

1 Introduction
For many decades, porphyrins have been the subject of intense researches due to their versatile and outstanding physico-chemical properties and also to their implication in essential natural processes, like photosynthesis, blood oxygen transport [1]. The progress in porphyrin chemistry has led to the synthesis of complex structures, adding new specifically designed fragments on the periphery of the porphyrin core. Conversely, the totally unsubstituted porphyrin called porphine has attracted much less attention despite its great fundamental interest. Recently, Senge published the first review on porphines, “Porphyrin (porphine) a neglected parent compound with potential” [2]. The conclusion was: “The true potential of porphines and their metal complexes is just emerging and hopefully the advances made in their synthesis will now spawn advances in their characterization and application.” Although several studies are reported on free base (H2P) and metallated porphines (MP) (in the solid state [3] or in solution [3g,4]), currently, the major part concerns theoretical studies owing to the structural simplicity and high symmetry of this macrocycle, resulting in less power/memory demanding calculations [5]. Though porphine is the common basic unit of porphyrins, its physico-chemical properties and electrochemical reactivity have been poorly explored principally due to its only recent commercial availability. Furthermore, despite its structural simplicity, its synthesis is not straightforward since the common Adler et al. [6] or Lindsey et al. [7] synthetic methods starting directly from pyrrole and formaldehyde do not work. However, a concise and efficient synthesis (three steps, 17% overall yield starting from pyrrole) of magnesium(II) porphine (MgP), was recently described by Lindsey et al. [8]. This magnesium(II) complex exhibits good solubility in common organic solvents contrary to the free base porphine. As its electrochemical properties had not yet been described in detail (only its first oxidation potential was mentioned [9]), our first contributions in this field were dedicated to the electrochemical behavior of MgP in CH2Cl2 [10], pyridine [11] and CH3CN [12].
In the preliminary synthesis of MgP according to Lindsey's procedure [8], we failed to reproduce the final purification step consisting in crystallization of the crude solution containing magnesium(II) porphine with an ethanol–water mixture. In our case, there was no other way to obtain MgP in a pure form than preparative chromatography. So, for practical purposes, a chromatography-free procedure was highly desirable.
Additionally, to the best of our knowledge, the electrochemical characterization of zinc(II) porphine (ZnP) has never been done before (only its first oxidation potential was mentioned [9a,9b]). Bearing in mind that MgP can oligomerize [10] and polymerize [12] upon oxidation, we were wondering if the zinc complex could behave similarly.
We, thus, report here a new method to purify MgP and ZnP by crystallization, their respective crystallographic structures as pyridine adducts and a comparison of their spectroscopic and electrochemical properties.
2 Results and discussion
2.1 Synthesis of MgP·(Py)2 and ZnP·(Py)2
As a result of their good solubility in a broad variety of organic solvents, a common issue with magnesium porphyrins stems from their final purification step, i.e. precipitation or crystallization [8,13]. Thus, one solution consists of the precipitation with water of the magnesium complex dissolved in a miscible organic solvent such as acetone, methanol, ethanol, DMF, DMSO, acetonitrile, THF. However, generally, the finely divided amorphous suspension obtained is difficult to filtrate on standard sintered glass frits since it has a trend to seal it. This behavior was also observed in our case with MgP. Besides, grease might be difficult to remove in these conditions. Inspired by the fact that several crystallographic structures of magnesium porphyrin–pyridine adducts were described [14], we found that the addition of an excess of pyridine into a solution of MgP in dichloromethane led during the evaporation of CH2Cl2 to the formation of shiny deep purple crystals. Upon addition of pyridine, the solution color changes from pink to deep purple, which evidences the formation of a pyridine-MgP adduct. These crystals were filtered, thoroughly washed with n-hexane and dried under vacuum at 45 °C. Starting from pure MgP, the crystallization yield was 79%. The interest of this method stems from the high purity of the pyridine-magnesium porphine MgP·(Py)2 obtained but also from a easy handling and removal from the sintered glass frits of the crystalline material. Furthermore, when starting with crude MgP, it is possible to synthesize pure MgP·(Py)2 in one or several crystallization steps, depending on the purity of the initial MgP. Ultimately, pure MgP can be totally recovered (100% yield) from pure MgP·(Py)2 by simple heating at 200 °C under vacuum (P ≈ 5.10−2 mbar) during 24 h. In agreement with the fact that metallated porphyrins are stable below 360 °C [15], we did not observe any degradation of MgP at this temperature.
Although ZnP is much less soluble in organic solvents than MgP, the same behavior was observed, i.e. addition of an excess of pyridine leads to ZnP·(Py)2 adduct in crystalline form.
