

1 Introduction
Sustainable Chemistry is becoming a wide field in Chemistry in general and in Organic Synthesis in particular. Among the 12 principles of Green Chemistry, one of the main subjects is the use of a green, recoverable and reusable heterogeneous catalyst [1].
Heteropolyacids (HPA) are well-defined molecular arrangements with remarkable and useful applications. Their main technological property is their reusability because of their solid state and the possibility of generating heterogeneous catalysis. Diverse electronic and molecular structures of HPA lead to their application in different areas such as medicine and materials science, among others; in the wide field of structural possibilities that the HPA have shown [2], the Wells-Dawson-type primary structure deserves to be mentioned. It has the formula [X2M18O62]-m and can be considered as the result of the fusion of two XM9O34 units, each one with a classic Keggin-type geometry [3]. Recently, we reported the use of the Wells-Dawson-structured HPA in the synthesis of diverse compounds such as flavones [4], coumarins [5], aryl and aralkyl cinnamates [6], quinoxaline derivatives [7] and azlactones [8], among others.
Moreover, 1,2,3,4-tetrahydroisoquinolines and derivatives are basic compounds inside many natural alkaloids, among them ecteinascidin, cherylline and latifine; many compounds of this type have shown diverse bioactivities such as anticonvulsant, antimicrobial, antitumor (against breast and prostatic cancer, and sarcoma), anti-HIV, antihypertensive, antiarrhythmic, antiherpes, bronchodilator, and as Parkinson disease therapeutic drug, among others [9–13].
In particular, N-sulfonyl-1,2,3,4-tetrahydroisoquinolines and N-sulfonyl-2,3,4,5-tetrahydro-1H-2-benzazepines are very important precursors for 1,2,3,4-tetrahydroisoquinoline and 2,3,4,5-tetrahydro-1H-2-benzazepine synthesis, respectively, although they also exist as substructures of diverse biologically active compounds [14]. Also, the introduction of sulfur atoms in heterocyclic compounds is very important since they provide molecules useful for crop protection [15].
The classic way for synthesizing these compounds involves a sulfonylaminomethylation of N-arakylsulfonamides [16] using various agents such as s-trioxane in acid media [17]. Among many of the synthetic strategies for the formation of an isoquinoline skeleton are the Pummerer [18], Bischler-Napieralski [19], Friedel-Craft [20], Horner [21] and Pictet-Spengler reactions [22–25].
The Pictet-Spengler strategy consists in the formation of an imino-intermediate from the condensation of an arylethylamine and a carbonyl compound and then an intramolecular aromatic electrophilic substitution for generating the isoquinoline structure. The reaction conditions can be modified in the way that an acyl or sulfonyl substituent in the nitrogen atom increases the reactivity of the electrophilic pair, favoring the advance of the reaction. In this way, many attempts were made modifying the Pictet-Spengler reaction conditions, including solvent-free conditions [26], Grignard reagents [27,28], and the use of diverse types of catalysts as superacids [29], BINOL-phosphoric acid [30], Preyssler heteropolyacids [31], and iridium salts [32].
In the present work we describe the preparation and characterization of the Wells-Dawson-type HPA with formula H6P2W18O62.24H2O (WD), and the 40% silica gel-supported analog (WD40); also we present their use as a recyclable acid catalyst in the synthesis of N-sulfonyl-1,2,3,4-tetrahydroisoquinolines and ring analogues. For example, Scheme 1 shows the synthesis of N-(3,4-dichlorobenzylsulfonyl)-1,2,3,4-tetrahydroisoquinoline using this procedure.
