

1 Introduction
The use of small-molecule organocatalysts in organic synthesis has flourished over the past decade [1–9]. However, the concept of organocatalysis has emerged as a discrete strategy for addressing modern-day challenges in chemistry. It has become widely appreciated that small-molecule organic catalysts can hold a wide range of practical advantages relative to macromolecular, precious metal or protic acidic catalysts, including air stability, low cost, commercial availability, relative non-toxicity, green nature and simple reaction conditions, and can promote a chemical reaction through different activation modes [1–9]. With the dramatic recent expansion of research efforts in organocatalysis throughout the world, synthetically useful transformations based on new reactivity concepts have been identified, often with no counterpart in the more established catalysis regimens [1–9]. Triarylmethyl chlorides (Ar3CCl) are an attractive class of small-molecule organocatalysts, which have been utilized to promote a few organic transformations by in situ formation of triarylmethyl carbocations [6–9]. It is noteworthy that the inherent instability of carbocations has precluded up to now their use in catalysis with decent turnover numbers [7].
The aldol condensation reaction has been widely applied for carbon–carbon bond formation in organic synthesis [10–12]. Among them, the cross-aldol condensation of cycloalkanones with arylaldehydes leading to α,α′-bis(arylidene)cycloalkanones [13], which have attracted much attention due to their use as precursors for the synthesis of pyrimidine derivatives [14], and their intriguing biological activities such as antiangiogenic [15], quinine reductase inducer [16], and cholesterol-lowering properties [17]. Generally, the aldol condensation reaction has been catalyzed by strong acids [18] and more likely by bases [19]; however, in these conditions, the reactions suffer from reverse and/or side reactions [20]. Moreover, some metal ions with different ligands [21–25] and solid acids [26,27] have been introduced as catalysts to replace strong acids or bases. But most of these methods are associated with one or more of the following drawbacks: moderate yields, unwanted side reactions, need for application of sealed ampoules or tubes, use of expensive, unavailable and toxic catalysts, and application of an additional energy (microwave). Although some catalysts (mostly Brønsted and Lewis acids) for the cross-aldol condensation of cycloalkanones with arylaldehydes leading to α,α′-bis(arylidene)cycloalkanones are known, newer catalysts continue to attract attention for their difference with the others, high novelty and effectiveness.
In this work, we introduce a high novelty in the cross-aldol condensation reaction between cycloalkanones and arylaldehydes, and perform the reaction using trityl chloride (Ph3CCl) as an efficient and homogeneous small-molecule organocatalyst under solvent-free conditions at 110 °C. Attractive points of this work are study of in situ formation of trityl carbocation with inherent instability from trityl chloride (Ph3CCl) which catalyzes the reaction, and suggestion of a plausible mechanism based on observations and the literature.
2 Results and discussion
At first, the condensation of cyclohexanone (1 mmol) with m-nitrobenzaldehyde (2.1 mmol) was selected as a model reaction (Scheme 1), and its behavior was studied in the presence of different molar ratios of Ph3CCl in the 100–120 °C range under solvent-free conditions. The results are displayed in Table 1. As it can be seen in Table 1, 10 mol% of the organocatalyst was sufficient to promote the reaction efficiently at 110 °C (Table 1, entry 3). Increasing the amount of Ph3CCl or the temperature decreased the reaction yield (Table 1, entries 4 and 6). The reaction was also examined without catalyst in which the product was obtained in low yield even after a long reaction time (Table 1, entry 1).
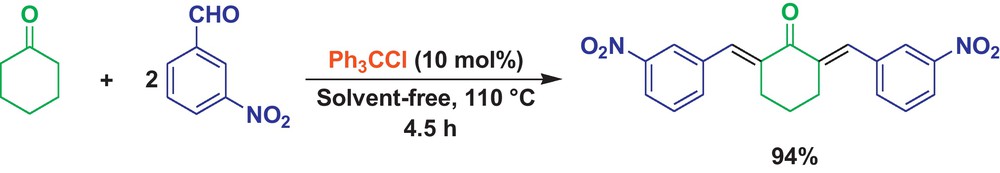
The cross-aldol condensation between cyclohexanone and m-nitrobenzaldehyde catalyzed by Ph3CCl.
