

1 Introduction
Since their discovery, X-ray related techniques have gained wide acceptance in materials science. Among them, X-ray fluorescence (XRF) [1–4] and X-ray diffraction (XRD) [5–8] are two well-established and powerful tools for a non-destructive analysis of materials with broad applications in science and industry. In medicine, numerous investigations of biological entities have also been performed with these techniques [9–15]. It is also possible to combine both XRF and XRD on the same experimental set-up to obtain simultaneously elemental and structural information on the sample. Furthermore, technical developments in X-ray laboratory sources, optics and detectors in recent years have allowed performing measurements at a local scale with micrometric X-ray beams (∼10 μm in diameter) to investigate possible heterogeneities. The fact that such measurements combining XRF and XRD are now available in the laboratory offers a significant advantage, albeit the weaker X-ray intensity, over similar experiments on synchrotron radiation facilities for which experimental access is scarce. XRD and XRF techniques can also be implemented at the hospital to help to characterize pathologies or for disease diagnosis through in vivo experiments [16].
In this contribution, we would like to underline through different kinds of XRF and XRD experiments, that chemical and structural characteristics of pathological calcifications [17–19] give invaluable information to clinicians. For example, most of the XRF experiments in medicine deal with trace elements which are intrinsic components of numerous biological systems, since a third of all known proteins contain metal cofactors as catalytic components [20]. Moreover, some trace elements are directly related to a specific disease (aluminum to Alzheimer's disease [21]; copper accumulation to Wilson disease [22]) or are present in therapeutic drugs or diagnostic agents (cis-platin in chemotherapy [23], gadolinium in magnetic resonance imaging [24] as well different kinds of nanoparticles [25]). After a brief description of the principles of XRF and XRD techniques, we will present a set of experimental data collected on such in-lab facilities.
2 Basics of XRD and XRF techniques
2.1 Interaction of X-rays with matter
X-rays considered as an electromagnetic radiation possess the duality of being both electromagnetic waves with short wavelength λ ranging typically from 0.1 to 1 Å and massless non-charged particles named photons with energies E from 1 to 100 keV. X-ray photons can interact with the electrons in matter by three different mechanisms as shown in Fig. 1: (i) the photo-electric effect when matter absorbs the incident X-ray photon and then emits electrons or X-ray photons, (ii) Compton scattering, where the scattering of the incident X-ray photon is accompanied by some energy transfer to the electron (inelastic scattering) and (iii) Thomson (elastic) scattering where the scattered X-ray photons have the same energy as in the incident beam. Note that at low energy, the photoelectric effect is more important than Compton scattering.
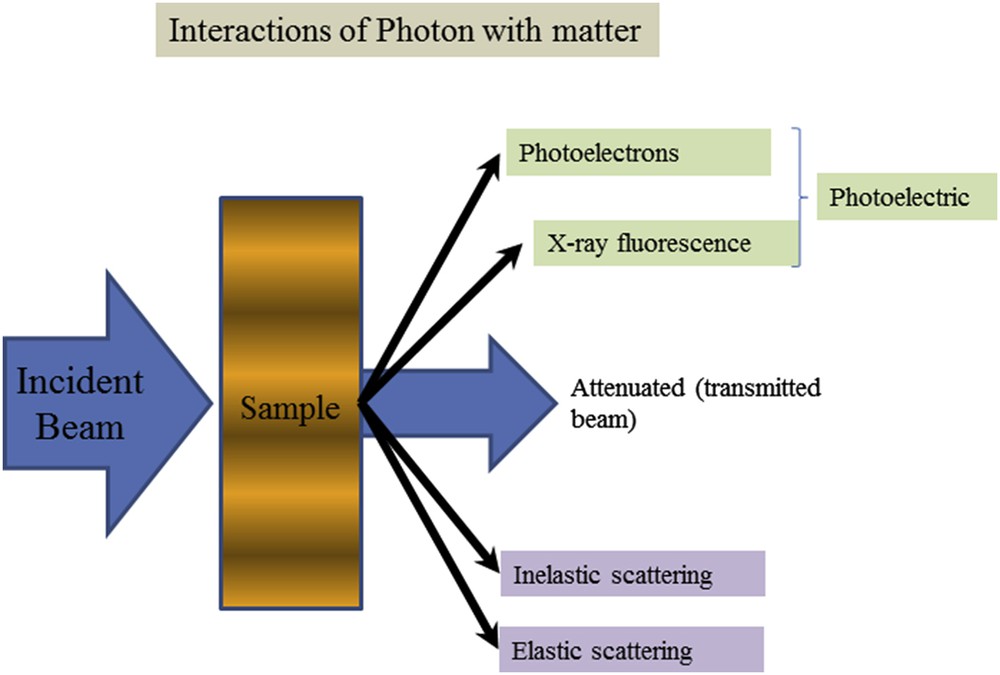
Schematic representation of the different interactions between X-rays and matter.
