, Emett et Teller (BET). Microscopie électronique à balayage (MEB) : nous avons utilisé un microscope de type ZEISS DSM 960A pour observer la morphologie des catalyseurs et définir les tailles des particules. L’image est obtenue à partir du rayonnement réfléchi par suite de l’interaction électron–matière, avec une résolution de l’ordre de 25 à 4 nm et un grandissement variant de 4× à 30 000×.
2.4 Tests catalytiques
Les tests catalytiques sont conduits sous atmosphère de O2 dans des tubes de Schlenk (volume total 20 cm3) munis de septum et d’un robinet latéral relié à deux burettes d’oxygène (de 100 cm3). Le degré d’avancement de la réaction est ainsi suivi par variation du volume à pression atmosphérique. Le montage est accompagné d’une pompe à vide afin d’évacuer l’air du système avant l’introduction du dioxygène. L’agitation est effectuée à l’aide d’un barreau aimanté. Un bain thermorégulé permet de maintenir une température constante.
2.5 Protocole expérimental
Après l’introduction du catalyseur phosphaté et du solvant dans le tube de Schlenk, ce dernier est connecté au montage de mesures volumétriques. La solution est alors dégazée à température ambiante, sous vide et sous agitation puis l’oxygène moléculaire est introduit à la pression d’une atmosphère, le tube est ensuite plongé dans le bain thermorégulé à la température désirée. Après, le substrat est ajouté à l’aide d’une seringue en une seule fois.
2.6 Analyse chromatographique
Afin d’identifier et de quantifier les produits des tests catalytiques, nous avons utilisé la chromatographie en phase gazeuse type Girdel équipé d’un détecteur à ionisation de flamme DELSI 30 avec une colonne capillaire en verre OV1701 (longueur : 50 m ; diamètre : 0,25 mm) couplé à un intégrateur ENICA 10 (Delsi instruments). Les conditions opératoires de CPG : gaz vecteur : hélium (40 cm3/min) ; hydrogène (20 cm3/min) ; air (300 cm3/min). Température initiale de four : 70 °C ; température finale : 200 °C ; injecteur : 200 °C ; détecteur 200 °C. Un programme de température croissant est adopté à 5 °C/min.
Dans le cas de deux isomères (1S, 2S, 5S) et (1R, 2R, 5R) de la 2-hydroxypinan-3-one, nous avons utilisé une chromatographie avec une colonne chirale afin de séparer les produits ayant un carbone asymétrique différent. Ainsi, la chromatographie en phase gazeuse est de type Shimadzu GC-17A équipé d’un détecteur à ionisation de flamme avec une colonne capillaire chirale TRA-5 (longueur : 30 m ; diamètre : 0,25 mm). Les conditions opératoires de CPG : température initiale : 80 °C pour dix minutes ; la vitesse de la rampe : 10 °C/min ; la température finale : 220 °C ; température d’injection : 220 °C, température du détecteur : 250 °C ; gaz vecteur : He (25 mL/min).
Les produits ont été identifiés et quantifiés par comparaison avec des étalons commerciaux.
3 Résultats et discussion
3.1 Caractérisation du catalyseur V/PN
3.1.1 Diffraction des rayons X sur poudre
L’analyse par diffraction aux rayons X des échantillons V/PN (1 mmol) calcinés à 400, 600 et 800 °C montre qu’au fur et à mesure que la température de calcination augmente, les raies de diffraction deviennent de plus en plus résolues et intenses ; ce qui traduit une amélioration de la qualité cristalline de l’apatite étudiée (Fig. 1).

Diagrammes de RX de (a) PN et de V/PN (1 mmol) calciné à (b) 400 °C, (c) 600 °C et (d) 800 °C.
Le diagramme V/PN (1 mmol) calciné à 800 °C est sensiblement identique à celui du PN seul. Ces deux échantillons semblent être constitués d’une seule phase. Les raies sont indexées dans le système hexagonal, groupe d’espace P63/m. Nous remarquons également sur ce diagramme que les raies de diffraction du vanadium, ou de son oxyde, ne sont pas visibles, probablement, en raison de leur dépôt en petites particules avec une grande dispersion [13,14], ou à cause de sa faible teneur [15].
Notons que l’augmentation de la teneur en vanadium dans le support PN conduit à la désorganisation de la structure apatitique sans apparition de la phase V2O5.
3.1.2 Étude par spectrométrie infrarouge à transformée de Fourier (IRTF)
Nous avons enregistré les spectres IRTF de V/PN (1 mmol) calciné à 400, 600 et 800 °C (Fig. 2).