The reaction of cylohexanone with m-nitrobenzaldehyde in the presence of different amounts of Ph3CCl at various temperatures.
| Entry | Mol% of Ph3CCl | Temperature (°C) | Time (h) | Yielda (%) |
| 1 | – | 110 | 8 | 19 |
| 2 | 7.5 | 110 | 6 | 76 |
| 3 | 10 | 110 | 4.5 | 94 |
| 4 | 12.5 | 110 | 4.5 | 92 |
| 5 | 10 | 100 | 4.5 | 83 |
| 6 | 10 | 120 | 4.5 | 90 |
a Isolated yield.
After optimization of the reaction conditions, the efficiency, the generality and the scope of the organocatalyst in the preparation of α,α′-bis(arylidene)cycloalkanones were assessed by the reaction of cycloalkanones (cyclopentanone, cyclohexanone and cycloheptanone) with different arylaldehydes including benzaldehyde as well as aldehydes containing electron-withdrawing substituents, electron-releasing substituents or halogens (Table 2). As Table 2 indicates, all reactions proceeded efficiently and the desired products were obtained in high to excellent yields and in logical reaction times (Table 2, entries 1–12 and 16–18). Trityl chloride also successfully catalyzed the reaction of cyclohexanone with p-dimethylaminobenzaldehyde (containing a basic group), heteroaromatic aldehydes and cinnamaldehyde (Table 2, entries 9 and 13–15). Thus, in this reaction, Ph3CCl was efficient and general catalyst.
The solvent-free cross-aldol condensation of cycloalkanones with aldehydes using Ph3CCl at 110 °C.
| Entry | n | R | Time (h) | Yielda (%) | M.p. (°C) | |
| Found | Reported | |||||
| 1 | 1 | C6H5 | 4 | 87 | 186-188 | 187-189 [13] |
| 2 | 1 | p-MeOC6H4 | 4 | 92 | 207-209 | 210-211 [25] |
| 3 | 1 | m-ClC6H4 | 5.5 | 79 | 174-176 | 173-175 [13] |
| 4 | 2 | C6H5 | 5.5 | 87 | 113-115 | 116-117 [22] |
| 5 | 2 | p-NO2C6H4 | 4 | 91 | 161-163 | 159 [21] |
| 6 | 2 | m-NO2C6H4 | 4.5 | 94 | 184-186 | 180-182 [13] |
| 7 | 2 | p-MeOC6H4 | 4 | 88 | 165-167 | 160-162 [25] |
| 8 | 2 | p-MeC6H4 | 5.5 | 76 | 169-171 | 170 [22] |
| 9 | 2 | p-Me2NC6H4 | 3.5 | 91 | 250-252 | 250-252 [30] |
| 10 | 2 | p-ClC6H4 | 4 | 90 | 143-145 | 147-148 [22] |
| 11 | 2 | m-ClC6H4 | 6 | 76 | 105-107 | 108-110 [13] |
| 12 | 2 | o-ClC6H4 | 5 | 84 | 104-106 | 103-105 [26] |
| 13 | 2 | 2-Thienyl | 5 | 92 | 141-143 | 142-143 [31] |
| 14 | 2 | 2-Furyl | 4.5 | 83 | 142-144 | 140-142 [13] |
| 15 | 2 | C6H5CH = CH | 4.5 | 86 | 176-178 | 179-180 [25] |
| 16 | 3 | C6H5 | 6 | 86 | 104-106 | 105-107 [13] |
| 17 | 3 | 3,4-(MeO)2C6H3 | 3.5 | 89 | 165-167 | 162-164 [13] |
| 18 | 3 | p-ClC6H4 | 4 | 87 | 129-131 | 131-133 [13] |
a Isolated yield.
Based on the literature reports related to the presented reaction shown in Scheme 2, our mechanistic proposal is that from the interaction of aldehyde and trityl chloride, tautomers I and II could be generated via a reversible reaction pathway [6–9].