2.2 Crystallographic structures of MgP·(Py)2 and ZnP·Py
2.2.1 Crystallographic structure of MgP·(Py)2
To our best knowledge, only two pyridine-magnesium porphyrins adducts have been described. The first one, the bis(pyridine)-octaethylporphyrin magnesium complex [14a] was reported in 1977 while the second one, the bis(pyridine)-tetraphenylporphyrin magnesium complex [14b], was published sixteen years later. Nevertheless, a functionalized pyridine adduct, the bis(4-methylpyridine)-magnesium porphyrin adduct was also described in 1984 by Mc Kee et al. [16]. Suitable crystals for X-ray diffraction studies of MgP·(Py)2 were obtained from slow diffusion of cyclohexane into a CH2Cl2/Py (4/1 v/v) solution containing MgP. This latter is the second example of metallated porphine crystallographic structure, the first one being the nickel(II) complex [17] and joins the very restricted family of magnesium porphyrin crystallographic structures since only 28 X-ray structures are recorded to date on CCDC database [11,14b,16,18]. Three-dimensional molecular views of MgP·(Py)2 are presented in Fig. 1. Selected data are also given in Table 1 and are compared with the bis(pyridine) adducts of MgOEP (MgOEP·(Py)2) [14a] and MgTPP (MgTPP·(Py)2) [14b]. The centrosymmetric macrocycle is almost planar (side view, Fig. 1), the Mg(II) ion, lying in the mean plane, formed by the 20 C and four equatorial N (NP) atoms of the porphine (mean planeP, d(Mg–NP) = 2.071(2) and 2.072(2) Å). The Mg–NPy (from pyridine) distance is 2.337(2) Å which is slightly shorter compared to the MgTPP·(Py)2 complex (d(Mg–NPy) = 2.3764(2) Å) but very similar to the MgOEP·(Py)2 adduct (d(Mg–NPy) = 2.3366(8) Å). Both pyridine ligands are lying in the same mean plane (front view, Fig. 1). The latter is nearly orthogonal to the porphine mean planeP with a dihedral angle of 88.45(6)° as a result of the absence of steric hindrance at the macrocycle periphery. This orthogonality is nearly kept in the case of the MgOEP·(Py)2 adduct (85.43°) but is clearly lost for the MgTPP·(Py)2 complex (71.83°). The RMS deviation from the planarity, i.e. the root mean square of the distances of the 24 carbon and nitrogen porphyrin ring atoms from the mean plane formed by these atoms, is very low for MgP·(Py)2 (0.0242 Å), increases slightly for MgOEP·(Py)2 (0.0319 Å) and significantly for MgTPP·(Py)2 (0.0810 Å). Thus, though MgTPP·(Py)2 is less substituted than MgOEP·(Py)2, this complex exhibits the most important degree of nonplanarity. As no additional solvent is included in the crystallographic cell of these porphyrins, it is also possible to directly compare their respective density. So, whereas MgOEP·(Py)2 and MgTPP·(Py)2 have similar density (1.257 and 1.262, respectively), MgP·(Py)2 exhibits a significant increase in this value (1.372), which could result from the absence of steric strains at the periphery of the porphine core and a higher magnesium/carbon ratio. Besides, each MgP·(Py)2 molecule interacts with two other ones through C(9) atoms (d(C(9)–C(9)) = 3.315(4) Å) leading to a 2D network (bottom, Fig. 1). These particular π-π interactions between carbon atoms of porphine cores are not observed in the other MgOEP·(Py)2 and MgTPP·(Py)2 complexes. Besides, adjacent porphyrins interact via π-stacking between the pyridine ligands, the distance between the pyridine centroïd Ctpyr of one molecule with the carbon of the second molecule located at its vertical Ctpyr…C13 is equal to 3.547(4) Å.
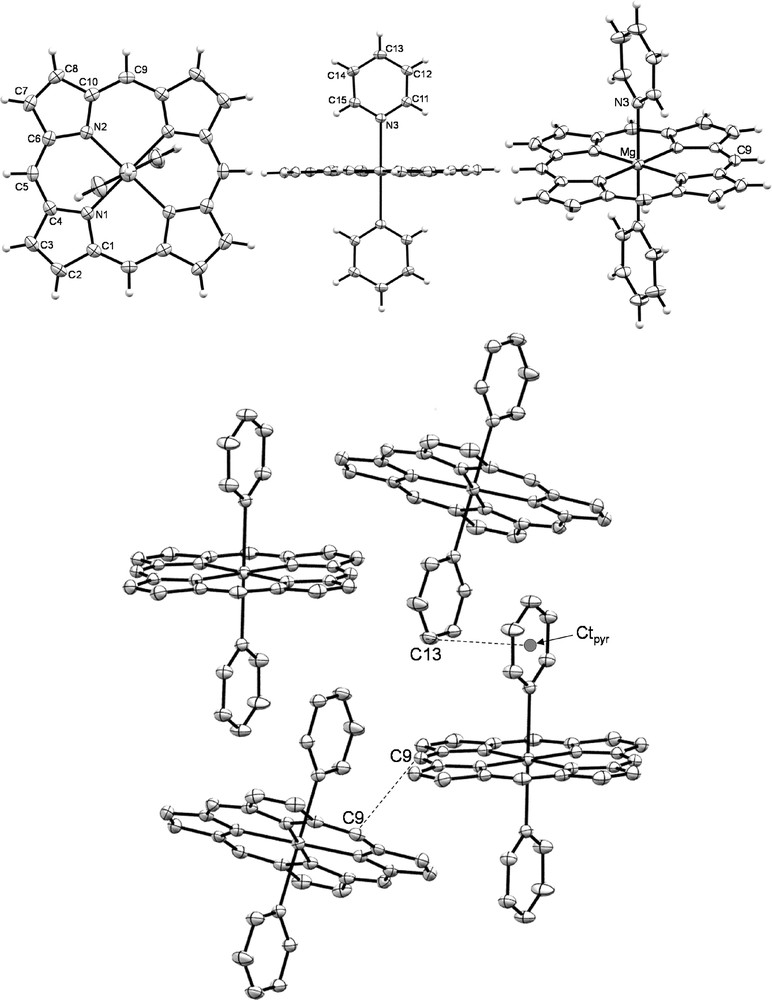
ORTEP views of MgP·(Py)2: front view (top left); side view (top center), general view (top right) and 2D network (bottom, H atoms have been omitted for clarity reasons) of MgP·(Py)2. Thermal ellipsoids are scaled to the 50% probability level.
Comparison of structural parameters extracted from the X-ray crystallographic structures of MgP·(Py)2, MgOEP·(Py)2[14a] and MgTPP·(Py)2[14b].
| MgP·(Py)2 | MgOEP·(Py)2 [14a] | MgTPP·(Py)2 [14b] | |
| d(Mg–NPy) (Å) | 2.337(2) | 2.3366(8) | 2.3764(2) |
| d(Mg–mean planeP) (Å)a | 0.000 | 0.000 | 0.000 |
| d(Mg–NP) (Å) | 2.071(2) < d < 2.072(2) | 2.041(1) < d < 2.0644(6) | 2.069(3) < d < 2.074(3) |
| mean planeP/mean planePy angle | 88.45(6)° | 85.43° | 71.83° |
| RMS deviation (Å)b | 0.0242 | 0.0319 | 0.0810 |
| density (g/m3) | 1.372 | 1.257 | 1.262 |
a The mean plane of the porphyrin (mean planeP) is calculated with the 20C and 4N atoms of the porphyrin ring.
b The RMS deviation corresponds to the root mean square of the distances of the 24 carbon and nitrogen porphyrin ring atoms from the mean plane formed by these atoms.