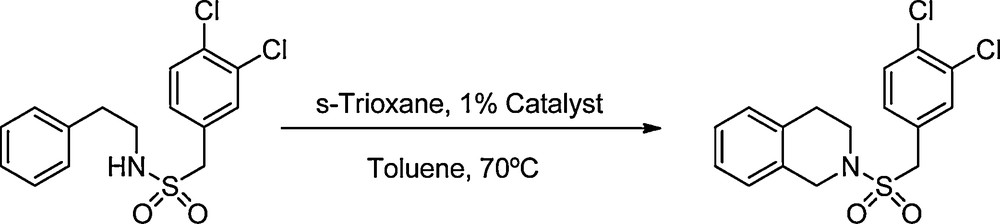
Synthesis of N-(3,4-dichlorobenzylsulfonyl)-1,2,3,4-tetrahydroisoquinoline.
2 Experimental
Chemicals were purchased from Aldrich, Fluka and Merck chemical companies and were freshly used after purification by standard procedures (distillation and recrystallization). All the reactions were monitored by TLC on precoated silica gel plates (254 mm). Flash column chromatography was performed with 230 to 400 mesh silicagel. All the yields were calculated from crystallized products. All the products were identified by comparison of physical data (mp, TLC and NMR) with those reported or with those of authentic samples prepared by the respective conventional methods using sulfuric acid as catalyst. Melting points of the compounds were determined in sealed capillary tubes and are uncorrected. The 1H-NMR and 13C-NMR spectra were obtained on a Bruker instrument 400 MHz model as CDCl3 solutions, and the chemical shifts were expressed in δ units with Me4Si (TMS) as the internal standard.
2.1 Preparation of catalyst
H6P2W18O62.24H2O (WD) was synthesized as described elsewhere [33] from an aqueous solution of α/β K6P2W18O62·10H2O salt. The WD silica-supported catalyst was obtained by wet impregnation of Grace Davison silica (Grade 59, specific area = 250 m2/g) with an aqueous solution of the synthesized WD. A catalyst containing 0.4 g/g of WD acid was prepared (0.4WDSiO2). Then, samples were dried at room temperature in a vacuum desiccator for 8 h.
2.2 Catalyst characterization
Bulk and supported catalysts were characterized by 31P MASNMR measurements. The 31P MAS-NMR spectra were recorded in Bruker MSL-300 equipment operating at frequencies of 121.496 MHz. A sample holder of 5 mm diameter and 17 mm in height was used. The spin rate was 2.1 kHz, and several hundreds of pulse responses were collected. Chemical shifts were expressed in parts per million with respect to 85% H3PO4 as an external standard for 31P-NMR.
FTIR spectra were obtained in Nicolet IR.200 equipment. Pellets in BrK and a measuring range of 400 to 4000 cm-1 were used to obtain the FT-IR spectra of the solid samples at room temperature.
Specific surface areas (SBET) of the catalysts were determined by nitrogen adsorption/desorption technique using Micromeritics ASAP 2020 equipment.
The acidic properties of samples were determined by potentiometric titration using a solution of n-butylamine in acetonitrile in a Metrohm 794 Basic Titrino apparatus. A 0.05 ml portion of n-butylamine (0.1 N), in acetonitrile, was added to a known mass of solid (between 0.1 and 0.05 g) using acetonitrile as solvent, and stirred for 3 h. Later, the suspension was titrated with the same base at 0.05 ml/min. The electrode potential variation was measured in a Metrohm 794 Basic Titrino apparatus with a double junction electrode.
2.3 Catalytic test
All catalysts were dried overnight prior to use. The reaction was performed in a round bottom flask, which was equipped with a condenser and immersed in an oil bath. In a typical test, N-phenylethyl-3,4-dichlorobenzylsulfonamide (1 mmol) and trioxane (3 mmol) and the bulk or silica-supported WD acid (1% mmol) in 2 ml toluene were heated to 70 °C with stirring for the indicated time (20–30 min). The solvent was evaporated and 10 ml of toluene was added. The catalyst was filtered and washed with more toluene.
The solution was dried over anhydrous Na2SO4. Filtration and concentration after the recrystallizations of acetone or ethyl acetate afforded the corresponding pure products.