The photo-electric effect is responsible for the XRF and Auger photoelectron phenomena whereas XRD is related to the Thomson scattering process. Note that photoelectron spectroscopy is usually subdivided according to the source of exciting radiation into X-ray Photoelectron Spectroscopy (XPS) which uses soft X-rays (with a photon energy of 200–2000 eV) to examine core-levels and Ultraviolet Photoelectron Spectroscopy (UPS) which is based on UV radiation (with a photon energy of 10–45 eV) to examine valence levels.
2.2 X-ray fluorescence
In the photoelectric absorption process, incident photons with quantum energy (hν) interact with an absorber atom and completely disappear. In their place, photoelectrons are ejected from one of the bound shells of the atom with energy equal to hν−Eb (where Eb represents the binding energy of the photoelectron in its original shell, see Fig. 2a). During the atomic relaxation, the filling of the inner shell vacancy can produce X-ray fluorescence radiation with characteristic energies of the atom and also other photoelectrons named Auger electrons [26,27].
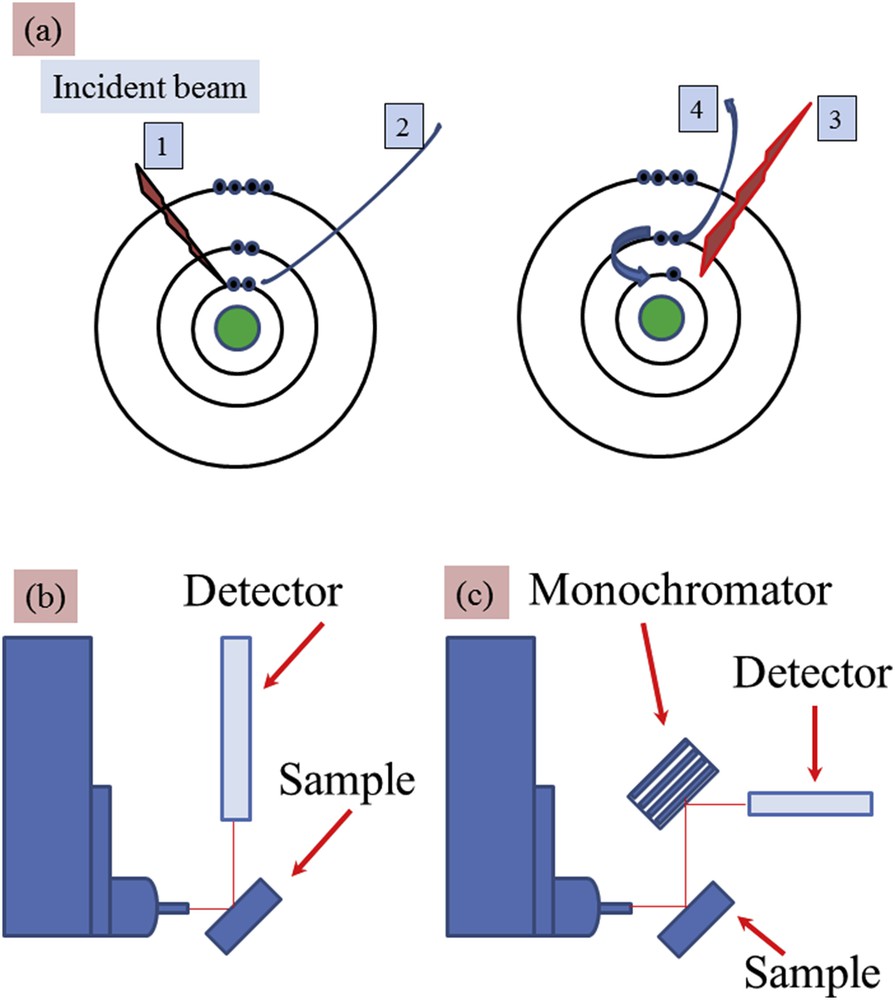
(a) Simple representation associated with the X-ray fluorescence process: 1 – incident X-ray photon, 2 – emitted photoelectron, 3 – X-ray fluorescence photon or 4 – Auger electron (b, c) schematic experimental set-ups corresponding to (b) energy-dispersive (ED) and (c) wavelength-dispersive (WD). For the ED experimental device, the energy resolution is given by using the detector (around 150–200 eV) while for the WD one, the energy resolution is defined by using the monochromator (around 1–2 eV).
The characteristic energies of the X-ray fluorescence radiation emitted from a sample enable us to identify unambiguously and quantify the different elements present. Such experiments can be performed on solid, liquid or thin-film samples. Finally, note that X-ray fluorescence can be also induced by gamma-emitting radioisotopes (241Am, 109Cd, 153Gd, and others), protons [28–31], electron sources and synchrotron light sources which produce highly intense, coherent, monochromatic X-ray beams [32–36].