2.2.2 Crystallographic structure of ZnP·Py
In contrast to magnesium(II) porphyrin pyridine adducts, numerous examples of X-ray crystallographic structures of zinc(II) porphyrins pyridine adducts are described. For example, to date 100 structures are reported when drawing a single pyridine molecule coordinated to ZnTPP as a substructure in CCDC. An interesting point stems from the rarity of the bis-pyridine-coordinated ZnTPP X-ray structures recorded in this database. Indeed, only two examples are reported among the 100 aforementioned. Thus, statistically, in the particular case of ZnTPP, the zinc(II) cation principally exhibits a pentacoordinated coordination sphere. A similar trend, though less pronounced, is also observed for ZnOEP with 11 examples when drawing one pyridine coordinated on ZnOEP as a substructure in CCDC. However among these, only one structure presents an additional coordinated pyridine.
Suitable crystals for X-ray diffraction studies of ZnP·Py were obtained from slow diffusion of cyclohexane into a solution of ZnP in CH2Cl2/Py (4/1 v/v). Three dimensional molecular views of ZnP·Py are given in Fig. 2. Selected data are also given in Table 2 and are compared with ZnTPP·Py [19] and ZnOEP·Py [20] adducts. Given the numerous examples of mono pyridine zinc(II) porphyrin adducts, it is, thus, not surprising to obtain a similar pentacoordinated coordination sphere around the zinc cation. Contrary to MgP·(Py)2, ZnP·Py macrocycle as well as its ZnOEP·Py and ZnTPP·Py homologues (see Table 2) deviate markedly from the planarity since the Zn(II) atom is clearly out of the mean plane formed by the 20 C and four N atoms of the porphyrin (mean planeP) (d(Zn–mean planeP) = 0.483 Å, side view, Fig. 2). As a result of an higher Zn–mean planeP distance than its ZnTPP·Py (0.435 Å) and ZnOEP·Py (0.396 Å) congeners, the Zn–N(porphyrin) (d(Zn–NP)) distances are the longer ones and are intermediate between 2.071(2) and 2.083(2) Å. However, the Zn–NPy (from pyridine) distance appears to be the shorter one (2.127(2) Å vs 2.2002 Å and 2.153 Å for ZnOEP·Py and ZnTPP·Py, respectively) in agreement with a stronger interaction of the pyridine with the zinc(II) atom in ZnP·Py. Contrary to ZnOEP·Py and ZnTPP·Py, the mean plane formed by the nitrogen and carbon atoms of the pyridine molecule (mean planePy) is almost orthogonal to the mean planeP (89.75(5)°). The RMS deviation from the planarity, i.e. the root mean square of the distances of the 24 carbon and nitrogen porphyrin ring atoms from the mean plane formed by these atoms, is 0.0873 Å, an intermediate value between those of ZnOEP·Py (0.0675 Å) and ZnTPP·Py (0.1153 Å). Similarly to MgP·(Py)2, ZnP·Py exhibits the higher density (1.516) compared with ZnOEP·Py (1.254) and ZnTPP·Py (1.438). Interestingly, each ZnP·Py unit interacts with four neighboring porphine molecules through π-π stacking (N(2)–C(23) = 3.177(5) Å; C(6)–C(24) = 3.345(5) Å; C(25)–C(18) = 3.374(4) Å) leading to a 2D network (Fig. 2, bottom).
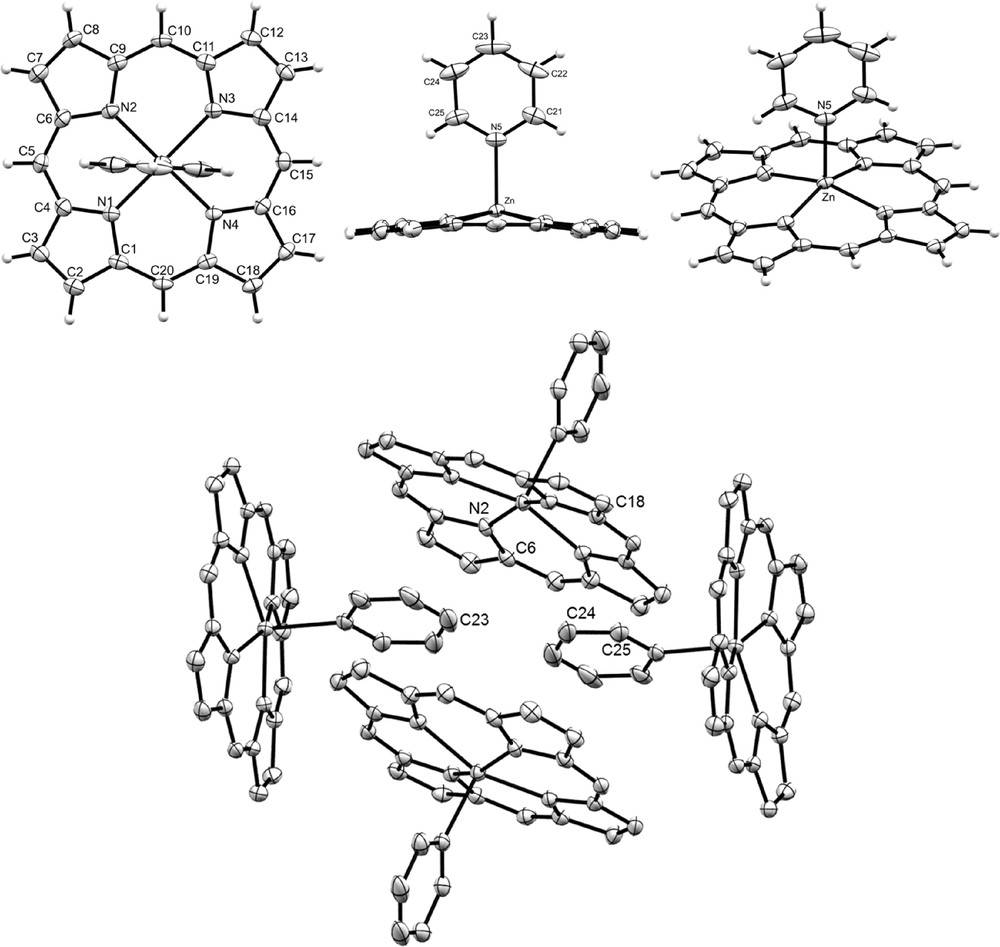
ORTEP views of ZnP·Py: front view (top left); side view (top center), general view (top right) and 2D network (bottom, H atoms have been omitted for clarity reasons) of ZnP·Py. Thermal ellipsoids are scaled to the 50% probability level.