2.4 Catalyst reuse
Stability tests of the bulk or silica-supported WD acid catalysts were carried out running four consecutive experiments, under the same reaction conditions. After each test, the catalyst was separated from the reaction mixture by filtration, washed with toluene (2 × 2 ml), dried under vacuum, and then reused.
2.5 Characterization data of selected compounds
N-(4-chlorobenzylsulfonyl)-1,2,3,4-tetrahydroisoquinoline (2a)
1H-NMR (CDCl3): δ = 2.8 (t, 2H, J = 5.9 Hz), 3.42 (t, 2H, J = 6.0 Hz), 4.21 (s, 2H), 4.35 (br s, 2H), 6.97 (br d, 1H, J = 8.7 Hz), 7.10 (br d, 1H, J = 8.7 Hz), 7.17 (ov m, 1H), 7.19 (ov m, 1H), 7.28 (br s, 4H).
13C-NMR (CDCl3): δ = 29.0, 43.7, 47.3, 56.7, 126.1, 126.5, 127.0, 127.4, 129.0, 131.9, 132.0, 133.3, 134.9.
N-(3,4-dichlorobenzylsulfonyl)-7-methoxy-1,2,3,4-tetrahydroisoquinoline (2c)
1H-NMR (CDCl3): δ = 2.76 (t, 2H, J = 6), 3.47 (t, 2H, J = 6), 4.16 (s, 2H), 4.34 (br s, 2H), 6.49 (d, 1H, J = 2.5 Hz), 6.77 (dd, 1H, J = 8.4 Hz, 2.6 Hz), 7.03 (d, 1H, J = 8.5 Hz), 7.18 (dd, 1H, J = 8.3 Hz, J = 2.0 Hz), 7.37 (d, 1H, J = 8.2 Hz), 7.40 (d, 1H, J = 2 Hz).
13C-NMR (CDCl3): δ = 28.0, 44.0, 47.6, 55.3, 56.4, 110.7, 113.6, 125.1, 129.1, 129.9, 130.1, 130.6, 132.4, 132.9, 133.2, 158.2.
N-(3,4-dichlorobenzylsulfonyl)-6,7-dimetoxy-1,2,3,4-tetrahydroisoquinoline (2d)
1H-NMR (CDCl3): δ = 2.74 (t, 2H, J = 5.9 Hz), 3.48 (t, 2H, 6 Hz), 4.16 (s, 2H), 4.28 (s, 2H), 6.43 (s, 1H), 6.59 (s, 1H), 7.19 (dd, 1H, J = 8.3 Hz, 2 Hz), 7.38 (d, 2H, J = 8.2 Hz).
13C-NMR (CDCl3): δ = 28.4, 43.8, 47.1, 56.5, 56.6, 108.7, 111.7, 123.7, 125.0, 129.1, 129.9, 130.6, 132.4, 132.8, 133.1, 147.9, 148.2.
N-(2-Methoxycarbonylethylsulfonyl)-6-chloro-1,2,3,4-tetrahidroisoquinoline (2e)
1H-NMR (CDCl3): δ = 2.82 (t, 2H, J = 7.4 Hz), 2.91 (t, 2H, J = 5.9 Hz), 3.32 (t, 2H, J = 7.4 Hz), 3.57 (t, 2H, J = 5.9 Hz), 4.44 (s, 2H), 7.08 (d, 1H, J = 2.2 Hz), 7.08 (d, 1H, J = 8.1 Hz), 7.17 (dd, 1H, J = 8.2, J = 2.1).
13C-NMR (CDCl3): δ = 28.2, 28.5, 43.2, 46.0, 46.7, 52.3, 126.2, 127.2, 130.4, 131.7, 132.2, 133.6, 170.9.
