

1 Introduction
A tandem reaction utilizing a single catalyst to mediate more than one transformation in a selective manner has been recognized as one of the best methods for the synthesis of complex skeletal organic compounds [1–6]. Tandem processes are very efficient in terms of work-up and catalyst utilization, and provide the advantage that intermediates do not require isolation where they are unstable or difficult to handle (e.g., toxic, volatile or prone to polymerize) [7,8].
Quinoxalines and pyrido[2,3-b]pyrazines are the subject of considerable interest from both academic and industrial perspectives because they are significant intermediates for the manufacturing of pharmaceuticals and advanced materials [9–18]. Conventionally, these heterocycles are synthesized by a double condensation reaction of 1,2-dicarbonyls with arene-1,2-diamines [19,20]. The use of α-hydroxy ketones instead of 1,2-dicarbonyls in this reaction, as an alternative route, has been recently developed [21–28]. They are oxidatively cyclized with diamines in the presence of transition metal oxides or oxygen to yield quinoxalines. Although these methods have some synthetic advantages individually, they are still plagued by some limitations such as expensive catalysts and protocols, excessive amounts of stoichiometric promoters, not readily available materials, use of organic solvents, and difficulty of recovery of high boiling solvents. Therefore, the development of a practical, efficient, cost-effective and greener alternative using a sustainable protocol is still of interest for the preparation of these compounds.
In recent years, polyethylene glycols (PEGs) have received increasing attention as benign reaction media in organic synthesis due to their unique properties, including very low volatility, biodegradability, high stability towards acids, bases, and high temperature, and availability in high quantities at low prices. Furthermore, their well-known low toxicity makes them greener versions of conventional halogenated solvents [29–33]. From the perspective of microwave chemistry [34–37], the polar nature of PEGs and their application in phase transfer catalysis make them attractive green reaction media in microwave-assisted organic reactions [38–44].
As part of our continuous efforts to develop sustainable processes for benzo[N,N]-heterocyclic condensation [45–52], herein, we introduce an oxidative polar paste of [(NH4)6Mo7O24·4H2O]–PEG as an efficient catalytic system for the tandem synthesis of quinoxaline derivatives from α-hydroxy ketones under microwave conditions. The advantage of the present protocol is the use of ammonium heptamolybdate tetrahydrate as a cheap, air stable and commercially available catalyst [53] and PEG 300 as the benign reaction medium under microwave irradiation in an open vessel without the aid of air, O2 or an inert atmosphere.
2 Experimental
2.1 General remarks
A laboratory microwave oven from Landgraf Laborsysteme HLL GmbH (model MW 3100, Langenhagen, Germany) equipped with a magnetic stirrer operating at 2450 MHz was used for all syntheses. Microwave heating was carried out using 420 W (70% of maximum power). The temperature inside the open vessel was monitored using a Precision Temperature Logger EBI-2 T Type 311 (possessing an external probe) from ebro® Electronic GmbH and Co KG. The recorded temperature after 15 min irradiation was 90 °C.
The 1H and 13C NMR spectra were recorded on a Bruker 500-MHz spectrometer using TMS as internal standard and CDCl3 as solvent. A Fisons instruments gas chromatograph 8000 connected to a mass detector (Trio 1000) with 70 eV was used.
2.2 General procedure for preparation of quinoxalines and pyrido[2,3-b]pyrazines under microwave irradiation
In a typical reaction, a mixture of arene-1,2-diamine (1 mmol), α-hydroxy ketones (0.5 mmol) and PEG 300 (0.2 g) was mixed thoroughly with 0.10 mmol of [(NH4)6Mo7O24·4H2O] for a few minutes in order to ensure complete homogeneity. The mixture was then inserted into the microwave chamber and the reaction was carried out in an open vessel maintaining the power at 420 W (70% of maximum power) for 15 min. The reaction mixture was washed with ethyl acetate (10 mL) and the mixture was filtered off. The resulting solid mixture was washed with dichloromethane (10 mL) and filtered. The combined organic medium was subjected to GC in order to find out the conversion and then removed with a rotary evaporator under reduced pressure to afford the product 1. The crude products were purified by column chromatography using ethyl acetate/hexane (2:8 v/v) [dichloromethane/ethanol (9:1 v/v) for products 1f–1j] to afford pure products for analytical measurements. The products were identified by comparison of their NMR and mass spectra with authentic samples.
3 Results and discussion
In an initial study, microwave-promoted oxidative cyclization of benzoin (0.5 mmol) with benzene-1,2-diamine (1 mmol) was chosen as a model reaction to afford the product 1a (Scheme 1). As all employed materials are non-volatile, the reactions were carried out under open-vessel microwave conditions (420 W).