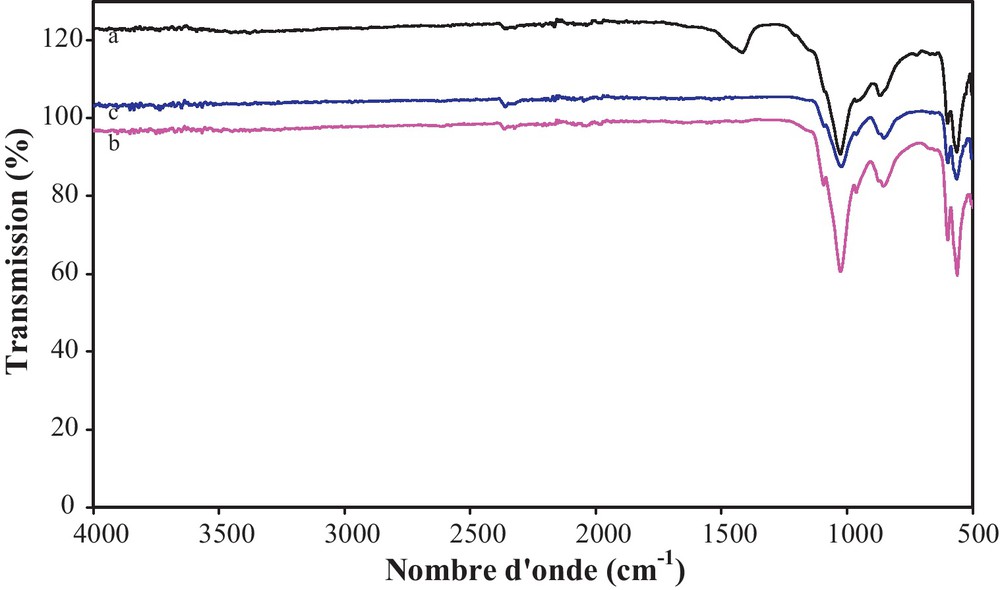
Spectres IRTF de V/PN (1 mmol) calciné à différentes températures : (a) 400 °C, (b) 600 °C et (c) 800 °C.
Le V/PN (1 mmol) a été caractérisé par diffraction des rayons X. Aucune nouvelle phase n’a été détectée. Cependant, la présence de V2O5 est observée par spectroscopie IR (Fig. 2). L’ensemble des spectres V/PN (1 mmol) calciné à 400, 600 et 800 °C met en évidence une bande intense avec un épaulement vers 870 cm−1. Cette bande est attribuée aux modes de vibration d’élongation antisymétrique des groupements V-O-V [16]. Les autres bandes de vibration de V2O5 sont masquées par celles du PN.
Par ailleurs, les spectres de ces échantillons montrent qu’au fur et à mesure que la température de calcination augmente, les bandes d’absorption deviennent bien résolues et celles de l’intervalle [1350–1600 cm−1] associées aux vibrations des ions carbonates, provenant de CO2 adsorbé sur l’apatite [17], diminuent en intensité ou disparaissent.
Tous les spectres IR de V/PN (1 mmol) (Fig. 2) montrent des bandes de vibration, situées vers 590 et 1000 cm−1 qui caractérisent les différents modes de vibrations des liaisons P-O de la structure apatitique [18,19]. En plus, la bande située à 875 cm−1 du groupement HPO42− [17] a été aussi détectée.
Il est intéressant de noter que l’absence des bandes à 3575 et à 630 cm−1, qui correspondent respectivement aux vibrations de valence symétriques (νs) et au mouvement vibrationnel des ions hydroxyles (νL) [20], montre que les structures du PN et de V/PN (1 mmol) sont similaires à celle de la fluoroapatite.
L’attribution de différentes bandes d’absorption IR de V/PN (1 mmol) calciné à 800 °C est donnée dans le Tableau 1.
Les bandes IR de V/PN (1 mmol) calciné à 800 °C.
| Mode de vibration | Position de la bande (cm−1) | Attribution |
| ν4 | 568 | Déformation antisymétrique des ions |
| 601 | PO43− | |
| νa | 860 | Élongation antisymétrique de V-O-V |
| ν1 | 983 | Élongation symétrique des ions PO43− |
| ν3 | 1035 | Élongation antisymétrique des ions |
| 1097 | PO43− |
3.1.3 Mesure de la surface spécifique et porosité
L’analyse des isothermes d’adsorption et désorption permet d’identifier les caractéristiques texturales du matériau étudié. Parmi les principaux paramètres qui peuvent être évalués, signalons la surface spécifique et le volume poreux spécifique.
Sur la Fig. 3 est reporté l’isotherme d’adsorption et désorption de l’azote sur le V/PN (1 mmol) calciné à 800 °C.

Isotherme d’adsorption et désorption de N2 à 77 K du V/PN (1 mmol) calciné à 800 °C.
Selon la classification de Brunauer, Deming, Deming et Teller (BDDT), cette isotherme est de type II avec une boucle d’hystérésis H1 pour des pressions relatives entre 0,85 et 1. Cela montre que le type des pores de solide est relativement large.
La surface spécifique de V/PN (1 mmol) a été calculée par la méthode de BET en utilisant des données d’adsorption dans la gamme de la pression relative de (P/P0) allant de 0,05 à 0,25. La valeur moyenne obtenue est d’ordre 7,18 m2/g, cette valeur est supérieure à celle de PN seul (S = 1,43 m2/g). Ainsi, la modification du PN par le vanadium entraîne l’augmentation de la surface spécifique, mais ; elle reste très faible en comparaison avec celle de la silice, l’alumine et l’hydrocalcite (100 à 300 m2/g).
La répartition poreuse du V/PN (1 mmol), responsable de la surface spécifique, a été étudiée par la méthode de Barrett, Jayer et Halenda (BJH). Cette technique détermine le rapport (volume/surface) de la distribution des pores en tenant compte du changement de l’épaisseur de la couche adsorbée et de la condensation dans les pores du solide. Cette étude a montré l’existence de pores de diamètre moyen d’environ 32 nm sur la surface de V/PN, ce qui indique que ces pores contribuent à la surface spécifique du fait que leur diamètre est inférieur à 80 nm. Le volume total des pores du V/PN mesuré par condensation de l’azote adsorbé est 0,0675 cm3/g. Cette valeur est supérieure à celle de PN seul qui ne dépasse pas 0,0055 cm3/g.