N-(3,4-dichlorobenzylsulfonyl)-2,3,4,5-tetrahydro-1-H-2-benzazepine (2g)
1H-NMR (CDCl3): δ = 1.49 (m, 2H), 3.60 (br t, 1H, J = 5.1 Hz), 3.84 (s, 2H), 4.45 (br s, 2H), 6.94 (dd, 1H, J = 8.7 Hz, J = 2.1 Hz), 6.95 (d, 1H, J = 2.1 Hz), 7.19 (br d, 1H, J = 7.2 Hz), 7.22 (ov m, 1H), 7.25 (ov m, 1H), 7.32 (d, 1H, J = 8.7 Hz).
13C-NMR (CDCl3): δ = 28.3, 52.3, 53.1, 58.3, 126.7, 128.6, 128.8, 129.2, 129.8, 130.0, 130.4, 132.4, 132.6, 132.9, 137.5, 141.9.
3 Results and discussion
3.1 Catalyst characterization
The 31P-NMR of Fig. 1 shows the characteristic only signal at –12.6 ppm of this class of Wells-Dawson structure [34]. The characteristic bands associated with the Wells-Dawson structure in the literature are present in the FTIR spectra of WD shown in Fig. 2: a band due to P–O stretching at 1091 cm−1; two bands attributed to W–O–W at 911 and 778 cm−1; and one band at 963 cm−1 corresponding to W = O [33]. The spectra of silica gel-supported acid (WD40) also shown in Fig. 2 exhibit the same bands, with a widening of the bands attributed to the loss of symmetry caused by the support media (Fig. 2).
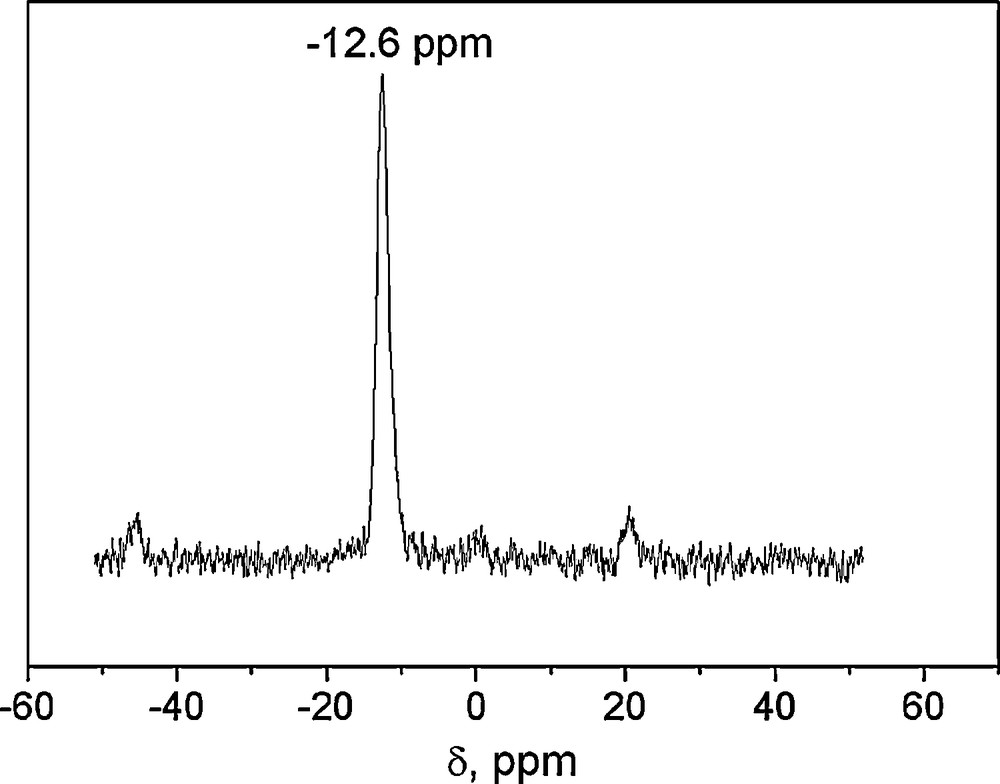
31P-RMN spectra of WD40.