XRF can be in general measured with two types of experimental set-ups: (i) energy-dispersive (ED-XRF) and (ii) wavelength-dispersive (WD-XRF) spectrometers (Fig. 2b). In the latter, the fluorescence radiation is diffracted by an analyzing single crystal optic just after the sample to select with a high sensitivity a very narrow wavelength or characteristic energy from an element of interest in the sample. In the ED-XRF mode, the detector allows us to detect simultaneously a broad range of characteristic energies possibly related to several elements, the selectivity depending on the energy resolution and counting rate performance of the detector.
Laboratory X-ray sources must provide sufficiently high energy radiation to be able to excite the characteristic energies of the broadest range of elements of the periodic table; they are electric generators producing X-rays emission radiation from a high-Z element anode like molybdenum, silver or tungsten. X-ray optics, crystalline monochromator or multilayer mirror is coupled with the generator to deliver a focusing monochromatic beam onto the sample that greatly improves the signal-to-noise ratio for measurements of the characteristic energies of elements present in the sample (Fig. 3a).
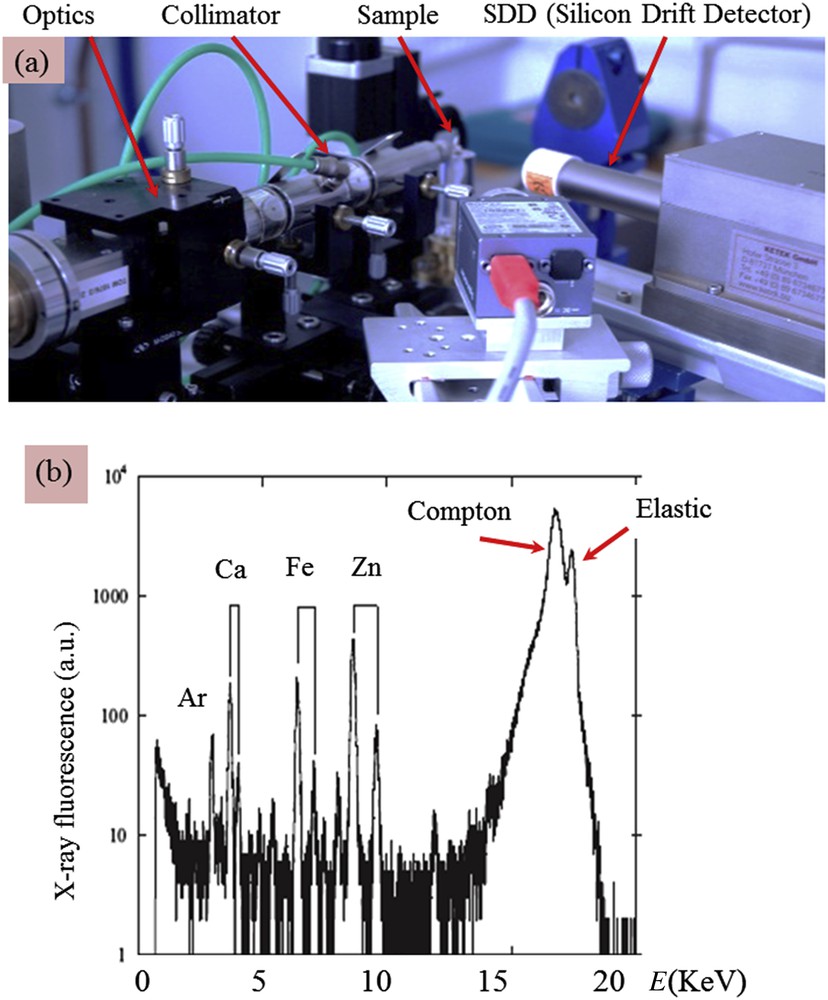
(a) Picture of the in-lab EDXRF experimental set-up at LPS with monochromatic X-ray Mo Kα radiation, the different elements are indicated by arrows (b) typical X-ray fluorescence measured for a pathological calcification (a kidney stone). The contributions of trace elements such Fe and Zn are clearly visible.