3.1.4 Microscopie électronique à balayage
Afin de mieux analyser la microstructure du PN et du V/PN (1 mmol) calciné à 800 °C, nous avons examiné le solide par MEB. Les images obtenues nous ont permis de comparer les microstructures de l’apatite modifiée par le vanadate à celle de l’apatite de référence (PN). Les micrographies obtenues sont représentées sur la Fig. 4.

Clichés de MEB, à différents agrandissements, de PN tout seul (colonne a) et V/PN (1 mmol) calciné à 800 °C (colonne b).
L’examen de ces images nous révèle que le PN est caractérisé par de microparticules en forme d’ovoïde ou de bâtonnet hexagonal, témoignant de la structure cristalline hexagonale du matériau (Fig. 4 colonne a). La présence d’agrégats au sein desquels les cristaux apparaissent imbriqués les uns dans les autres traduit l’existence de macles. Les agglomérats ont des dimensions très variables. Les plus gros peuvent atteindre des tailles de l’ordre de 100 μm. Pour V/PN (1 mmol) calciné à 800 °C, un net changement de la morphologie est constaté (Fig. 4 colonne b). Les grains se présentent alors sous forme de particules irrégulières et hétérogènes, formant de nombreux agglomérats de tailles différentes variant de 10 à 100 μm. L’examen de leur surface indique aussi qu’ils sont composés de petites particules fortement compactées de quelques micromètres de diamètre.
3.2 Oxydation catalytique de cycloalcanones α-substituées par le système V/PN/O2
3.2.1 Effet de la teneur en vanadium dans le PN
Nous avons préparé une série de catalyseurs V/PN avec des concentrations différentes en vanadate d’ammonium (0,5 ; 1 ; 1,5 ; 2 et 2,5 mmol) (voir la partie expérimentale). Tous les échantillons obtenus ont été calcinés à 800 °C.
Les matériaux préparés ont été testés dans la coupure oxydante de la liaison C-C de la 2-méthylcyclohexanone, 1, dans un mélange acide acétique/eau, à 60 °C et à pression atmosphérique du dioxygène. Le Tableau 2 donne les résultats préliminaires obtenus.
Effet de la concentration en vanadium sur le rendement en 6-oxoheptanoïque 2a.
| Test | Conc. de V dans PN (mmol) | O2 cons./Subst. cons. (mol/mol) | t (h) | Conversion (%) | Rendement en 2 (%) |
| 1 | 0,5 | 0,9 | 10 | 93 | 91 |
| 2 | 1 | 1,01 | 8 | 99 | 98 |
| 3 | 1,5 | 1,04 | 8 | 98 | 97 |
| 4 | 2 | 1,05 | 8 | 98 | 96 |
| 5 | 2,5 | 1,1 | 8 | 99 | 98 |
a Conditions : substrat (0,8 mmol), AcOH/H2O (4,5/0,5 ; mL), V/PN (1 mmol) (0,1 g), T = 60 °C, p(O2) = 0,1 MPa.
Il apparaît que la concentration de 1 mmol en vanadium est la valeur optimale. L’augmentation de la concentration du vanadate d’ammonium modifiant le PN conduit à l’amélioration du rendement en 2 après huit heures de réaction (98 %). Cependant, une concentration de vanadate supérieure à 1 mmol ne varie quasiment pas le rendement et le temps de la réaction (tests 3, 4 et 5). Nous avons montré précédemment (vide supra) que la structure apatitique se détruit lorsqu’on modifie le PN par une concentration de vanadate supérieure à 1 mmol. En plus, une grande teneur de vanadium pourrait conduire à la formation des espèces polymériques du vanadium, ces derniers sont moins actifs que leurs homologues monomériques [21]. Ainsi, ces deux facteurs pourraient expliquer le fait que l’activité de V/PN (1 mmol) n’a pas été améliorée à des concentrations élevées en vanadium.
3.2.2 Influence de la masse du catalyseur
Nous avons déjà montré dans le cas du PN tout seul que la masse du catalyseur influence le rendement et la vitesse de la réaction [22]. De même, nous avons testé quatre masses différentes de V/PN (1 mmol) dans l’oxydation de la 2-méthylcyclohexanone. Les résultats obtenus sont mentionnés dans le Tableau 3.
Oxydation de la 2-méthylcyclohexanone par le système ‘V/PN/O2/AcOH/H2O’ : effet de la masse de V/PN (1 mmol)a.
| Test | Masse de V/PN (g) | O2 cons./Subst. cons. (mol/mol) | Conversion (%) | Rendement en 2 (%) |
| 6 | 0,01 | 0,55 | 45 | 43 |
| 7 | 0,05 | 0,6 | 55 | 53 |
| 2 | 0,1 | 1,01 | 99 | 98 |
| 8 | 0,5 | 1,01 | 99 | 99 |
| 9b | – | – | – | – |
| 10c | 0,1 | – | – | – |
a Conditions : substrat (0,8 mmol), AcOH/H2O (4,5/0,5 ; mL), temps de réaction (8 h), T = 60 °C, p(O2) = 0,1 MPa.
b Réaction conduite sans catalyseur, après 24 heures de réaction.
c Réaction effectuée en l’absence du dioxygène, après 24 heures de réaction.
Nous remarquons qu’une faible quantité de V/PN (1 mmol) est capable de couper la liaison C-C de la 2-méthylcyclohexanone. Cependant les rendements sont moyens (tests 6 et 7). Une masse de 0,1 g conduit à la formation sélective du cétoacide ; le rendement est de 98 %, après huit heures de réaction (test 2). À titre de comparaison, le PN tout seul donnait un rendement de 20 % dans les mêmes conditions [22]. Notons qu’une grande quantité de V/PN n’améliore pas les résultats (test 8).
