

1 Introduction
Proteasome is a large multicatalytic proteinase complex that plays an important role in several critical cellular functions, including the processing of proteins involved in cell cycle progression and gene expression [1,2]. The 26S form of the proteasome, responsible for the ATP-dependent degradation of poly-ubiquitinated proteins, is composed of one copy of the catalytic core, known as the 20S subunit, and two copies of the 19S regulatory subunit. The X-ray crystal structure of the 20S subunit from an archaebacterial proteasome revealed that it is a barrel-shaped structure made up of four stacked rings [3]. The two outer rings are each composed of seven identical α-subunits and the two inner rings are each composed of seven identical β-subunits. The catalytic nucleophile is the hydroxyl group of the N-terminal threonine (Thr) of the β-subunit; thus, this enzyme is the first reported member of the threonine proteinase class. The proteasome from higher organisms has the same quaternary structure [4]; however, the seven α and seven β subunits are distinct from one another. The 20S proteasome has been shown to possess multiple catalytic activities, including chymotryptic, tryptic, and peptidylglutamyl-like activities; thus the complex is capable of cleaving most peptide bonds. However, among its various activities, the “chymotrypsin-like” activity of the proteasome has emerged as the biological function of greatest interest. Increased levels of this enzyme and subsequent protein breakdown have been implicated in many disease states including muscular dystrophy, cachexia accompanying cancer, malnutrition, lupus and cancers such as acute leukemia [5]. Proteasome also controls levels of proteins that are critical for cell cycle control, including p53, p27 and cyclin B, and is responsible for the activation of the transcription factor NF-κB through the degradation of its regulatory subunit IκBα [6]. Thus, the development of proteasome inhibitors has emerged as an attractive target for cancer therapy [7].
Over the last years, our laboratories reported on several novel, potent, and selective inhibitors of the chymotrypsin-like activity of proteasome [8–10]. This effort culminated in a potent, selective, and orally bioavailable dipeptide boronic acid inhibitor compound 1 (CEP–18770, delanzomib, Fig. 1) that entered clinical trials for the treatment of myeloma [11]. While our effort was in progress, bortezomib (compound 2, Fig. 1), a structurally different dipeptide boronic acid, was approved by the US FDA for the treatment of multiple myeloma and mantle cell lymphoma, validating the concept of the targeted proteasome inhibition in cancer therapy (for a review on bortezomib, see ref. [12], for an excellent Perspective on development of bortezomib from bench to bedside in record time, see ref. [13]).
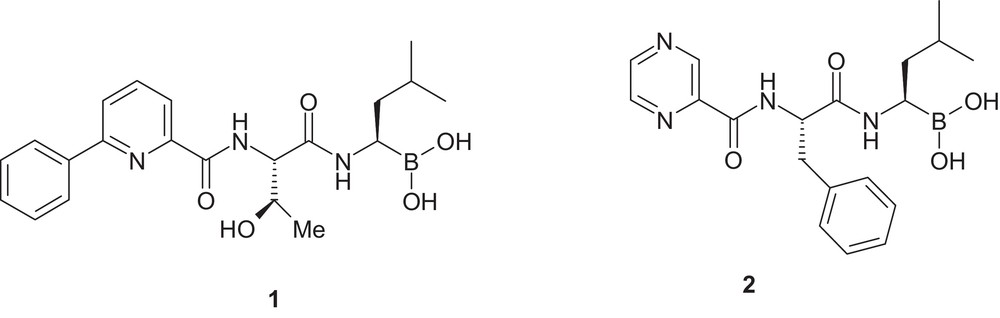
Chemical structures of compounds 1 and 2.
Recently, the US FDA approved carfilzomib containing an epoxy-ketone enzyme reactive group on a tetrapeptide backbone (compound 3, Fig. 2) as the second proteasome inhibitor for clinical use [14].