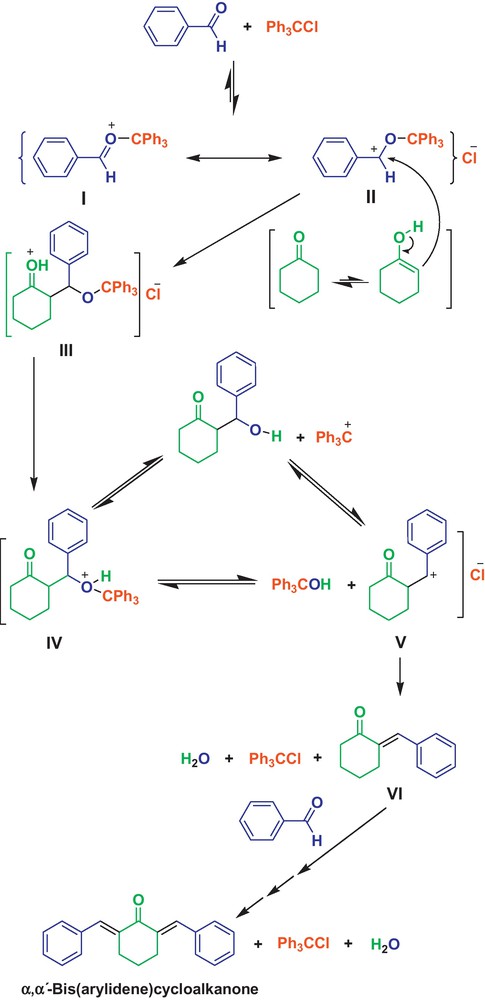
The proposed mechanism for the cross-aldol condensation reaction using Ph3CCl.
In order to validate our proposal, benzaldehyde was reacted with trityl chloride (in ratio of 1/1), and then IR, 1H NMR spectra of the resulting interacted aldehyde functional group of proposed tautomers I and II in the reaction mixture were compared with those in benzaldehyde as follows.
According to the obtained FT-IR spectrum of the resulting reaction mixture, stretching vibration of the carbonyl of the aldehyde functional group slighlty shifts to lower frequency because of the increased dipole moment character of carbonyl functionality: IR (nujol): νmax (cm–1) of CO in benzaldehyde (1705) decreased to (1697) in the reaction mixture (Fig. 1).
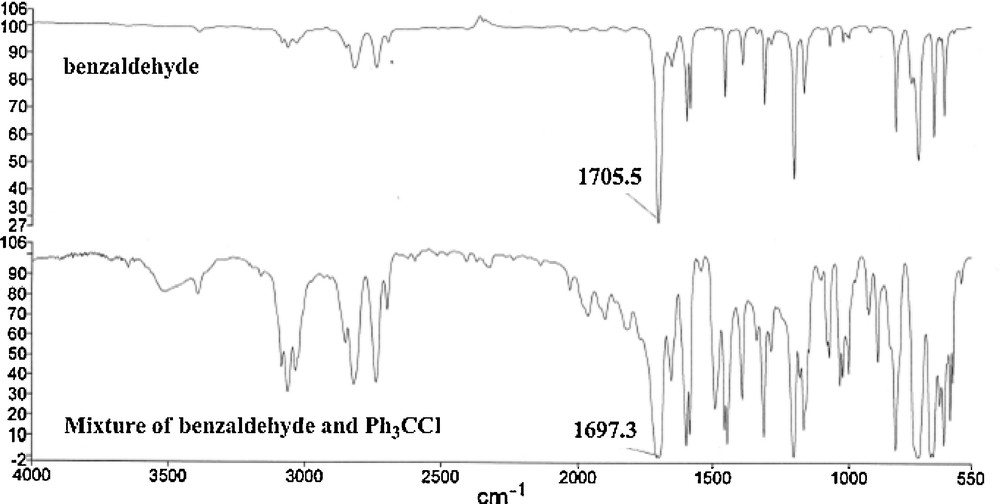
IR spectra of benzaldehyde as well as the mixture of benzaldehyde and Ph3CCl (in ratio of 1/1).
Accordingly, in the 1H NMR spectra, the aldehyde hydrogen as well as carbonyl carbon were deshielded, which is in agreement with the increased single bond character of carbonyl functional group observed in the FT-IR spectra: 1H NMR (300 MHz, CDCl3): δ (ppm) of the aldehydic hydrogen (9.78) increased to (10.04) in the reaction mixture (Fig. 2).