In Fig. 3b, we can see clearly the contributions of Ar (Kα = 2958 eV), Ca (Kα = 3691 eV, Kβ = 4012 eV), Fe (Kα = 6404 eV, Kβ = 7058 eV) and Zn (Kα = 8638 eV, Kβ = 9572 eV). Other contributions exist coming from inelastic process (Compton scattering) as well as the elastic scattering related to the energy of the incident photons generated by the molybdenum anode. Finally, the absence of a signal at low energy (typical elements below Al) cannot contribute to the spectrum due to air absorption, the high difference between the incident energy of the Mo Kα and the binding energy of the electrons of low Z elements which induces a low probability of photoionization, and the out-of-range (below 1 keV) sensitivity of the silicon drift detector.
XRF is a representative multi-element technique for the analysis of trace elements. This technique constitutes a simple and rapid procedure of analysis for a large number of samples, and has high sensitivity and low-detection limits, enabling the determination of element concentrations at trace levels.
2.3 X-ray diffraction
At the atomic level, each atom of the sample defines a scattered wave built from an incident X-ray beam. Wavelengths of X-rays being of the order of inter-atomic distances in the matter, the spatial periodic configuration of atoms causes phase interferences between scattered waves, thus giving rise to the diffraction pattern. XRD was discovered in 1913 by von Laue and by Bragg father and son [37] who established the well-known Bragg formula, 2d sin θ = n λ (n is a positive integer), which connects the wavelength λ of the incident X-rays, the atomic lattice interplanar distance d and the deviation angles θ corresponding to the constructive interferences of scattered waves. From an experimental point of view, the XRD pattern is measured through a collection of diffraction peaks at different Bragg angles 2θ, their angular positions and their intensities being intimately related to the spatial arrangement of the atoms. In Fig. 4, the different cases of diffraction from a single crystal (long range order periodic atomic arrangement), a polycrystalline powder (random orientation of small crystals) and a powder of crystallites with a preferential orientation (texture) are shown to illustrate the corresponding diffraction patterns.