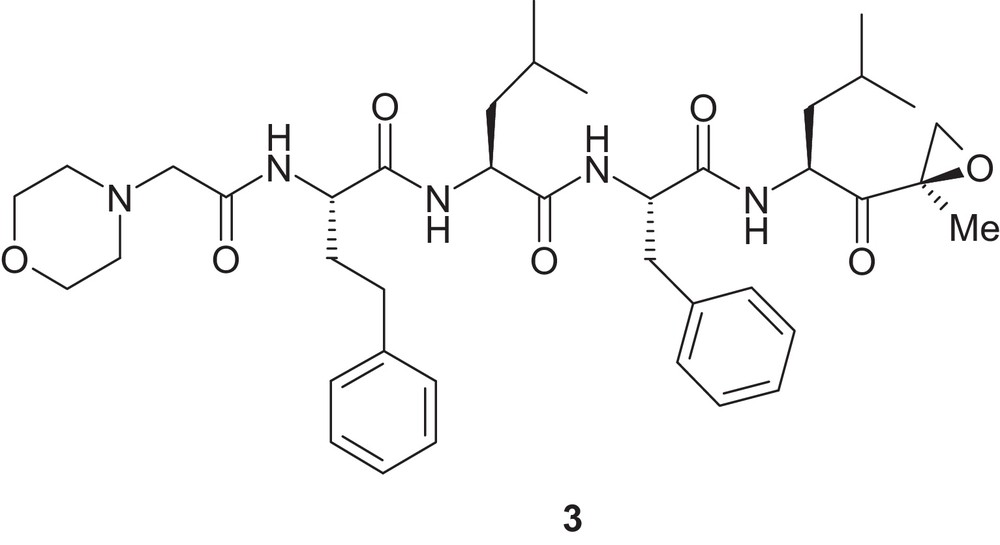
Chemical structure of compound 3.
Approval of compound 3 encouraged us to explore additional enzyme reactive group(s) in parallel to the boronic acid moiety. Edwards et al. reported a series of tri-peptidyl α-keto-2-oxazoline as potent inhibitors of human neutrophil elastase (HNE), a serine protease [15]. Thus, we explored whether an α-keto-2-oxazoline moiety could act as an enzyme reactive group for proteasome, a threonine protease. Herein, we report on our efforts based on the tri-peptide derived compound 4 (Fig. 3).
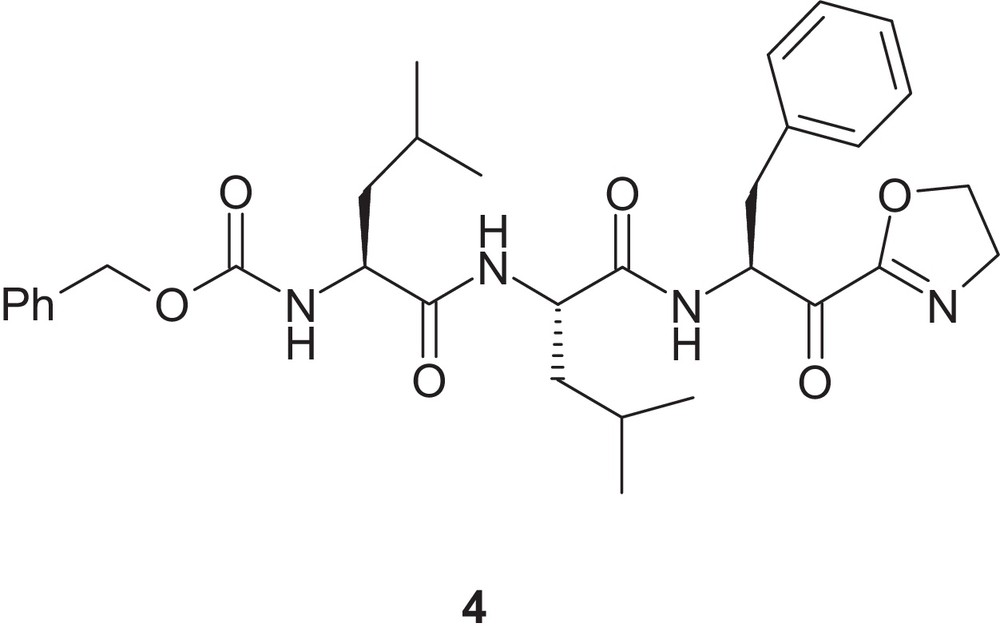
Chemical structure of compound 4.
2 Chemistry
Compound 5 [16] (Scheme 1) was coupled with compound 6 to generate compound 7 that was subsequently de-protected to produce compound 8. Coupling of pre-formed dipeptide Cbz-Leu-Leu-OH with compound 8 afforded compound 9, which in the subsequent step underwent an internal cyclization reaction in presence of Burgess reagent—(methoxycarbonylsulfamoyl)triethyl ammonium hydroxide, inner salt—[17], generating compound 10. Compound 10, on oxidation with Dess–Martin reagent, produced compound 4. It should be mentioned that the route described above was different from that reported by Edwards et al. (not shown) [15]. In their work, the group converted a terminal aldehyde moiety to a cyanohydrin moiety, which in turn was converted into an α-hydroxyimidate moiety. Treatment of the α-hydroxyimidate intermediate with 2-aminoethanol produced the corresponding α-hydroxy-2-oxazoline; subsequent Swern oxidation produced the final α-keto-2-oxazoline (ca. 3% overall yield in last two steps) [15c]. Thus, the overall synthetic yield in our scheme was a significant improvement over that of the previous one.