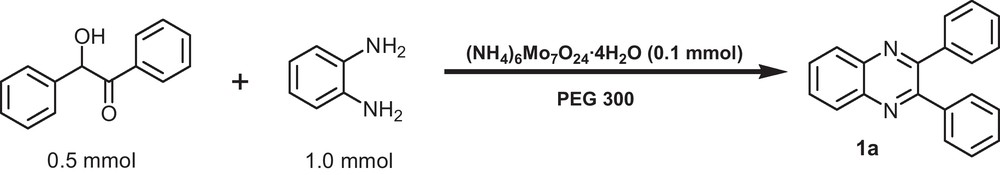
Oxidative cyclization of benzoin with benzene-1,2-diamine using [(NH4)6Mo7O24·4H2O] as catalyst in PEG 300.
To optimize the reaction conditions, the effects of catalyst loading, the PEG amount and the irradiation time were investigated (Table 1). The microwave-promoted reaction in the absence of PEG or (NH4)6Mo7O24·4H2O or both of them afforded the product 1a in low yield (Table 1, entries 1, 5, 10). In contrast, the yield of 1a in the presence of catalyst and PEG sharply increased to show their synergistic cooperation in this tandem process (Table 1, entries 2–4).
Optimization of the reaction conditions on the model reaction.
| Entry | Catalyst (mmol) | PEG 300 (g) | Conditions | Time (min) | Conversion (%) |
| 1 | – | – | MWIa | 15 | 36 |
| 2 | 0.05 | 0.50 | MWI | 15 | 87 |
| 3 | 0.10 | 0.50 | MWI | 15 | 93 |
| 4 | 0.15 | 0.50 | MWI | 15 | 90 |
| 5 | 0.10 | – | MWI | 15 | 46 |
| 6 | 0.10 | 0.70 | MWI | 15 | 82 |
| 7 | 0.10 | 0.10 | MWI | 15 | 54 |
| 8 | 0.10 | 0.20 | MWI | 15 | 100 |
| 9 | 0.10 | 0.20 | MWI | 10 | 72 |
| 10 | – | 0.20 | MWI | 15 | 23 |
| 11 | 0.10 | 0.20 | 90 °C | 15 | 100 (81)b |
| 12 | 0.10 | 0.20 | 25 °C | 300 | 10 |
a Microwave irradiation / 420 W / 90 °C
b Although quantitative conversion was observed, the selectivity of the reaction for the target product 1a is 81%.
In the study of catalyst loading, the optimum amount of (NH4)6Mo7O24·4H2O was found to be 0.1 mmol of catalyst per 0.5 mmol of benzoin (Table 1, entries 2–4). The optimum amount of PEG was 0.2 g per 0.1 mmol of catalyst. Higher and lower amounts of PEG gave the lower yield of 1a (Table 1, entries 3, 6–8).
In the absence of microwave irradiation, lower yield of 1a (81%) was obtained at 90 °C for 15 min (Table 1, entry 11), due to the formation of some by-products such as benzimidazole homologues of 1a. Enhanced selectivity under microwave heating compared to the conventional one, may be in fact due to the “non-thermal microwave effects” in this reaction [54].
This indicates that a significant difference in terms of selectivity persist between experiments performed under microwave heating and conventional thermal heating. In other words, the acceleration of the reaction induced by microwave irradiation, leads to no decomposition products through a very clean reaction within a short reaction time.
Accordingly, a convenient combination of catalyst and PEG led to quantitative yield of 1a within 15 min microwave irradiation (Table 1, entry 8).
To expand the generality of this novel catalytic method, various arene-1,2-diamines and α-hydroxy ketones were tested under the optimized conditions (Scheme 2) and the results are presented in Table 2. Most of the reactions took 15 minutes to complete (i.e. 100% conversion) under microwave conditions, although the yields were highly dependent on the substrate used. To establish the scope and limitations of catalyst for quinoxaline formation, the condensation between 4-nitrobenzene-1,2-diamine and 4,4′-dimethoxybenzoin was considered. The presence of the methoxy group in 4,4′-dimethoxybenzoin reduces the electrophilicity of the carbonyl carbon through resonance and the strong electron-withdrawing property of the nitro group in 4-nitrobenzene-1,2-diamine decreases the nucleophilicity of the amine group [24,45–50]. Accordingly, the combination of these substrates provided the corresponding product 1i in 48% yield (Table 2, entry 9).
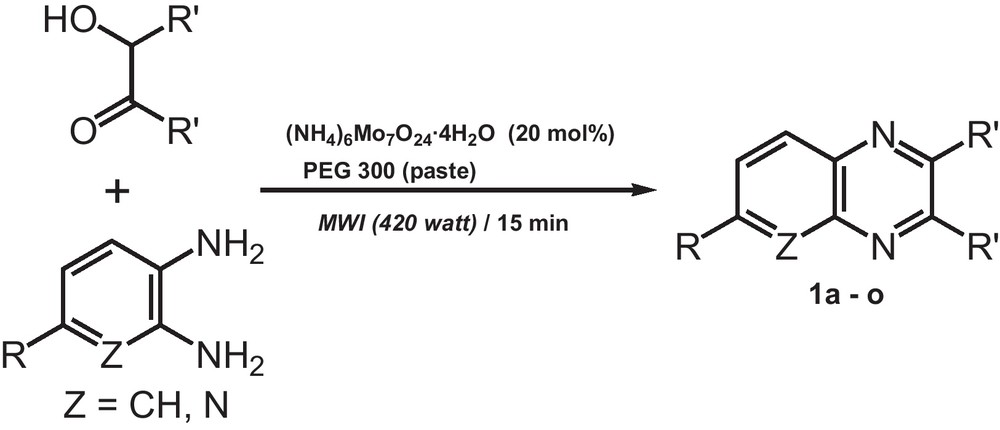
Access to various benzo[N,N]-heterocycles from α-hydroxy ketones via microwave-induced polar paste catalyst system.