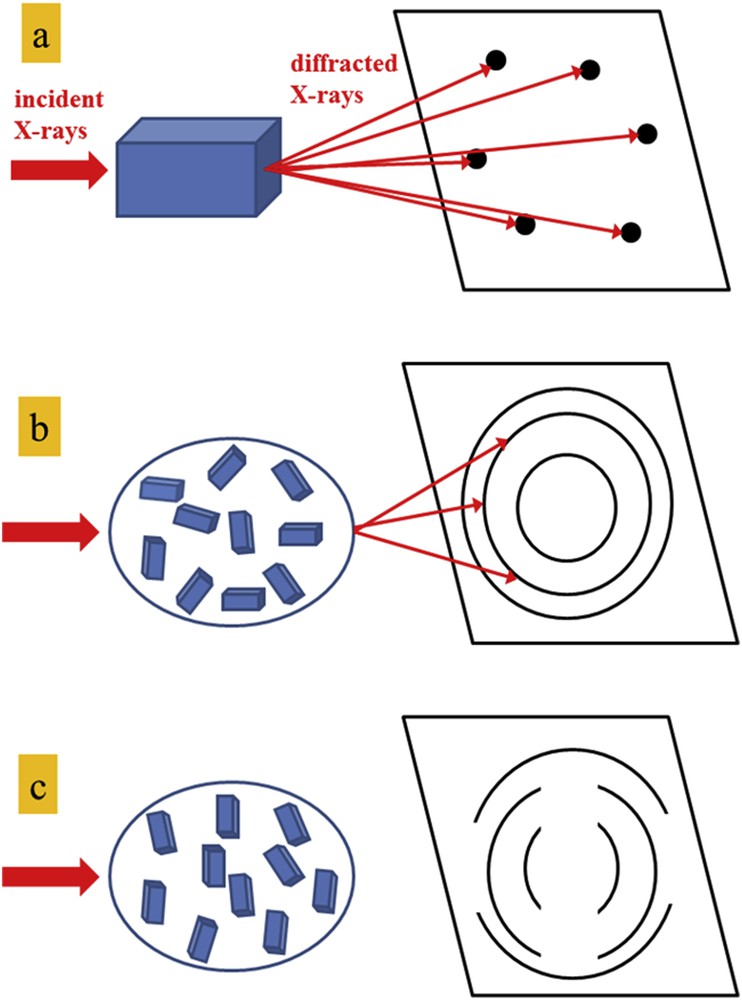
Schematic representation of the diffraction patterns on a plane detector from a single crystal (a), a polycrystalline powder (b) and a preferential orientated powder (c).
XRD can bring different kinds of information on the sample such as the identification of the crystallographic phases, its microstructure or its texture [38–41]. Generally, XRD is used for phase identification by comparing the values of diffracted peaks with the ones tabulated in a data base [42] and can also determine the structure of new compounds. Recently, we have observed stone formation in rats subjected to diets containing l-choline tartrate. Using powder and single-crystal XRD, it is possible to determine the crystallographic structure of completely new chemical phase [43–45]. It is also possible to follow the formation and reactivity of a phase or observing changes in lattice constants under stress/change as, for example, to study prosthesis under mechanical stress [46–50].
2.4 In-lab micro-XRD and XRF
X-ray micro-diffraction (μXRD) is well adapted to investigate the local variations of crystallographic properties such as the crystalline phase identification, the microstructure or the preferential orientation. It is also valuable to combine such structural information with a local elemental analysis by μXRF. The main concern to perform experiments at a micrometric scale on a lab X-ray source is to obtain an X-ray microbeam sufficiently intense. Recent developments of instrumentation in X-ray optics and detectors allow optimizing the detection of low-intensity signals with a monochromatic Cu Kα X-ray microbeam with dimensions typically of 20 μm in diameter [51]. In Fig. 5, the experimental setup includes a multilayer mirror associated with a high-brilliance rotating-anode generator delivering a well-defined beam of sufficient intensity to measure μXRD and μXRF signals within a short time (time scale from 1 to 10 min according to the sample studied). Detectors using recently developed technology, such as hybrid-pixel detectors are well suited to measure the low-intensity signals. High-resolution motorized translation stages allow performing area mapping studies on materials.