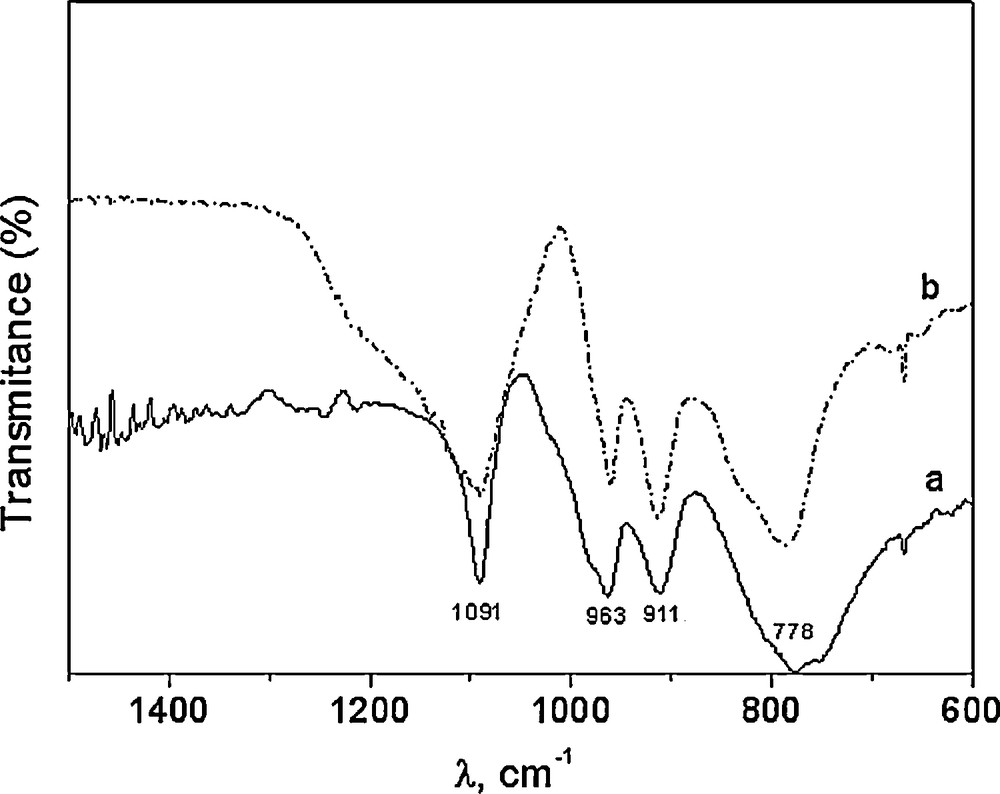
FTIR spectra of WD (a) and WD40 (b).
To determine the acid properties of bulk and supported acids we used potentiometric titration (Fig. 3). In order to interpret the results, it is suggested that the initial electrode potential (E) indicates the maximum acid strength of the surface sites, and the values (meq/g solid), where the plateau is reached, indicate the total number of acid sites. The acid strength of surface sites can be assigned according to the following ranges: very strong site, E > 100 mV; strong site, 0 < E < 100 mV; weak site, –100 < E < 0 mV; and very weak site, E < –100 mV. The maximum acid strength of the acid sites for WD is 827 mV. The magnitude for this value indicates very strong acid sites for bulk solid acid, very similar to the bulk Keggin HPA. The values of meq base/g solid where the plateau is reached indicate three different acid sites. Similarly, the supported acid shows a low decrease in the maximum acid strength, 801 mV for WD40. The support (silica) acid strength was tested, with an initial potential of 25 mV. When PW is supported on silica, the number of acid sites is very similar to that of bulk WD (Fig. 3).