Il est à noter qu’en l’absence du catalyseur ou de l’oxygène moléculaire, l’oxydation de 1 n’a pas eu lieu après 24 heures de réaction (tests 9 et 10) (Fig. 5).
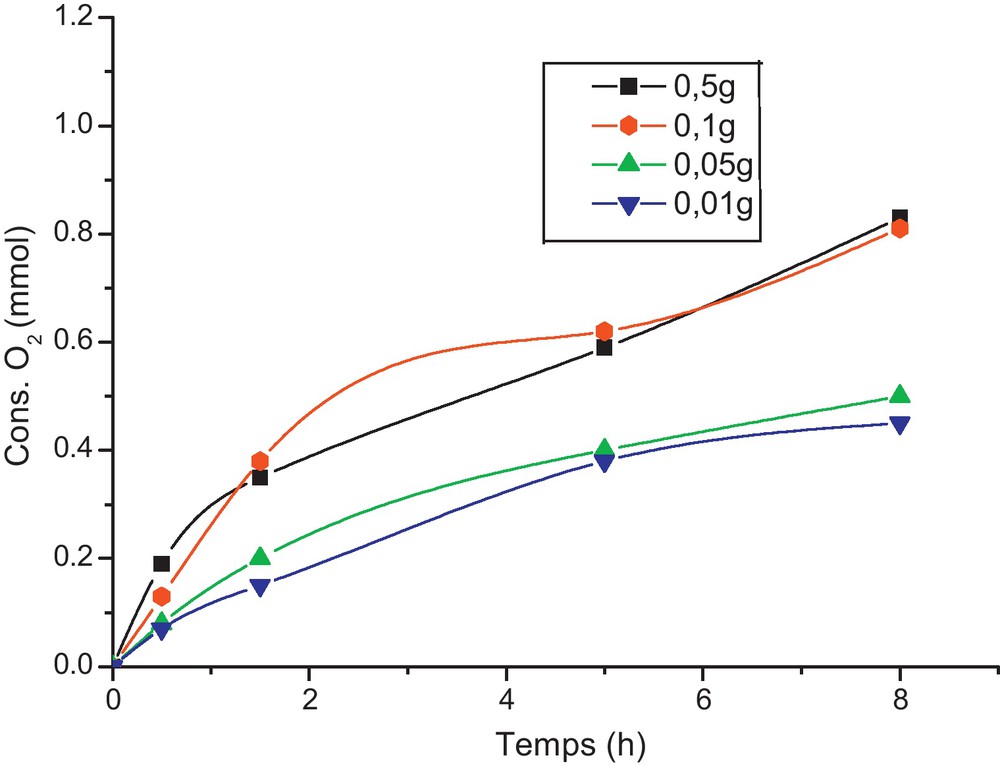
Suivi de la consommation du dioxygène lors de l’oxydation de la 2-méthylcyclohexanone par le système ‘V/PN/O2/AcOH/H2O’.
Le profil de toutes les courbes, même pour les faibles quantités de V/PN, ne présente pas un temps de latence comme dans le cas du PN tout seul [22].
3.2.3 Effet du solvant
Il est à noter qu’en catalyse hétérogène solide/liquide, l’influence du solvant est un facteur essentiel. En plus de son rôle comme activant ou inhibiteur de la réaction, dans le cas d’un catalyseur supporté, le solvant peut provoquer un décrochage partiel ou total de la phase active ; le phénomène de lessivage.
D’après le Tableau 4, les résultats semblent difficiles à interpréter en basant uniquement sur le moment dipolaire et la constante diélectrique des solvants. Les bons rendements en cétoacide, 2, ont été obtenus en présence des solvants polaires protiques (AcOH/H2O et MeOH). Cependant, la réaction est complètement inhibée dans le cas des solvants apolaires (hexane et toluène). Toutefois, le catalyseur est actif en présence de MeCN, CH2Cl2 et THF (solvants polaires aprotiques). Dans le solvant mixte AcOH/H2O, l’activité catalytique du V/PN (1 mmol) devient très intéressante. Ce mélange offre deux avantages : l’acidité qui favorise vraisemblablement l’énolisation de la 2-méthylcyclohexanone [23] et l’activation de la surface du catalyseur par les molécules d’eau [24,25].
Oxydation de la 2-méthylcyclohexanone par le système ‘V/PN/O2’ : effet du solvanta.
| Solvant (mL) | AcOH/H2O (4,5/0,5) | MeOH (5)b | MeCN (5) | CH2Cl2 | THF | Toluène (5) | Hexane (5) |
| Rendement en 2 (%) | 98 | 86 | 80 | 70 | 38 | 0 | 0 |
| Temps (h) | 8 | 24 | 24 | 24 | 24 | 24 | 24 |
| Cons. O2 mmol | 0,81 | 0,73 | 0,7 | 0,58 | 0,31 | 0 | 0 |
a Conditions : substrat (0,8 mmol), V/PN (1 mmol) (0,1 g), T = 60 °C, p(O2) = 0,1 MPa.
b Estérification in situ du cétoacide en 6-oxohexanoate de méthyle.
3.2.4 Étude du recyclage de V/PN
L’intérêt d’un catalyseur hétérogène réside dans sa réutilisation plusieurs fois sans perte de l’activité catalytique. Pour cette étude, après la fin du premier test catalytique, le V/PN (1 mmol) a été récupéré par filtration, lavé par l’acétone, séché à 150 °C et calciné à 800 °C durant une heure.