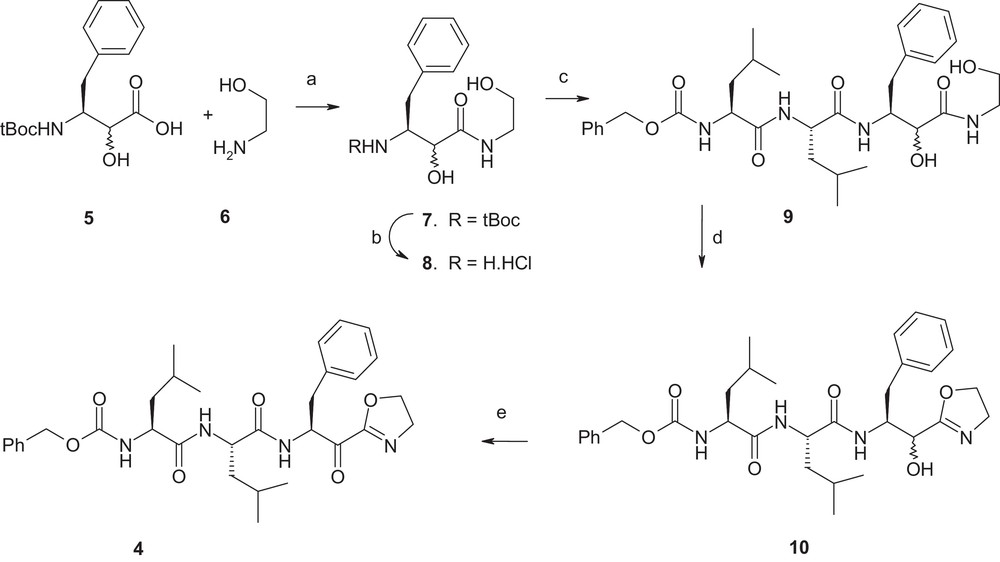
Reagents and conditions: a: HOBt, BOP, NMM, DMF, 0 °C to room temperature, 2 h, 65%; b: 4 N HCl in dioxane, room temp., quantitative; c: Cbz–Leu–Leu–OH, HOBt, BOP, NMM, DMF, 0 °C to room temperature, 2 h, 60%; d: Burgess reagent (freshly opened bottle), THF, heat, 1.5 h, 45–50%; e: Dess–Martin periodinane, methylene chloride, 0 °C to room temperature, 2 h, 65%.
3 Biology and discussion
Compound 4 was assayed for the chymotrypsin-like activity of the human liver proteasome following the published procedure [11]. In the assay, compound 4 displayed ca. 90% inhibition at 10 μM concentration; however, its inhibitory activity dropped to 40% at 1 μM concentration. It should be noted that the corresponding compound with a boronic acid enzyme reactive group displayed an IC50 in the low nanomolar range (1–2 nM). While our work was going on, the literature reported that the threonine residue, which happens to be the N-terminus of the proteasome catalytic subunits, irreversibly attaches to compound 3 via the formation of a morpholino ring. Normally, the alcohol side chain of the threonine attacks amide bonds, but when presented with compound 3, it preferentially reacts with the ketone portion of the epoxy-ketone group. This sets the epoxide group amenable to nucleophilic attack by the amino group of the N-terminus, leading to a morpholino ring adduct (scheme not shown) [18]. It appears that a similar type of transformation involving compound 4 has not been possible due to the presence of a less reactive oxazoline ring next to the keto group. On the other hand, while in a serine protease the hydroxyl group interacting with an inhibitor belongs to a primary alcohol moiety, in a threonine protease, the hydroxyl group is a part of a secondary alcohol moiety (due to the presence of the neighboring methyl group). Thus, a non-favorable steric interaction might be at play, giving rise to low potency. The emergence of the clinical compound 1 at this stage prohibited any further expansion of this series.
4 Conclusion
In this manuscript, we described our exploration involving compound 4 to uncover whether an α-keto-2-oxazoline moiety could be a viable potent enzyme reactive group able to inhibit the chymotrypsin-like activity of human liver proteasome. The compound inhibited the chymotrypsin-like activity of proteasome at higher dose (90% inhibition at 10 μM), while activity dropped at a lower dose.
Acknowledgments
The author thanks Dr. Satish Mallya for the assay data. Drs. James C. Kauer and Mohamed Iqbal provided their support and encouragement during the course of this research.
Vous devez vous connecter pour continuer.
S'authentifier