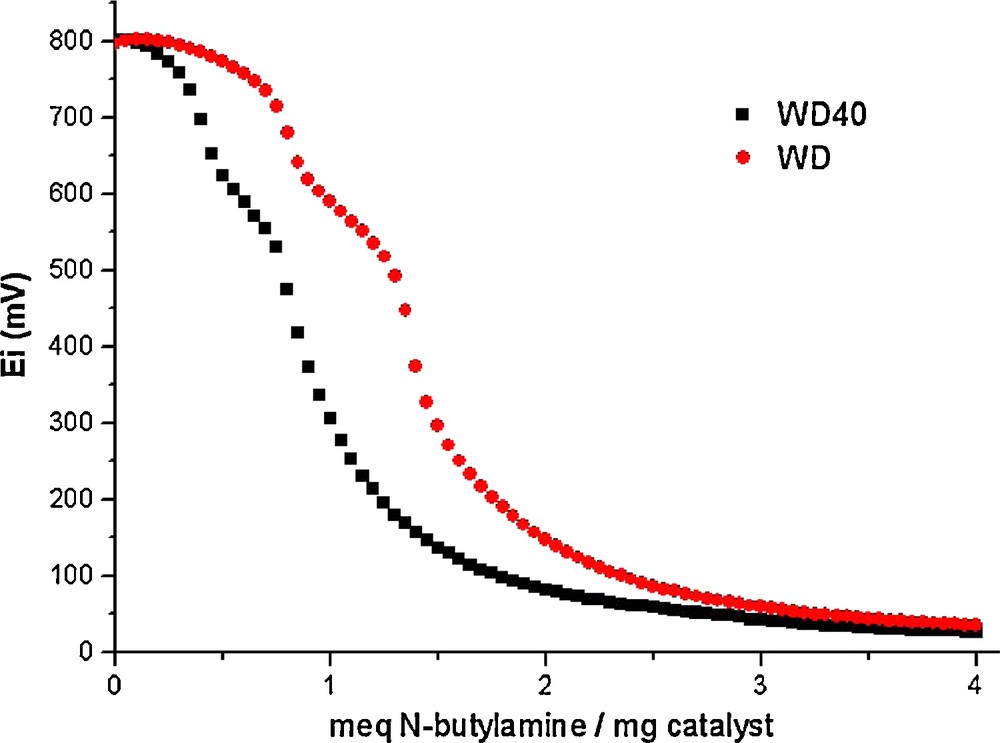
Potentiometric titration of WD and WD40.
The surface area of the supported acid WD40 sample determined from N2 adsorption–desorption isotherms using the BET method, BET surface, pore and micropore volume, and pore diameter are shown in Table 1. All properties of bulk HPA are lower; in all cases the values are close to the experimental error of the methods used. This result shows a reduction of the specific surface area attributed to particle agglomeration, while no significant changes in porosymmetry appear. The comparison with a bulk acid BET surface value of 1.9 m2/g indicates a big increase in the specific surface area.
Textural parameters.
| WD40 | Support | |
| BET surface (m2/g) | 245.7 | 268.41 |
| Pore volume (cm3/g) | 1.02 | 1.123 |
| Micropore volume (cm3/g) | 0.014 | 0.012 |
| Pore diameter (Å) | 166.82 | 167.28 |
3.2 Catalytic tests
Optimum reaction conditions were examined employing N-phenylethyl-3,4-dichlorobenzylsulfonamide as test reaction substrate with s-trioxane in toluene at 70 °C for 2 h. Without the presence of catalyst no reaction was observed. Similarly, no reaction occurred when the support (silica gel) was used as catalyst, under the same reaction conditions. Then the acids WD and WD40 were tested (Table 2, entry 3) with very good results.First, the influence of the reaction temperature on the N-(3,4-dichlorobenzyl-sulfonyl)-1,2,3,4-tetrahydroisoquinoline synthesis was investigated using both bulk (WD) and supported catalyst (WD40), and the results are illustrated in Table 2. In order to obtain the optimal temperature, five temperatures (25, 50, 70, 90 and 100 °C, Table 2) were tested.The tested experimental reaction conditions were: N-phenylethyl-3,4-dichlorobenzylsulfonamide, 1 mmol; s-trioxane, 3 mmol; toluene, 2 mL; 0.01 mmol catalyst; 30 min. No reaction was observed at 25 °C (entry 1 in Table 2).