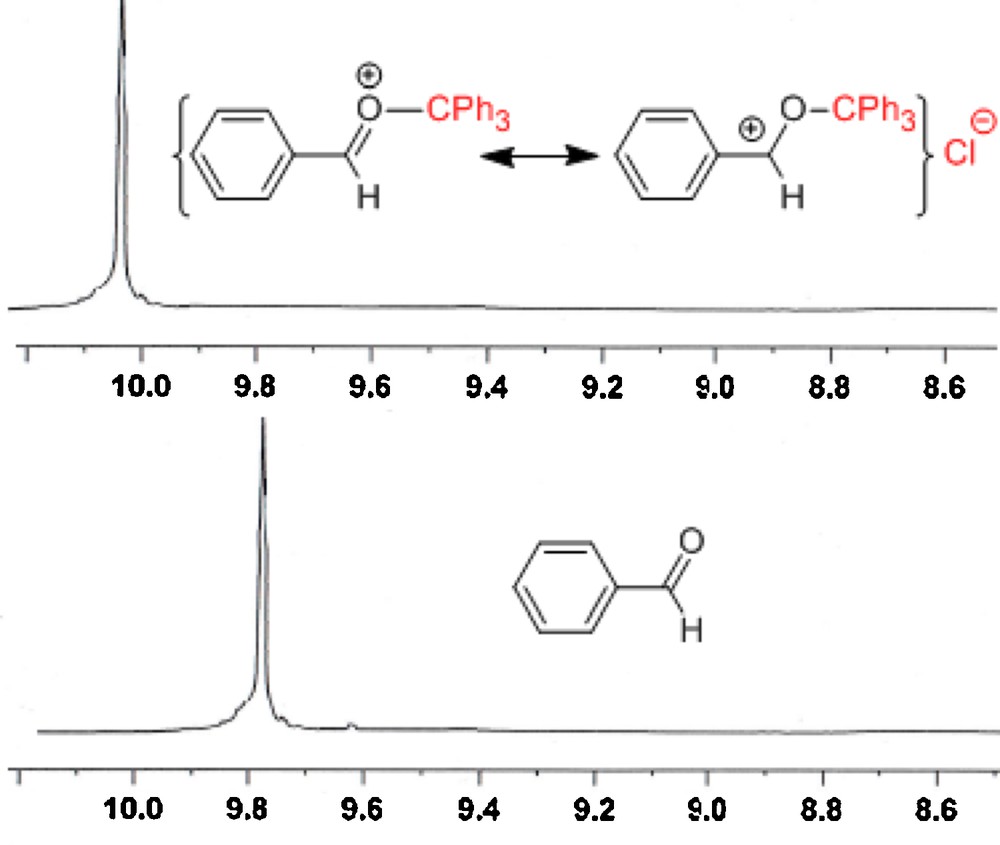
1H NMR spectra of the formyl hydrogen of benzaldehyde, separately as well as in the mixture of benzaldehyde and Ph3Cl (in ratio of 1/1).
UV (n-hexane): λmax (nm) of absorption of benzaldehyde and trityl chloride appears at 240 and 222, respectively; however, λmax of the absorbtion of the complexes of benzaldehyde and trityl cation was observed at 245 (Fig. 3).
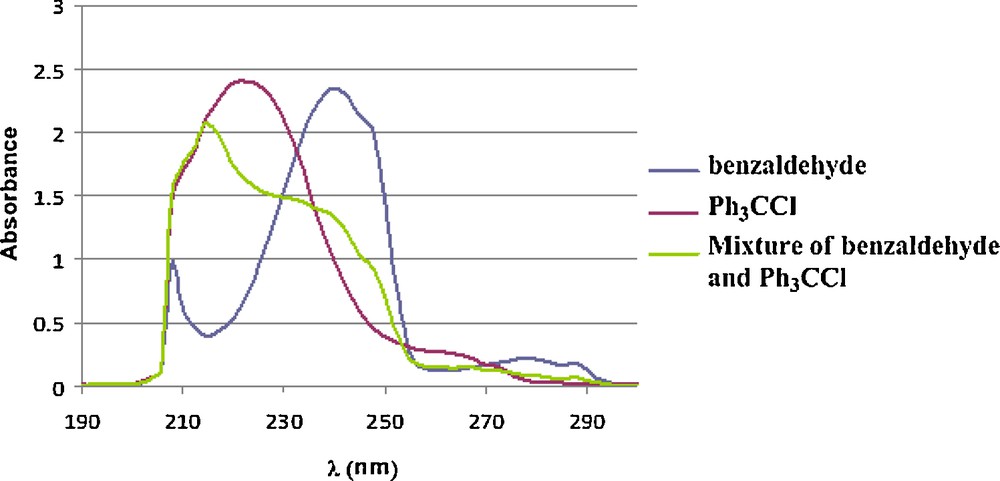
UV spectra of benzaldehyde, Ph3CCl and the complexes of benzaldehyde and Ph3CCl (in ratio of 1/1).