Comparison of structural parameters extracted from the X-ray crystallographic structures of ZnP·Py, ZnOEP·Py [20] and ZnTPP·Py.[19].
| ZnP·Py | ZnOEP·Py [20] | ZnTPP·Py [19] | |
| d(Zn–NPy) (Å) | 2.1305(1) | 2.2002(5) | 2.153(4) |
| d(Zn–mean planeP) (Å)a | 0.483 | 0.396 | 0.435 |
| d(Zn–NP) (Å) | 2.0737(1) < d < 2.0820(1) | 2.0602(5) < d < 2.0751(5) | 2.065(4) < d < 2.076(4) |
| mean planeP/mean planePy angle | 89.75(5)° | 85.07° | 70.11° |
| RMS deviation (Å)b | 0.0873 | 0.0675 | 0.1153 |
| density (g/cm3) | 1.516 | 1.254 | 1.438 |
a The mean plane of the porphyrin (mean planeP) is calculated with the 20 C and 4 N atoms of the porphyrin ring.
b The RMS deviation corresponds to the root mean square of the distances of the 24 carbon and nitrogen porphyrin ring atoms from the mean plane formed by these atoms.
2.3 NMR characterization of MgP·(Py)2 and ZnP·(Py)2
NMR spectra of MgP·(Py)2 and ZnP·(Py)2 were recorded in deuterated acetone and are compared with MgP, ZnP and pyridine in the same solvent. Partial 1H NMR spectra are presented in Fig. 3. Surprisingly, contrary to ZnP·Py crystals grown from slow diffusion of cyclohexane in a CH2Cl2–pyridine mixture, NMR characterization of the crystals synthesized by dissolving ZnP in a CH2Cl2/pyridine mixture followed by rapid evaporation of CH2Cl2/pyridine mixture, filtration, washing with n-hexane and drying for 24 h under vacuum at 45 °C, reveals a bis-pyridine adduct (ZnP·(Py)2) in deuterated acetone.
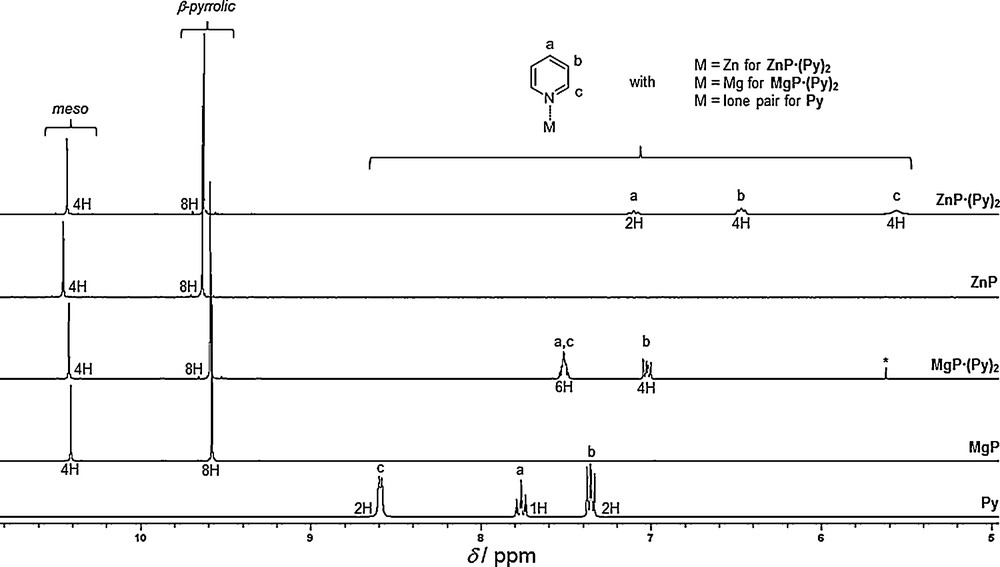
Partial 1H NMR spectra, from top to bottom, of ZnP·(Py)2, ZnP, MgP·(Py)2, MgP and Pyridine (Py) (300 MHz, 298 K, CD3COCD3). The peak marked by an asterisk corresponds to CH2Cl2 traces.