Effect of temperature on N-(3,4-dichlorobenzylsulfonyl)-1,2,3,4-tetrahydroisoquinoline yields (%).
| Entry | Temperature (°C) | Yield (%) WD | Yielda (%) WD40 |
| 1 | 25 | – | – |
| 2 | 50 | 38 | 49 |
| 3 | 70 | 80 | 82 |
| 4 | 90 | 78 | 77 |
| 5 | 100 | 57 |
a Isolated yield.
A temperature increase leads to a higher N-(3,4-dichlorobenzyl-sulfonyl)-1,2,3,4-tetrahydroisoquinoline yield. For example, the yield of N-(3,4-dichlorobenzylsulfonyl)-1,2,3,4-tetrahydroisoquinoline for a reaction time of 30 min at 50 °C was only 38 and 49% (entry 2, Table 2) for bulk and supported catalysts, respectively, whereas at 70 °C the yields were 80 and 82%, respectively (entry 3, Table 2). Finally at 100 °C the reaction yields were considerably lower (57 and 57% respectively entry 5), due to the several unidentified side products that were detected by TLC.
By comparing the conversion values of supported and bulk catalysts at different temperatures, it was observed that the difference between them was not as high as expected considering that the amount of catalyst employed was always the same and the supported ones have a well-dispersed active phase. So, these results could be explained in terms of pseudoliquid behavior of the bulk catalyst, as we described before. The difference with the supported catalysts can be explained in terms of diffusion of reagents and products inside and outside crystallites in the supported catalysts. We assume that the difference in conversion between bulk and supported catalysts can be explained considering that they have microporous and mesoporous structures respectively, with 0.5 and 2.5 nm pore radius, so the diffusion factor in the whole reaction has a different influence on both systems, as we have already reported [35].
For this reason, 70 °C was employed as the ideal temperature to continue with the analysis of other reaction variables. Table 3 shows the result for N-(3,4-dichlorobenzylsulfonyl)-1,2,3,4-tetrahydroisoquinoline synthesis as a function of reaction time using WD and WD40 catalysts at a reaction temperature of 70 °C. The experimental reaction conditions were: N-phenylethyl-3,4-dichlorobenzylsulfonamide, 1 mmol; s-trioxane, 3 mmol; toluene, 2 ml; WD40, 0.01 mmol; and 70 °C. It can be observed that the yields of 3,4-dichlorobenzylsulfonyl-1,2,3,4-tetrahydroisoquinoline increased with the reaction time up to approx. 30 min and then remained at a constant level.
Effect of time of reaction on N-(3,4-dichlorobenzylsulfonyl)-1,2,3,4-tetrahydroisoquinoline yields (%).
| Entry | Reaction time (min) | Yielda (%) WD | Yielda (%) WD40 |
| 1 | 15 | 48 | 70 |
| 2 | 30 | 80 | 82 |
| 3 | 45 | 83 | 83 |
| 4 | 60 | 81 | 82 |
a Isolated yield.
Table 4 displays the effect of the amount of catalyst (WD40) on the yield of N-(3,4-dichlorobenzylsulfonyl)-1,2,3,4-tetrahydroisoquinoline in the reaction. The experimental reaction conditions were: N-phenylethyl-3,4-dichlorobenzylsulfonamide, 1 mmol; s-trioxane, 3 mmol; toluene, 2 mL mmol; 30 min; 70 °C, and a variable amount of WD40 catalyst (0.1%, 1%, 2% and 5%). It can be seen that the yields increased from 33% to 82% when the amount of WD40 increased from 0.1% to 1% (Table 4, entries 1 and 2). No relevant changes of reaction yields were observed with further increase in the amount of WD40 (5%), (83%, Table 4, entry 4). Thus 1% of WD40 is a suitable amount in this reaction.