Evaluation of the reaction scope.
| Entry | R | R’ | Z | Product | Conversion (%) | Yield (%)a | TOF (h−1)b |
| 1 | H | Ph | CH | 1a | 100 | 98 | 20 |
| 2 | Me | Ph | CH | 1b | 100 | 90 | 20 |
| 3 | Cl | Ph | CH | 1c | 100 | 94 | 20 |
| 4 | NO2 | Ph | CH | 1d | 83 | 72 | 17 |
| 5 | H | Ph | N | 1e | 100 | 91 | 20 |
| 6 | H | 4-(MeO)-Ph | CH | 1f | 100 | 93 | 20 |
| 7 | Me | 4-(MeO)-Ph | CH | 1g | 100 | 84 | 20 |
| 8 | Cl | 4-(MeO)-Ph | CH | 1h | 100 | 92 | 20 |
| 9 | NO2 | 4-(MeO)-Ph | CH | 1i | 60 | 48 | 12 |
| 10 | H | 4-(MeO)-Ph | N | 1j | 55 | 35 | 11 |
| 11 | H | Me | CH | 1k | 100 | 88 | 20 |
| 12 | Me | Me | CH | 1l | 100 | 81 | 20 |
| 13 | Cl | Me | CH | 1m | 100 | 89 | 20 |
| 14 | NO2 | Me | CH | 1n | 100 | 80 | 20 |
| 15 | H | Me | N | 1o | 100 | 79 | 20 |
a Yields refer to those of pure isolated products characterized by 1H NMR, 13C NMR and MS spectral analyses which is consistent with literature values [46–51].
b TOF: turnover frequency = moles of converted substrate (α-hydroxy ketone)/(moles of catalyst × reaction time in h).
The turnover frequency (TOF) for this catalytic process, which is defined as mole of converted substrate per mole of catalyst per hour, is listed in Table 2. In most cases, moderate to good values of TOF in the range of 11 to 20 h−1 were achieved.
There are recent reports of carrying out this reaction in the presence of other catalysts under various conditions. The data presented in Table 3 show the promising features of this protocol in terms of reaction rate, product yield, cost-effectiveness, catalyst availability and eco-friendliness when compared with other protocols.
Literature results for the synthesis of 1a under various conditions.
| Entry | Catalyst/Conditions | Time | Yield (%)c | [References] |
| 1 | MnO2 (10 equiv.)/MSa 4 Å/CH2Cl2/reflux | 45 min | 75 | [21] |
| 2 | Pd(OAc)2 (2 mol %)/MSa 3 Å/toluene/Et3N/THF/reflux | 24 h | 74 | [22] |
| 3 | MnO octahedral MSa (50 mg)/toluene/reflux | 8 h | 99d | [24] |
| 4 | Fe–molybdophosphoric acid (50 mg)/O2/CH2Cl2/reflux | 3 h | 95 | [25] |
| 5 | TiO2 (1 equiv.)/TEMPO (10 mol%)/MWIb (150 W)/sealed vial/150 °C | 10 min | 81 | [27] |
| 6 | Ru/C (10 mol%)/Me-β-cyclodextrin (300 mg)/O2/H2O/75 °C | 24 h | 96 | [28] |
| 7 | (NH4)6Mo7O24·4H2O (20 mol%)/PEG 300 (0.2 g)/MWIb (420 W)/90 °C | 15 min | 98 | This work |
a MS: molecular sieves.
b MWI: microwave irradiation.
c Yields refer to those of pure isolated products.
d GC yield (%).
4 Conclusions
In conclusion, we have developed a novel and benign tandem oxidation protocol for the microwave-induced direct synthesis of quinoxaline and pyrido[2,3-b]pyrazine derivatives from α-hydroxy ketones and arene-1,2-diamines. Due to the wide range of functional groups tolerated via this approach, various benzo[N,N]-heterocycles were attained in moderate to excellent yields. The use of (NH4)6Mo7O24·4H2O as an inexpensive air stable catalyst and PEG 300 as a green paste can provide an alternative route to expensive protocols and reduce the harmful effects of conventional organic solvents on the environment.
Besides, open-vessel microwave irradiation without the aid of air, O2 or an inert atmosphere leads to reaction safe conditions and rate enhancement. This catalytic procedure offers general reactivity and is illustrated by the cost-effective, energy-efficient and rapid preparation of condensed benzo[N,N]-heterocyclic compounds making the procedure a powerful tool from an environmental vantage point.