Due to the high symmetry of these porphine derivatives, 1H NMR spectra are very simple. Their respective meso and β-pyrrolic singlets have similar chemical shifts (Table 3). The structure of the solid-state metalloporphine adducts of MgP·(Py)2 and ZnP·(Py)2 is preserved in the deuterated acetone solution since the corresponding coordinated pyridine signals are shielded compared with the simple pyridine molecule (Fig. 3). These significant high field shifts as well as the integration of these signals agree well with the coordination of two pyridine molecules on the magnesium(II) and zinc(II) metals undergoing the influence of the shielding cone of these porphines. Interestingly, this influence is more pronounced in the case of ZnP·(Py)2, in agreement with a stronger interaction of nitrogen atoms with zinc(II) cation. Attribution of the pyridine signals was solved by means of 2D techniques (1H-1H COSY, 1H-13C HSQC).
1H NMR chemical shifts of porphine compounds and pyridine (300 MHz, CD3COCD3, 298 K).
| Proton signals δ (ppm) | ZnP·(Py)2 | ZnP | MgP·(Py)2 | MgP | Py |
| meso | 10.43 | 10.45 | 10.42 | 10.41 | |
| β-pyrrolic | 9.63 | 9.64 | 9.59 | 9.58 | |
| Ha | 7.10 | 7.51 | 7.76 | ||
| Hb | 6.47 | 7.02 | 7.35 | ||
| Hc | 5.56 | 7.51 | 8.59 |
2.4 Electrochemical characterization of MgP·(Py)2 and ZnP·(Py)2
As the electrochemical behaviors of MgP·(Py)2 and ZnP·(Py)2 were not, to our best knowledge examined before, we studied these pyridine adducts in DMF with tetraethylammonium hexafluorophosphate (TEAPF6) as the supporting electrolyte. Although ZnP·(Py)2 is poorly soluble in common organic solvents, contrary to MgP·(Py)2, its solubility was sufficient in DMF for electrochemical studies. Their cyclic voltammograms (CV) are presented in Fig. 4 and are compared with MgP and ZnP which have been synthesized by the removal of coordinated pyridine molecules from MgP·(Py)2 and ZnP·(Py)2 (Section 3). At first sight, there is no difference between the CVs of ZnP·(Py)2 and ZnP and between MgP·(Py)2 and MgP. This result is not surprising since DMF is known to be a strongly coordinating solvent. Thus, these pyridine adducts are instantaneously and totally dissociated in solution as previously reported for octaethylporphyrin magnesium pyridine adduct in other coordinating solvents [21]. Besides, in DMF, UV-vis absorption spectra of ZnP·(Py)2 and ZnP as well as those of MgP·(Py)2 and MgP are identical, confirming this full pyridine dissociation. Usually, metalloporphyrins with a non-electroactive metal, like Mg(II) or Zn(II), undergo reversibly two ring-centered reductions and two ring-centered oxidations delivering, respectively, the π-anion radical and π-dianion, and the π-cation radical and π-dication [22]. This common behavior is indeed observed for ZnP·(Py)2 and ZnP in the negative potential range, with two successive monoelectronic reduction waves at E1/2 = −1.44 V and E1/2 = −1.81 V (peaks R1 and R2, respectively, Fig. 4). For MgP·(Py)2 and MgP, the first reduction peak is reversible (E1/2 = −1.48 V, peak R1) whereas the second one is not clearly distinguished due to the proximity of the solvent reduction though a reduction at Epc ≈ −1.98 V (peak R2, Fig. 4) is discernible. In the positive potential direction, all the oxidation peaks of the metallated porphines are irreversible. Moreover, the first oxidation peak (O1) is significantly higher than the first mono-electronic and reversible reduction peaks (R1). This marked difference in intensity (ratio ipa(O1)/ipc(R1) = 3.5 and 2.5 for ZnP·(Py)2 and MgP·(Py)2, respectively), also observed on rotating disk electrode, is indicative of a multiple electron transfer (n > 2) coupled with chemical reactions. This behavior agrees well with the high reactivity of the electrogenerated cation radicals, as already reported for MgP in other solvents [10–12]. Besides, this superior ratio for ZnP·(Py)2 compared with MgP·(Py)2 can be correlated with a higher first oxidation potential leading to enhanced reactivity of the initially formed cation radical. Remarkably, the experimentally determined HOMO-LUMO gap, i.e. the potential difference between the first oxidation and the first reduction (2.27 and 2.23 V for ZnP·(Py)2 and MgP·(Py)2, respectively) falls into the diagnostic criterion values (2.15 ± 0.15 V) reported for TPP and OEP derivatives [22]. Thus, the primary electron transfers of ZnP·(Py)2 and MgP·(Py)2 only affect the macrocycle (Table 4).