Le V/PN (1 mmol) est réutilisé trois fois successives dans l’oxydation de la 2-méthylcyclohexanone. Les résultats obtenus sont illustrés sur la Fig. 6.
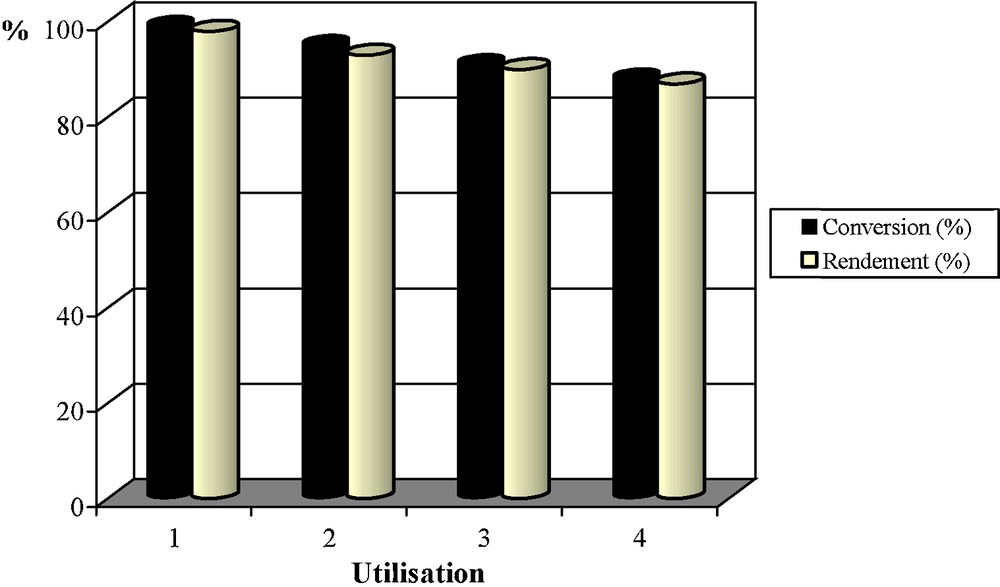
Recyclage du système ‘V/PN/O2/AcOH/H2O’ dans la coupure oxydante de la 2-méthylcyclohexanone (voir conditions dans le Tableau 2, test 2).
Nous constatons une légère diminution de l’activité catalytique du V/PN (1 mmol) d’un test à l’autre. L’analyse chimique de la solution après le premier test catalytique révèle un relargage de 12,4 % de vanadium (diminution de la teneur massique initiale en vanadium pour le V/PN natif de 0,22 à 0,19 %). Donc, le lessivage des entités métalliques n’est pas trop important. Il est à préciser que l’on ne détecte pas de lessivage après les premier et deuxième tests. Lors des essais suivants, la conversion de la 2-méthylcyclohexanone reste importante ainsi que les sélectivités en acide 6-oxoheptanoïque qui sont globalement maintenues dans le système catalytique. L’analyse élémentaire et la caractérisation physicochimique des matériaux récupérés après chaque test sont en cours.
3.2.5 Coupure oxydante de différentes α-cycloalcanones par le système ‘V/PN/O2/AcOH/H2O’
Le système catalytique ‘V/PN/O2/AcOH/H2O’ a été étendu à d’autres cétones cycliques α-substituées. Le Tableau 5 regroupe les résultats obtenus.
Coupure oxydante des cétones cycliques α-substituées par le système ‘V/PN/O2/AcOH/H2O’a.
| Test | Substrat | Cons. O2 (mmol) | Temps (h) | Conversion (%) | Produit (rendement [%]) |
| 11 | 0,83 | 8 | 97 | ||
| 12 | 0,78 | 12 | 92 | ||
| 2 | 0,81 | 8 | 99 |
a Conditions : substrat (0,8 mmol), AcOH/H2O (4,5/0,5 ; mL), V/PN (1 mmol) (0,1 g), T = 60 °C, p(O2) = 0,1 MPa.
Ces résultats expérimentaux montrent clairement l’efficacité du système ‘V/PN/O2/AcOH/H2O’ dans la coupure oxydante de différentes cycloalcanones α-substituées.
D’ailleurs, ce système a montré sa supériorité par rapport à ‘PN/O2’ [22] ; dans le cas de la 2-phénylcyclohexanone où le rendement en cétoacide est d’ordre 90 % avec un temps de réaction qui ne dépasse pas 12 heures (test 12). Généralement, les rendements et les vitesses de réaction sont améliorés en présence de V/PN/O2, ce qui confirme l’utilité de la modification du PN par le vanadium. Nous avons constaté que la consommation de O2 ramenée au nombre de moles de substrat transformé est proche de la stoechiométrie, ce qui indique que le système ‘V/PN/O2/AcOH/H2O’ est régiosélectif.
3.3 Oxydation des α-hydroxycétones par les systèmes PN/O2 ou V/PN/O2
Dans ce paragraphe, nous présentons les résultats d’oxydation de différentes α-hydroxycétones (appelés aussi α-cétols) par les systèmes ‘PN/O2’ ou ‘V/PN/O2’. Les études antérieures effectuées au sein de notre équipe ont montré que la 2-hydroxycyclohexanone, 7, n’est pas un intermédiaire clé lors de la réaction d’oxydation de la cyclohexanone en acide adipique. D’autres α-cétols ont été testés pour une étude comparative entre les performances des deux systèmes catalytiques : ‘PN/O2’ et ‘V/PN/O2’. Aussi, nous avons envisagé des applications dans le domaine de la chimie fine, notamment dans la valorisation des substances naturelles dérivées du pin.