Picture of the in-lab μXRD/μXRF instrument at LPS with monochromatic X-ray Cu Kα radiation, the different elements of the setup are indicated.
3 Randall's plaque, XRD and XRF
Crystallization may be induced in metastable supersaturated solutions by spontaneous nucleation when a certain degree of supersaturation is reached (homogeneous nucleation) or by an additional support made of seed crystals or polymeric substances (heterogeneous nucleation). One of the main challenges for urology in the 21st century is related to a heterogeneous nucleation namely the Randall's plaque (RP) [52–55]. These subepithelial calcifications of the renal papilla (Fig. 6) act as an anchor for calcium oxalate monohydrate crystals and considerable efforts have been made over the last decades to identify the biochemical parameters responsible for this ectopic calcification [56–65]. More precisely, a detailed description of the interface area between a RP and a whewellite (CaC2O4·H2O, calcium oxalate monohydrate, COM) stone at the mesoscopic scale shows randomly distributed COM crystals trapped in the carbonated apatite of RP [17]. S.C. Kim et al. [66] have observed that stone formation is in fact proportional to the papillary surface coverage by RP. It is also worthwhile to note that such calcification of the renal papilla may eventually become large enough to occlude the tubular lumina and obliterate the tubular epithelium [67].

Randall's plaques observed at the mesoscopic scale. (a, b, and c) Randall's plaques made of calcium phosphate apatite (RP, red arrows) are positioned on the kidney stone (KS, blue arrows) constituted of calcium oxalate monohydrate. Calcium phosphate apatite can be present within the tubular lumen (1 in b), while other tubules are empty with calcified walls (2 in b). In some cases (d), Randall's plaques are found at the surface of kidney stones made of calcium oxalate dehydrate (KS, blue arrows).
This deposit is also present at the surface of kidney stones [68,69]. In that case, even if other phosphate phases such as whitlockite or brushite can be found as minor components (less than 5%), calcium phosphate apatite (CA) as well as amorphous carbonated calcium phosphate (ACCP) are the major components of most RPs. In order to improve our knowledge on the chemical composition of RP present at the tip of the papilla, we use a technique specific to synchrotron radiation namely, X-ray absorption spectroscopy [70,71]. Such a technique had already been used in several investigations regarding pathological and physiological calcifications [72–76]. In the case of Randall's plaque, such experiments give for the first time direct structural evidence of the presence of ACCP as a major constituent [77].
Based on these previous achievements, we have performed XRD and XRF experiments on a set of RP extracted using a stereomicroscope from seven human KSs and one kidney stone made of calcium phosphate apatite (used in this study as a reference compound) [78]. As it can be observed in Fig. 7, XRD diagrams correspond to a relatively fine (002) diffraction peak (at 2θ = 26°) and several poorly resolved lines constituting a broad peak between 2θ = 30° and 2θ = 35°. From these observations, it is quite easy to conclude that RP are made of nanocrystalline particles displaying the usual anisotropy along the c axis [79–84].