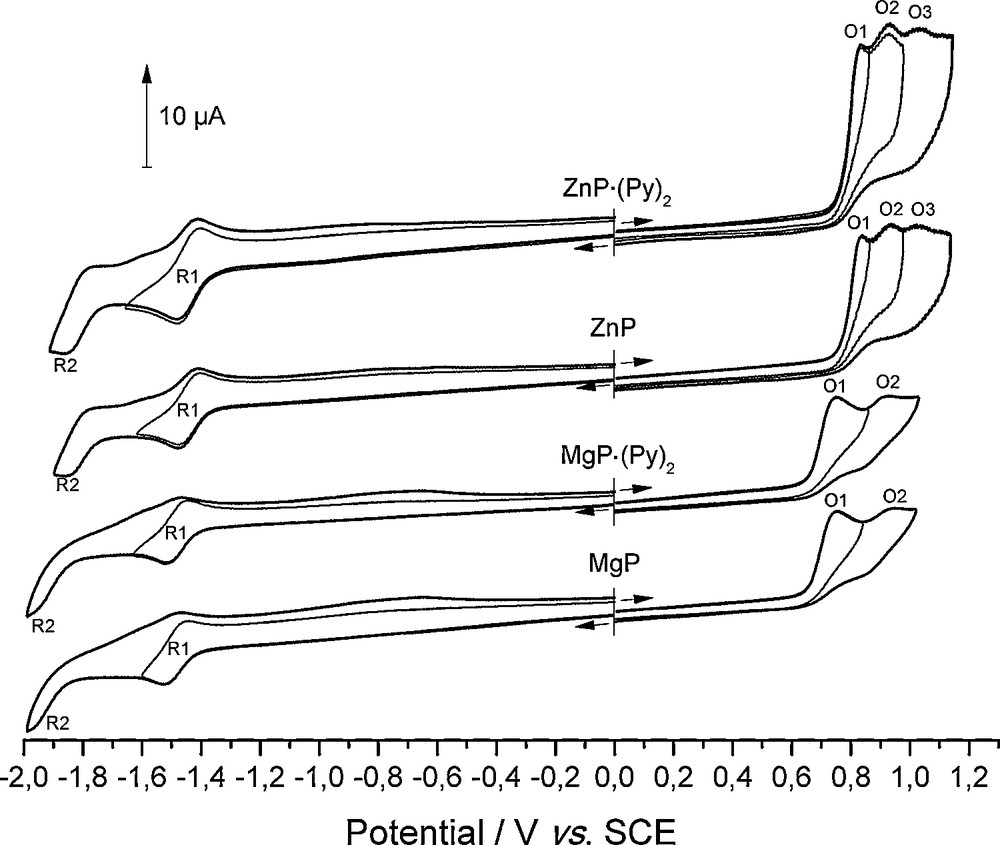
Cyclic voltammograms of ZnP·(Py)2, ZnP, MgP·(Py)2 and MgP (c ≈ 5 × 10−4 M, DMF 0.1 M TEAPF6, RT, WE: Pt Ø = 2 mm, CE: Pt, RE: SCE, ν = 100 mV s−1).
Potential values (E1/2 or Ep for irreversible systems) for ZnP·(Py)2, ZnP, MgP·(Py)2 and MgP (in V vs SCE). When redox systems are reversible, ΔEp = (Epa - Epc) values are given in brackets.
| 2nd reduction peak R2 | 1st reduction peak R1 | 1st oxidation peak O1 | 2nd oxidation peak O2 | 3rd oxidation peak O3 | ΔE(ox1/red1) = (Epa(ox1)–E1/2(red1)) | |
| ZnP·(Py)2 | −1.81 (100) | −1.44 (75) | 0.83 | 0.93 | 1.03 | 2.27 |
| ZnP | −1.81 (100) | −1.44 (75) | 0.83 | 0.93 | 1.03 | 2.27 |
| MgP·(Py)2 | ≈ −1.98 | −1.48 (75) | 0.75 | 0.93 | 2.23 | |
| MgP | ≈ −1.98 | −1.48 (75) | 0.75 | 0.93 | 2.23 |
In order to obtain more information about the oxidation of ZnP and ZnP·(Py)2, spectroelectrochemical experiments were performed in DMF 0.1 M TEAPF6. Solutions of these zinc complexes were electrolyzed on a large area platinum spiral and electrolyses were stopped for an amount of electricity corresponding to two electrons transferred per porphine molecule. For purpose of comparison, these experiments were reproduced with MgP·(Py)2 and MgP. No difference on the evolution of the UV-vis spectra between ZnP and ZnP·(Py)2 and between MgP·(Py)2 and MgP was observed. Consequently, only the UV-vis evolutions during the electrolysis of ZnP·(Py)2 and MgP·(Py)2 are presented in Fig. 5. After each electrolysis, the platinum working electrode was totally covered by a black/purple solid material, in agreement with an electropolymerization process occurring during the oxidation of these metallated porphines, as already established for MgP [10,12]. On both spectra, two major trends are noted:
- • the progressive decrease in the initial bands at 307 and 532 nm for ZnP·(Py)2 and at 309 and 536 nm for MgP·(Py)2;
- • the emergence of one band at 440 and 441 nm for ZnP·(Py)2 and MgP·(Py)2, respectively.
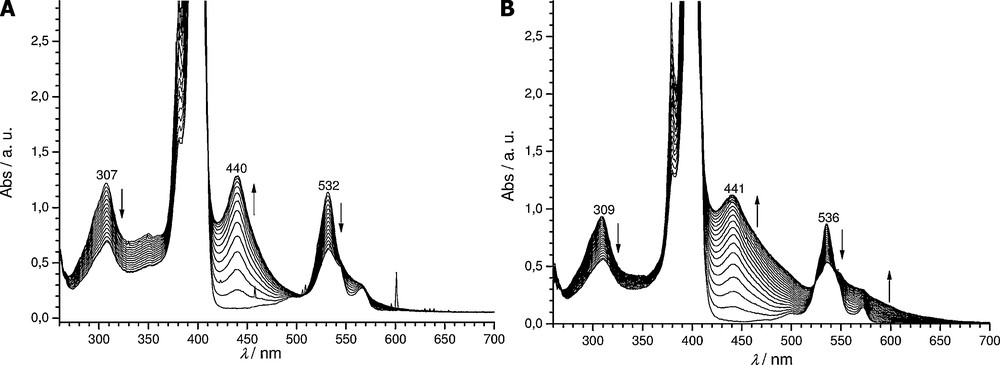
Electrolyses of 5 × 10−4 M solutions of ZnP·(Py)2 (A) and MgP·(Py)2 (B) followed by UV-vis absorption spectroscopy (l = 1 mm, 0.1 M TEAPF6 in DMF, Eapp = 0.83 and 0.75 V for ZnP·(Py)2 and MgP·(Py)2, respectively, -2.0 electrons, WE: Pt spiral, CE: Pt, RE: SCE).