3.3.1 Oxydation de la 2-hydroxycyclohexanone
La coupure oxydante de la 2-hydroxycyclohexanone a été effectuée à pression atmosphérique de dioxygène (régime statique) et en présence d’une quantité catalytique de PN seul ou de PN modifié au vanadium (Schéma 2 et Tableau 6).

Oxydation de la 2-hydroxycylohexanone par les systèmes ‘PN/O2’ ou ‘V/PN/O2’a.
| Test | Catalyseur (g) | O2 cons./Subst. cons. (mol/mol) | t (h) | Conversion (%) | Rendement en 8 (%)b |
| 13 | PN (0,5 g) | 0,9 | 24 | 88 | 86 |
| 14 | V/PN (0,1 g) | 1,01 | 0,5 | 100 | 99 |
| 15c | PN (0,5 g) | – | 24 | – | – |
| 16c | V/PN (0,1 g) | – | 24 | – | – |
| 17d | – | – | 24 | – | – |
a Conditions : substrat (0,8 mmol), AcOH/H2O (4,5/0,5 ; mL), T = 60 °C, p(O2) = 0,1 MPa.
b Résultats obtenus après estérification par le MeOH.
c Réaction conduite en l’absence du dioxygène.
d Réaction effectuée sans catalyseur.
La consommation du dioxygène a été suivie à l’aide d’un montage permettant des mesures volumétriques (voir la partie expérimentale). En l’absence de PN ou de V/PN (1 mmol), les réactions n’ont pas eu lieu (test 17). De même, dans un milieu dépourvu de dioxygène, la 2-hydroxycyclohexanone n’est pas convertie après 24 heures de réaction (tests 15 et 16).
Les résultats du Tableau 6 montrent que la coupure oxydante de la 2-hydroxycyclohexanone est régiosélective avec le PN ou le V/PN (1 mmol). Dans le cas du PN, l’oxydation de 7 conduit sélectivement à l’adipate de méthyle, 8, avec un rendement de 86 %, après 24 heures de réaction (test 13). Dans le solvant AcOH/H2O, l’acide carboxylique obtenu doit être estérifié afin de pouvoir être quantifié sur les colonnes retenues. L’estérification est effectuée en ajoutant du MeOH aux mélanges réactionnels en présence de H+ pendant 24 heures.
L’oxydation de 7 par le système ‘PN/O2/MeOH’ fournit directement le diméthyle adipate avec un rendement de 54 %.
Avec le catalyseur V/PN (1 mmol), la conversion de 7 est quantitative et la sélectivité est très élevée pendant un temps trop court (test 14).
3.3.2 Oxydation de différents α-cétols aromatiques en présence du système ‘PN/O2’ ou ‘V/PN/O2’
Les résultats expérimentaux de la coupure oxydante de la liaison C(O)-C(OH)H de différents α-cétols étudiés sont regroupés dans le Tableau 7 ci-après.
Coupure oxydante de différents α-cétols par les systèmes ‘PN/O2/AcOH/H2O’ ou ‘V/PN/O2/AcOH/H2O’a.
| Test | Substrat | Catalyseur | O2 cons./Subst. cons. (mol/mol) | Conversion (%) (t en h) | Produits (rendement [%]) | |
| PhCOOH 13 | ||||||
| 18 | PN | 1,06 | 98 (24) | (98) | ||
| 19 | V/PN | 1 | 100 (0,5) | (100) | ||
| PhCOOH | PhCOH | |||||
| 20 | PN | 0,67 | 88 (24) | (88) | (88) | |
| 21 | V/PN | 0,70 | 90 (12) | (90) | (90) | |
| PhCOOH | ||||||
| 22 | PN | 0,40 | 30 (24) | (30) | ||
| 23 | V/PN | 0,60 | 55 (24) | (55) | ||
| PhCOOH | C6H10(=0) | |||||
| 24 | PN | 0 | 0 (24) | (0) | (0) | |
| 25 | V/PN | 0,50 | 50 (24) | (50) | (50) |
a Conditions : substrat (0,8 mmol), AcOH/H2O (4,5/0,5 ; mL), PN (0,5 g), V/PN (1 mmol) (0,1 g), T = 60 °C, p(O2) = 0,1 MPa.
En présence de PN, la 2-hydroxyacétophénone réagit facilement pour conduire sélectivement à l’acide benzoïque, 13 (98 %) (test 18). Avec le catalyseur V/PN (1 mmol), le même rendement en acide benzoïque est obtenu pendant un temps trop court (test 19). Pour les deux catalyseurs, la coupure oxydante est régiosélective.
D’après la littérature, l’oxydation de la 2-hydroxy-2-phénylacétophénone (benzoïne) 10, dans le mélange acide acétique/eau, conduit à la formation de l’acide benzoïque, du benzaldéhyde et du benzile (Schéma 3) [26].
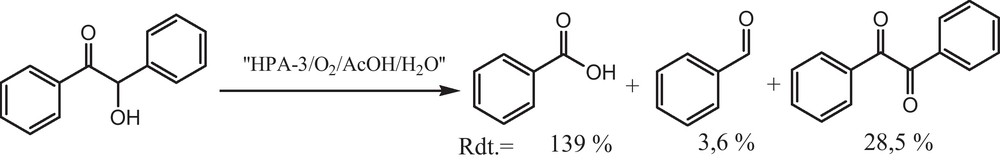
Oxydation de la benzoïne [26].