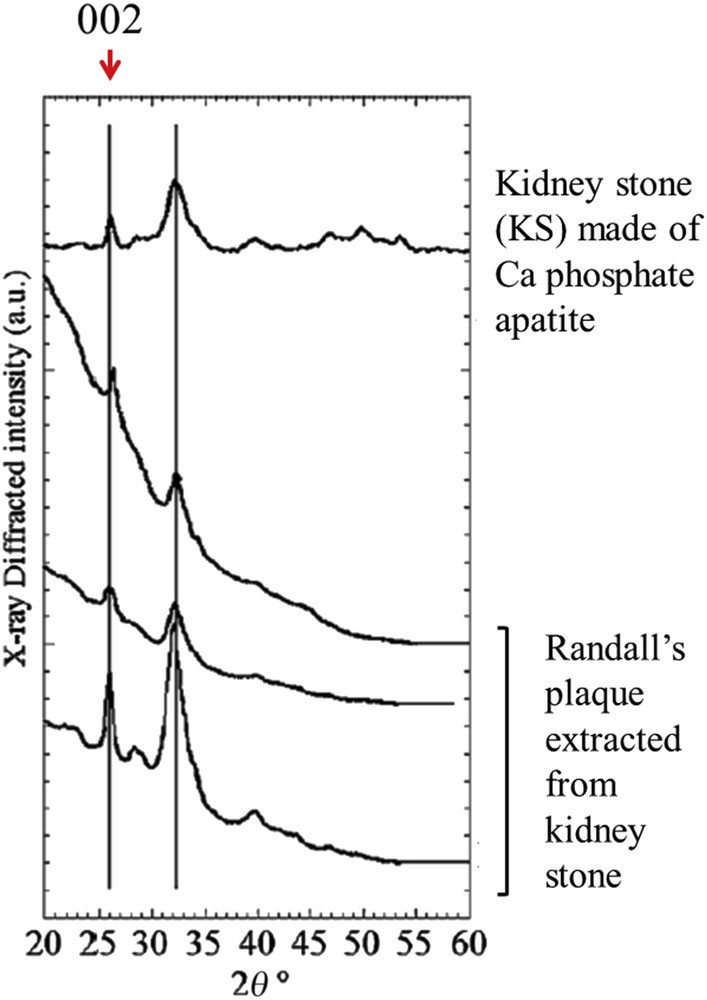
X-ray diffraction diagrams collected with monochromatic copper radiation (λ = 1.542 Å) for different RP and a KS made of apatite used as a biological apatite reference sample in this study (after Ref. [78]). Measurements were performed with an X-ray photosensitive image plate.
More interesting results have been collected by XRF (Fig. 8). The contributions of different trace elements such as Zn, Pb and Sr have been measured. These elements have divalent cations and an ionic radius close to that of Ca2+ in line with the Goldschmitt rules which are associated with insertion processes [85]. Such data are in line with the fact that apatitic calcium phosphates offer a wide range of ion substitutions which can modify the physical–chemical and biological properties of apatites [86–88]. The quantitative analysis provides evidence that Zn levels are dramatically increased in RP by comparison to the ones corresponding to kidney stones of the same composition. The amount of Zn (μg/g) is between 15 and 32 in kidney tissue, around 0.1 in urine, 1000 in CA kidney stones compared to 5600 in RP (up to 10,000 in some cases) [78]. Such a high level of Zn suggests that formation of calcified deposits within the medullar interstitium is clearly a pathological process involving a tissue reaction in agreement with the decrease of the kidney function [79–93].
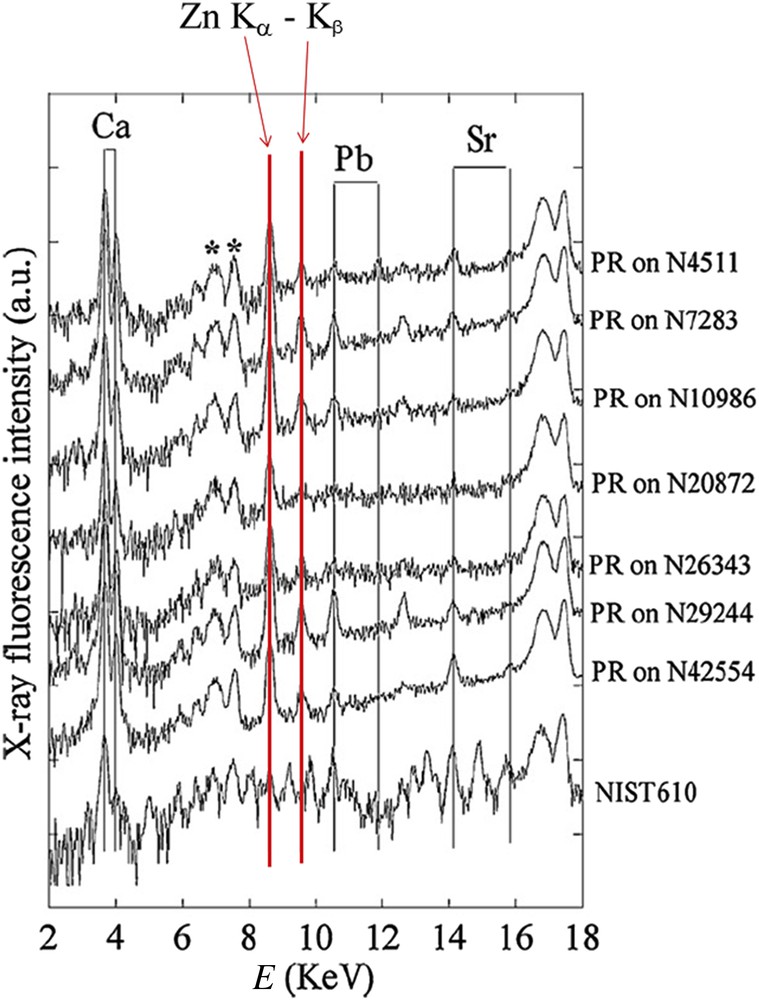
Typical X-ray fluorescence spectra collected for the RP and calibration compound NIST610. For RP, we can clearly see the contributions of Ca (EKα = 3.691 keV, EKβ = 4.012 keV), Zn (EKα = 8.638 keV, EKβ = 9.572 keV), Pb (ELα = 10.551 keV, ELβ = 12.613 keV) and Sr (Eα = 14.165 keV, EKβ = 15.835 keV) (after Ref. [78]). Measurements were performed with a germanium detector.
4 Hyperoxaluria, hypercalciuria and XRF
As reported for a long time, weddellite (CaC2O4·2H2O, calcium oxalate dehydrate) is mainly related to hypercalciuria while whewellite is associated with hyperoxaluria [94,95]. In a previous investigation [33], we have noticed that Zn and Sr contents were higher in weddellite (290 μg/g and 125 μg/g, respectively) than in whewellite (42 μg/g and 61 μg/g, respectively) stones, suggesting significant differences in the urine, including its content of Zn and Sr, which is known to follow calcium pathways, seems