As previously reported for the zinc(II) porphine model, i.e. 5,15-ditolyl-10-phenyl zinc(II) porphyrin [23] and for MgP [10] and as confirmed by MALDI-TOF mass spectrometry analyses of the electrolyzed solutions, these new Soret bands correspond to the signature of meso-meso dimers (ZnP)2 and (MgP)2. Indeed, chemical or electrochemical oxidation of magnesium and zinc porphyrins having at least one unsubstituted meso position is known to lead to meso-meso linked porphyrin dimers with their specific 30 to 40 nm spaced double Soret bands features [24]. It should be noted that the highest energy Soret band of the meso-meso dimer is located at nearly (within a few nm) the same wavelength than the monomer one. As MgP·(Py)2 and ZnP·(Py)2 Soret band maxima appear at 400 and 401 nm in DMF, respectively, and considering that their corresponding dimer highest energy Soret band absorbs at the same wavelength, the wavelength gap between both Soret bands is 41 and 39 nm, respectively. Interestingly, the Soret band at 440 nm (during the oxidation of ZnP·(Py)2) is narrower than the one at 441 nm (corresponding to the oxidation of MgP·(Py)2). This behavior could stem from a lower solubility of the zinc derivatives compared with the magnesium ones as already observed for the monomer. Indeed, the adsorption on the electrode surface of the less soluble zinc oligomers would continuously maintain a low concentration of these products in the electrolyzed solution.
3 Experimental
3.1 X-ray crystallographic study
CCDC-912742 & 912743 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request.cif.
Crystal data for MgP·(Py)2 [C30H22MgN6], red prism crystal: 0.18 × 0.10 × 0.08 mm3, M = 490.85 g/mol, monoclinic, space group C2/c (No. 15), Z = 4, a = 12.7579(9) Å, b = 15.0501(12) Å, c = 12.3850(8) Å, β = 92.071(4)°, V = 2376.5(3) Å3, Dcalcd = 1.372 g/cm3, μ = 0.108 mm−1, T = 115(2) K, F(000) = 1024, Mo Kα radiation (λ = 0.71073 Å), mixture of ϕ rotations and φ scans, 4122 reflections collected on a Nonius Kappa CCD diffractometer (index ranges: h = ±16; k = –19/17; l = ±16) measured in the range 1.0° ≤ θ ≤ 27.49°, with 2695 independent (Rint = 0.0323) and 2053 observed reflections [I ≥ 2σ(I)], 169 refined parameters, R indices for observed reflections: R1 = 0.0623, wR2 = 0.1091, R indices for all data: R1 = 0.0892, wR2 = 0.1225, goodness of fit = 1.168, Δρ = 0.283/−0.248 e.Å3.
Crystal data for ZnP·(Py) [C25H17ZnN5], red prism crystal: 0.15 × 0.10 × 0.05 mm3, M = 452.81 g/mol, monoclinic, space group P21/c (No. 14), Z = 4, a = 9.5746(4) Å, b = 14.6935(6) Å, c = 14.6410(6) Å, β = 105.542(2)°, V = 1984.44(14) Å3, Dcalcd = 1.516 g/cm3, μ = 1.261 mm−1, T = 115(2) K, F(000) = 928, Mo Kα radiation (λ = 0.71073 Å), mixture of ϕ rotations and φ scans, 7847 reflections collected on a Nonius Kappa CCD diffractometer (index ranges: h = ±12; k = −18/19; l = ±18) measured in the range 1.0° ≤ θ ≤ 27.49°, with 4495 independent (Rint = 0.0305) and 3688 observed reflections [I ≥ 2σ(I)], 280 refined parameters, R indices for observed reflections: R1 = 0.0426, wR2 = 0.0787, R indices for all data: R1 = 0.0609, wR2 = 0.0877, goodness of fit = 1.143, Δρ = 0.534/−0.350 e.Å3.
3.2 X-ray equipment and refinement
Diffraction data were collected on a Nonius KappaCCD diffractometer equipped with a nitrogen jet stream low-temperature system (Oxford Cryosystems). The X-ray source was graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) from a sealed tube. The structure was solved by direct methods using the SIR92 [25] program and refined with full-matrix least-squares on F2 using the SHELXL97 [26] program with the aid of the WIN-GX [27] program suite. All non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atoms attached to carbon atoms were included in calculated positions and refined as riding atoms.
3.3 Reagents and instrumentation
Magnesium porphine (MgP) and zinc porphine (ZnP) were synthesised according to known procedures [8]. Our data (1H NMR, 13C NMR, UV-vis absorption, and MALDI-TOF mass spectrum) were consistent with those described in reference [8]. Tetraethylammonium hexafluorophosphate (TEAPF6, Fluka puriss., electrochemical grade, ≥ 99.0%), pyridine (99%+, Acros) were used as received. CH2Cl2 (Carlo Erba 99.5%) was distilled over P2O5; DMF (SDS, Carlo Erba, purity (GC) 99.9%) was distilled under vacuum on CaH2.
UV-vis absorption spectra were obtained with a Varian UV–vis spectrophotometer Cary 50 scan using quartz cells (Hellma). In spectroelectrochemical experiments, a UV–vis immersion probe (Hellma, l = 1 mm) was connected through a fibre optic to the same spectrophotometer.
High-resolution mass spectra (HRMS) were recorded on a MicrOTOF Q Bruker instrument in ESI (positive mode) at the Plateforme d’analyse chimique et de synthèse moléculaire de l’université de Bourgogne (PACSMUB).
1H and 13C NMR spectra were measured on a BRUKER 300 MHz spectrometer (Avance III Nanobay). The reference was the residual non-deuterated solvent.
Elemental analyses (C, H, N, S) were carried out on a Flash EA 1112 Thermo Electron analyser.
All electrochemical manipulations were performed using Schlenk techniques in an atmosphere of dry oxygen-free argon at room temperature (T = 20 ± 3 °C). The supporting electrolyte was degassed under vacuum before use and then dissolved to a concentration of 0.1 mol/L. Voltammetric analyses were carried out in a standard three-electrode cell, with an Autolab PGSTAT 302 N potentiostat, connected to an interfaced computer that employed Electrochemistry Nova software. The reference electrode was a saturated calomel electrode (SCE) separated from the analysed solution by a sintered glass disk filled with the background solution. The auxiliary electrode was a platinum wire separated from the analysed solution by a sintered glass disk filled with the background solution. For all voltammetric measurements, the working electrode was a platinum disk electrode (Ø = 2 mm). In these conditions, when operating in DMF (0.1 M TEAPF6), the formal potential for the Fc+/Fc couple was found to be +0.44 V vs SCE.