Dans notre cas, la coupure oxydante de la benzoïne par le système ‘PN/O2’ ou ‘V/PN/O2’ conduit à l’acide benzoïque et au benzaldéhyde avec des rendements en acide voisin de 90 %, après 12 heures de réaction (test 21). Il apparaît qu’avec le PN tout seul les performances catalytiques sont nettement inférieures ; le rendement de 88 % en 13 n’est atteint qu’après 24 heures de réaction (test 20).
La coupure oxydante de deux α-cétols tertiaires : 2-hydroxy-2-méthylpropiophénone, 11, et 1-hydroxycyclohexylphénylcétone, 12, conduit à la formation de l’acide benzoïque et de la cétone correspondante (Schéma 4).
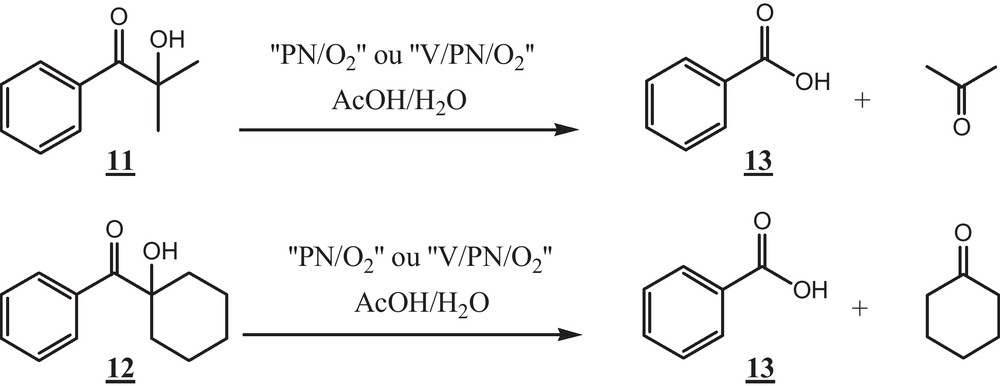
Par ailleurs, la coupure oxydante de la 2-hydroxy-2-méthylpropiophénone, 11, par le PN tout seul conduit à la formation de l’acide benzoïque avec un rendement faible de 30 % (test 22). Nous soulignons que l’acétone terminale n’est pas facilement oxydable contrairement à la cyclohexanone [27]. De même, la réactivité de 11 (cétol tertiaire) est inférieure à celle des cétols primaires et secondaires.
En présence du système PN, la 1-hydroxycyclohexylphénylcétone 12 ne réagit pas (test 24). Cependant, en présence de V/PN (1 mmol) ce substrat est converti partiellement en acide benzoïque (50 %) et cyclohexanone (50 %), après 24 heures de réaction (test 25).
3.3.3 Application en chimie fine
Dans l’objectif de valoriser les substances naturelles, nous avons mis au point la synthèse diastéréosélective du 3-acétyl-2,2-diméthylcyclobutaneacétique (appelé aussi l’acide pinonique). Ce composé, à condition d’être optiquement pur, permet de synthétiser des acides aminés et les peptides correspondants dotés de motifs cyclobutane chiraux [28]. De tels produits isolés à partir de sources naturelles présentent en effet d’intéressantes propriétés biologiques et ont des applications thérapeutiques [29,30].
La plupart des méthodes utilisées pour l’obtention de l’acide pinonique (dérivé de l’α-pinène), à partir de la coupure oxydante de la double liaison endocyclique de l’alcène [31], sont stoechiométriques [32–34]. Ces méthodes n’aboutissent pas toujours à la stéréosélectivité recherchée. Néanmoins, quelques processus catalytiques ont été décrits. Ainsi, Moglioni et al. ont montré que le (-)-α-pinène peut être converti en acide (-)-cis-pinonique en présence de periodate de sodium et de quantités catalytiques de RuCl3 [35]. Récemment, Brégeault et al. ont pour la première fois synthétisé l’acide pinonique en présence de l’oxygène moléculaire et avec des quantités catalytiques de ‘H6[PMo9V3O40], aq’ (HPA-3) dans des conditions homogènes douces [26]. En effet, le HPA-3 catalyse la coupure oxydante de (1S, 2S, 3S) 2-hydroxypinan-3-one (ou son énantiomère) pour donner l’acide pinonique avec un rendement de 100 % et la diastéréosélectvité est excellente (99/1) [26]. Cependant, aucun exemple de la catalyse hétérogène n’a été décrit pour la synthèse de l’acide pinonique.
Les résultats obtenus avec les différents α-cétols, nous ont encouragé à tester l’activité catalytique de nos systèmes ‘PN/O2/AcOH/H2O’ et ‘V/PN (1 mmol)/O2/AcOH/H2O’ dans l’oxydation de la liaison C-C de deux énantiomères commerciaux de la 2-hydroxypinan-3-one, 14 (1S, 2S, 3S) et 15 (1R, 2R, 3R) (Schéma 5).