These results confirm that tautomer forms I and II are present in the reversible reaction media. Moreover, the cationic tautomers I and II have been introduced by Kusumoto et al. for the first time [28]. These tautomer forms act as activated aldehyde, and then react with cyclohexanone providing III, which converts to IV by proton transfer. Intermediate IV can be converted to V by removing Ph3COH in a reversible reaction. Afterward, V and Ph3COH produce intermediate VI, Ph3CCl and H2O. VI accordingly reacts with another activated aldehyde by Ph3CCl to afford the corresponding α,α′-bis(arylidene)cycloalkanone. The suggested mechanism is confirmed by the literature [29], and also by the fact that Ph3CCl was completely recovered unchanged and Ph3COH could not be identified after completion of the reaction as it could be observed on TLC by comparison with pure authentic samples. In another study, to demonstrate that Ph3CCl cannot convert to Ph3COH and HCl by the water produced during the reaction, and consequently HCl is not the real catalyst of the process, the solvent-free condensation of cyclohexanone with 3-nitrobenzaldehyde was examined in the presence of the expected amount of HCl produced by the reversible reaction of Ph3COH with H2O, at 110 °C, in which the product was obtained in 48% within 8 h. The reaction was also tested using Ph3COH wherein the reaction yield was 19% after 8 h. These evidences showed that HCl is not produced from Ph3CCl in these conditions, and that Ph3CCl is the real catalyst of this transformation.
3 Conclusion
In summary, we have shown that organocatalyst trityl chloride, by in situ generation of carbocationic system, efficiently catalyzed the cross-aldol condensation reaction. The promising points for the presented protocol are simple and clean reaction profile, efficiency, generality, high yields, reaction in solvent-free and homogeneous media without side reactions, economic availability, non-toxicity as well as low cost of the catalyst, and good compliance with the green chemistry protocols which makes it an attractive procedure for the synthesis of α,α′-bis(arylidene)cycloalkanones as an important class of organic compounds.
4 Experimental
4.1 General
All chemicals were purchased from Merck or Fluka Chemical Companies. All known compounds were identified by comparison of their melting points and spectral data with those reported in the literature. Progress of the reactions was monitored by TLC using silica gel SIL G/UV 254 plates. The 1H NMR (500 MHz) and 13C NMR (125 MHz) were run on a Bruker Avance DPX, FT-NMR spectrometer. UV spectra were run on a T8 UV-VIS spectrometer, PG instruments Ltd. Melting points were recorded on a Büchi B-545 apparatus in open capillary tubes.
4.2 General procedure for the cross-aldol reaction
A mixture of cycloalkanone (1 mmol), aldehyde (2.1 mmol) and trityl chloride (0.028 g, 0.1 mmol) in a test tube connected to a reflux condenser, was stirred at 110 °C, and the reaction progress was monitored by TLC. After completion of the reaction, the mixture was cooled to room temperature, petroleum ether (10 mL) was added to it, refluxed and stirred for 3 min, and filtered to separate the catalyst. The solid residue (crude product) was purified by recrystallization from EtOH (95%) or column chromatography on silica gel eluted with n-hexane/EtOAc (1/5) to give the pure α,α′-bis(arylidene)cycloalkanone.