Bulk electrolyses were performed in a three-compartment cell with glass frits of medium porosity with an Amel 552 potentiostat coupled with an Amel 721 electronic integrator. A platinum wire spiral (l = 53 cm, Ø = 1 mm, S = 16.65 cm2) was used as the working electrode, a platinum plate as the counter electrode and a saturated calomel electrode as the reference electrode. Electrolyses were followed by UV-vis absorption measurements.
3.4 Synthesis of MgP·(Py)2
An amount of 50.0 mg of MgP (1.50 × 10−4 mol) is dissolved in 12 mL of CH2Cl2. Then, 500 μL of pyridine (6.195 × 10−3 mol) are added to this solution. CH2Cl2 is then evaporated yielding deep purple shiny crystals in the remaining pyridine. These crystals are filtered and thoroughly rinsed with n-hexane. Crystals are then dried at 45 °C under vacuum (P ≈ 5.10−2 mbar) for 24 h. 58.4 mg of pure MgP·(Py)2 crystals are obtained (79% yield).
This experimental procedure can be repeated one or several times for purification of crude MgP.
MgP·(Py)2: HRMS (ESI/TOF) m/z calcd for C20H13N4Mg: 333.0985; found: 333.0983 [M–2Py + H]+; 1H NMR (CD3COCD3, 300 MHz, 298 K): δ (ppm) 10.42 (s, meso, 4H), 9.59 (s, β-Pyrr, 8H), 7.55–7.47 (m, o- and p-Py, 6H), 7.05-7.00 (m, m-Py, 4H); 13C NMR (CD3COCD3, 75 MHz, 298 K): δ (ppm) 150.6 (α-Pyrr), 149.7 (o-Py), 136.8 (p-Py), 133.1 (β-Pyrr), 124.4 (m-Py), 106.4 (meso); (λmax (DMF)/nm (log ɛ) 309 (4.26), 380 (4.67), 400 (5.79), 500 (3.28), 536 (4.23) 572 (3.46); Elemental analysis (Found: C, 70.81; H, 4.75; N, 16.52%. Calc. for C20H12N4Mg·2 C5H5N·H2O: C, 70.85; H, 4.69; N, 16.37%).
MgP of higher purity than the starting one can be recovered by heating at 200 °C under vacuum for 24 h (100% yield from MgP·(Py)2).
3.5 Synthesis of ZnP·(Py)2
The experimental procedure performed for MgP·(Py)2 was reproduced on 50.0 mg of ZnP (1.338 × 10−4 mol) yielding 56.9 mg of pure ZnP·(Py)2 crystals (80% yield).
ZnP·(Py)2: HRMS (ESI/TOF) m/z calcd for C20H12N4Zn: 372.0348; found: 372.0357 [M–2Py]+; 1H NMR (CD3COCD3, 300 MHz, 298 K): δ (ppm) 10.43 (s, meso, 4H), 9.63 (s, β-Pyrr, 8H), 7.17–7.05 (m, p-Py, 2H), 6.53-6.40 (m, m-Py, 4H), 5.56 (br s, o-Py, 4H); 13C NMR (CD3COCD3, 75 MHz, 298 K): δ (ppm) 150.7 (α-Pyrr), 147.8 (o-Py), 137.0 (p-Py), 133.1 (β-Pyrr), 124.1 (m-Py), 105.5 (meso); (λmax (DMF)/nm (log ɛ) 307 (4.26), 350 (4.04), 381 (4.65), 401 (5.70), 496 (3.29), 532 (4.22) 566 (3.50); Elemental analysis (on ZnP·Py crystals) (Found: C, 64.92; H, 3,71; N, 15,09%. Calc. for C20H12N4Zn·C5H5N·0.5H2O: C, 65.02; H, 3.93; N, 15.16%).
ZnP of higher purity than the starting one can be recovered by heating at 200 °C under vacuum for 24 h (100% yield from ZnP·(Py)2).
4 Conclusion
Addition of a pyridine excess into a concentrated CH2Cl2 solution of MgP and ZnP affords MgP·(Py)2 and ZnP·(Py)2 adduct crystals. This treatment is, thus, a new method for purification of these metallated porphines. X-ray crystallographic structures of MgP·(Py)2 and ZnP·Py are described for the first time. These crystals are grown from slow cyclohexane diffusion into a CH2Cl2/Py (4/1 v/v) solution of MgP and ZnP. The magnesium porphine adduct is hexacoordinated, hence a totally plane structure, whereas the zinc porphine adduct is pentacoordinated, leading to a cone-type conformation. As confirmed by NMR analyses in CD3COCD3 pyridine adducts are kept in solution. However, in DMF, these adducts are totally dissociated as observed in electrochemistry and in UV-vis absorption analyses. In cyclic voltammetry, the metallated porphines exhibit irreversible oxidation peaks in agreement with the high reactivity of the electrogenerated cation radical. Controlled potential electrolysis at the first oxidation peak, followed by UV-vis absorption spectroscopy, leads to the formation of meso-meso linked dimers and oligomers while a black/purple deposit is observed on the platinum electrode surface. Our efforts are now focused on the characterization of the ZnP-based polymer, which may have interesting applications in molecular devices such as non-linear optical devices, sensors or photoactive species.
Acknowledgments
The authors would like to thank the Centre national de la recherche scientifique, the Conseil régional de Bourgogne and the Université de Bourgogne for financial support. The authors are grateful to Sophie Dalmolin for technical support and to Dr Fanny Chaux for carrying out the ESI-MS analyses.