Effect of amount of catalyst on N-(3,4-dichlorobenzylsulfonyl)-1,2,3,4-tetrahydroisoquinoline yields (%).
| Entry | Catalyst amount (%) | Yielda (%) |
| 1 | 0.1 | 33 |
| 2 | 1 | 82 |
| 3 | 2 | 82 |
| 4 | 5 | 83 |
a Isolated yield.
The reusability of the catalysts was investigated in the sequential reaction of N-phenylethyl-3,4-dichlorobenzylsulfonamide with s-trioxane. At the end of each run the catalyst was removed, washed with toluene, dried in vacuum at 40 °C and reused. The results, which are summarized in Table 5, showed that the catalyst was reused four consecutive runs and no appreciate loss of its catalytic activity was observed. Performance of the reactions under the same conditions using WD40 as a catalyst showed 78%, 79%, 77%, and 77% of N-(3,4-dichlorobenzylsulfonyl)-1,2,3,4-tetrahydroisoquinoline yield, respectively. In the five cycle the conversion of substrate was 90% and the maximal yields 75% in 40 min.
Catalyst reuse in N-(3,4-dichlorobenzylsulfonyl)-1,2,3,4-tetrahydroisoquinoline yields (%).
| Entry | Cycle | Yielda (%) |
| 1 | 0 | 82 |
| 2 | 1 | 78 |
| 3 | 2 | 79 |
| 4 | 3 | 77 |
| 5 | 4 | 77 |
| 6 | 5 | 75b |
a Isolated yield.
b The reaction is not complete in 30 min (conversion 90%) and the yields of 75% was obtained in a reaction time of 40 min.
An additional test was performed in order to evaluate the possible catalyst solubilization. A WD40 sample was refluxed in toluene for 2 h, filtered and dried in vacuum till constant weight. The activity of the so-treated catalyst was the same as that of the fresh catalyst (81% in 30 min). The refluxed toluene was used as solvent for attempting the reaction without adding the catalyst. After 4 h no product was detected and the starting material was quantitatively recovered.
Using the optimized conditions, sulphonamide, 1 mmol; s-trioxane, 3 mmol; toluene, 2 mL; WD40, 0.01 mmol; and 70 °C, several N-sulfonyl-1,2,3,4-tetrahydroisoquinolines and N-sulfonyl-2,3,4,5-tetrahydro-1H-2-benzazepines were prepared (Table 6). The yield of the reaction was comparable for both catalysts. In all the experiments, the desired products were obtained with high selectivity. When the sulphonamide and s-trioxane were treated with the WD40 catalysts for 30 min, the tetrahydroquinolines and ring analogues were obtained with very good yields (59%–95%).
Preparation of N-sulfonyl-1,2,3,4-tetrahydroisoquinolines and ring analogues catalyzed by WD40a.
| Entry | Product | Yield (%) |
| 1 | 33 | |
| 2 | 83 | |
| 3 | 64 | |
| 4 | 88 | |
| 5 | 59 | |
| 6 | 95 | |
| 7 | 84 | |
| 8 | 80 |
4 Conclusions
The described procedure for the synthesis of N-sulfonyl-1,2,3,4-tetrahydroisoquinolines and N-sulfonyl-2,3,4,5-tetrahydro-1H-2-benzazepines using a supported heteropolyacid with Wells-Dawson primary structure results in a clean and useful alternative; the advantages of this methodology are operative simplicity, use of a reusable and noncorrosive solid acid catalyst, soft reaction conditions, low reaction times, and good yields. The use of a solid acid catalyst instead of the usual soluble acid catalyst (sulfuric, hydrochloric, etc.) contributes to a reduction in waste generation by allowing an easy separation and recovery without any loss of its catalytic activity.
Acknowledgements
We thank Universidad Nacional de La Plata, CONICET and ANPCyT for financial support. GB, HT and GR are members of CONICET.