4.3 Selected spectral data of the products
2,5-Bis(3-chlorobenzylidene)cyclopentanone (Table 2, entry 3): 1H NMR (500 MHz, CDCl3): δ = 3.01 (s, 4H), 7.29–7.33 (m, 4H), 7.39–7.42 (m, 2H), 7.43 (s, 2H), 7.51 (t, J = 1.2 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ = 27.0, 128.6, 129.6, 130.5, 130.7, 132.5, 135.3, 137.1, 138.6, 196.0.
2,6-Bis(3-chlorobenzylidene)cyclohexanone (Table 2, entry 11): 1H NMR (500 MHz, CDCl3): δ = 1.69 (quintet, J = 5.7 Hz, 2H), 2.78 (t, J = 5.7 Hz, 4H), 7.30–7.37 (m, 6H), 7.41 (s, 2H), 7.62 (t, J = 1.2 Hz, 2H); 13C NMR (125 MHz, CDCl3): 23.7, 27.9, 126.3, 127.9, 129.3, 130.4, 130.9, 133.2, 135.8, 137.8, 188.9.
2,6-Bis(2-chlorobenzylidene)cyclohexanone (Table 2, entry 12): 1H NMR (500 MHz, CDCl3): δ = 1.75 (quintet, J = 5.8 Hz, 2H), 2.78 (t, J = 5.8 Hz, 4H), 7.28 (m, 4H), 7.34 (d, J = 3.4, 2H), 7.45 (d, J = 3.3 Hz, 2H), 7.89 (t, J = 1.2 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ = 23.2, 28.7, 126.8, 130.8, 130.9, 131.0, 134.5, 135.0, 135.5, 138.3, 189.2.
(2E,6E)-2,6-Bis((E)-3-phenylallylidene)cyclohexanone (Table 2, entry 15): 1H NMR (500 MHz, CDCl3): δ = 1.85 (quintet, J = 5.8 Hz, 2H), 2.81 (t, J = 5.8 Hz, 4H), 6.97 (d, J = 15.4 Hz, 2H), 7.10 (dd, 2H, J = 15.4, 1.2 Hz), 7.28 (m, 2H), 7.36 (m, 4H), 7.48–7.54 (m, 6H); 13C NMR (125 MHz, CDCl3): δ = 22.5, 27.8, 124.7, 124.8, 127.5, 129.3, 135.9, 136.8, 137.3, 141.4, 188.3.
(2E,7E)-2,7-Dibenzylidenecycloheptanone (Table 2, entry 16): 1H NMR (500 MHz, CDCl3): δ = 2.03 (br, s, 4H), 2.75 (br, s, 4H), 7.37 (t, J = 7.2 Hz, 2H), 7.43 (s, 2H), 7.45 (m, 4H), 7.51 (d, J = 7.5 Hz, 4H); 13C NMR (125 MHz, CDCl3): δ = 28.5, 29.3, 128.7, 128.9, 129.9, 136.1, 136.4, 142.2, 199.9.
(2E,7E)-2,7-Bis(3,4-dimethoxybenzylidene)cycloheptanone (Table 2, entry 17): 1H NMR (500 MHz, CDCl3): δ = 2.05 (br, s, 4H), 2.77 (br, s, 4H), 3.91 (s, 6H), 3.96 (s, 6H), 6.94 (d, J = 8.5 Hz, 2H), 7.05 (s, 2H), 7.12 (d, J = 8.5 Hz, 2H), 7.39 (s, 2H); 13C NMR (125 MHz, CDCl3): 28.7, 29.1, 56.3, 56.4, 111.5, 113.3, 123.1, 129.2, 135.9, 140.5, 149.2, 149.7, 199.9.
(2E,7E)-2,7-Bis(4-chlorobenzylidene)cycloheptanone (Table 2, entry 18): 1H NMR (500 MHz, CDCl3): δ = 2.00 (br, s, 4H), 2.71 (br, s, 4H), 7.38 (s, 2H), 7.41–7.45 (m, 8H); 13C NMR (125 MHz, CDCl3): δ = 28.4, 29.1, 129.2, 131.1, 134.6, 134.7, 135.0, 142.4, 199.3.
Acknowledgements
The authors gratefully acknowledge financial support of this work by the Research Council of Payame Noor University.