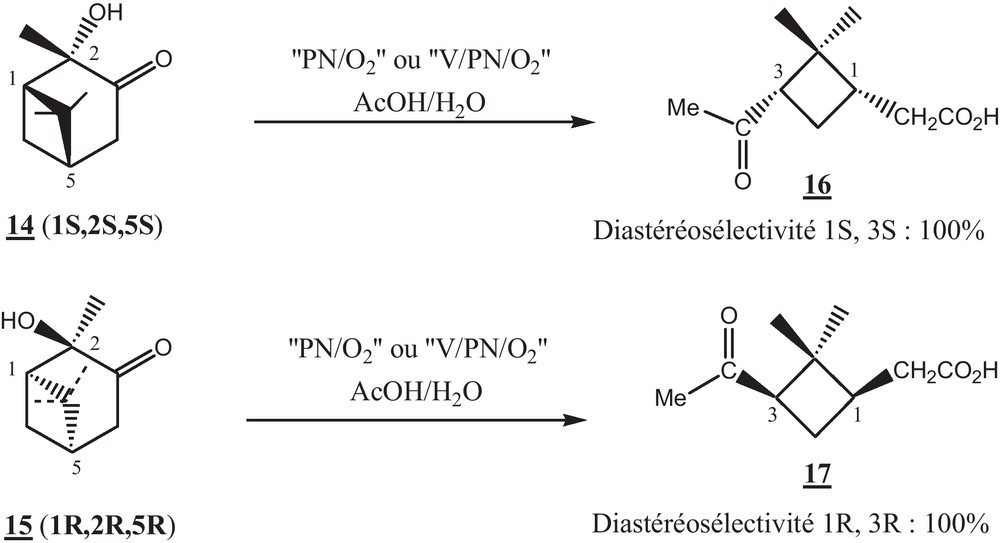
La coupure oxydante de ces deux énantiomères 14 et 15 conduit à la formation d’un seul isomère de l’acide pinonique 16 et 17, respectivement. Ces derniers ont été identifiés par la chromatographie en phase gazeuse équipée d’une colonne chirale (voir la partie expérimentale). D’après les travaux antérieurs, dans le milieu acide, l’acide pinonique subit une épimérisation au niveau du carbone no 3 (no 1 dans le cétol, voir Schéma 5) inclus dans le cycle à quatre chaînons et lié au groupement acyle du groupement pinonique, ce qui conduit à la formation de deux diastéréo-isomères de configuration (1S, 3S) et (1S, 3R) [36] (Tableau 8).
Oxydation de la 2-hydroxypinan-3-one [14 (1S, 2S, 3S) et 15 (1R, 2R, 3R)] par les systèmes ‘PN/O2/AcOH/H2O’ ou ‘V/PN/O2/AcOH/H2O’a.
| Test | Catalyseur (g) | O2 cons./Subst. cons. (mol/mol) | t (h) | Conversion (%) | Rendement (diastéréosélectivité) (%) |
| 26 | PN (0,5) | 0,8 | 24 | 90 | 90 (100) |
| 27 | V/PN (0,1) | 0,8 | 15 | 93 | 92 (100) |
| 28 | PN (0,5) | 0,78 | 24 | 91 | 90 (100) |
| 29 | V/PN (0,1) | 0,81 | 15 | 93 | 91 (100) |
a Conditions : substrat (0,8 mmol), AcOH/H2O (4,5/0,5 ; mL), T = 60 °C, p(O2) = 0,1 MPa.
Cependant, dans le cas du système ‘PN/O2/AcOH/H2O’ l’α-cétol tertiaire 14 conduit à la formation d’un seul diastéréo-isomère ; l’acide pinonique 16 (1S, 3S) avec un bon rendement en 24 heures (test 26). De même, la coupure oxydante de l’énantiomère 15 par le système ‘PN/O2’ conduit à la formation diastéréosélective de l’acide pinonique 17 (1R, 3R) (test 27). Dans le cas du système ‘V/PN/O2/AcOH/H2O’, la diastéréosélectivité n’est pas affectée mais la vitesse de la réaction est améliorée. Ainsi, la 2-hydroxypinan-3-one (1S, 2S, 5S) 14 et son énantiomère 15 (1R, 2R, 5R) conduisent à l’acide pinonique 16 (1S, 3S) et 17 (1R, 3R) respectivement après 15 heures de réaction (tests 28 et 29).
4 Conclusion
Nous avons développé de nouveaux systèmes catalytiques propres mettant en œuvre le phosphate naturel ; ‘PN/O2/AcOH-H2O’ ou ‘V/PN/O2/AcOH-H2O’, dans la coupure oxydante de la liaison C-C des cycloalcanones α-substituées et des α-cétols. Les conditions opératoires ne sont pas exigeantes, elles sont de mise en œuvre simple.
Nous avons démontré que la modification du PN par le vanadium améliore les performances catalytiques du système. L’oxydation de la 2-hydroxypinane-3-one, réputée exigeante, par les deux systèmes est aussi très efficace. Cela ouvre des perspectives intéressantes pour une préparation plus sélective de l’acide adipique moyennant un passage forcé de la cyclohexanone à la 2-hydroxycyclohexanone.
Du fait de la prise de conscience des dangers de pollution de l’environnement, les systèmes catalytiques ‘PN/O2’ et ‘V/PN/O2’ s’avèrent très prometteurs. Il en est ainsi de PN pour l’oxydation régiosélective par le dioxygène dans la mesure où les processus deviennent hautement sélectifs et les problèmes de séparation facile à résoudre. De plus, les très faibles quantités de matière de catalyseur utilisées permettent d’envisager des systèmes propres et économiques. Cependant, il est encore très difficile de rentrer dans l’intimité du mécanisme catalytique et d’envisager un ensemble de réactions permettant d’interpréter les résultats.
Remerciement
Ce travail est partiellement supporté par le comité mixte franco-marocain (PICS No 562). M.D. remercie le CNRST du Maroc pour l’attribution de la Bourse d’Excellence.