to be increased in kidney stones associated with hypercalciuric states [33]. A simple question arises: what is the Zn content in kidney stones made of whewellite for which a phase conversion has occurred, considering that crystalline conversion is observed for weddellite, which is able to transform to whewellite. Crystalline conversion is suspected when the morphological type of the stone is type II, typical for the weddellite structure with large specific bipyramidal crystals while the infrared spectrum clearly demonstrates that whewellite is the main component of the corresponding areas of the stone. By contrast, stones initially made of whewellite exhibit a type I morphology, very useful for assessing hyperoxaluric states as a driving force of stone formation. In the case of crystalline conversion, the initial stone morphology is preserved for a long time, allowing easy detection of a conversion process by comparison between the morphology and composition of the stone. Because crystalline conversion from weddellite to whewellite implies a dissolution-crystallization process, it was expected that all stones mainly composed of whewellite, irrespective to the initial phase, should contain significantly less Zn and Sr than weddellite stones. To answer this question, a new set of measurements have been performed (Table 1). The ratio calculated from the areas of the respective fluorescence peaks of each element allows us to carry out a semi-quantitative analysis to compare the relative ratio of two elements in each sample. As we can see, even if a large modulation of the Zn/Ca ratio is observed in this series, the statistical analysis of these measurements is able to distinguish between native whewellite and whewellite coming from weddellite as assessed by the morphology of the stones. Studies based on XRF confirm our the first results suggesting a different biological environment for whewellite and weddellite crystallization, weddellite being associated with a Zn-rich environment, partly favored by an inflammation process [33,96]. Thus, whewellite resulting from weddellite conversion by a dissolution-crystallization process (type-II morphology) preserves the initial environment and presents a high content of Zn (and at a less extent strontium) by comparison to native whewellite (type-I morphology). The Ca/Zn and Ca/Sr ratios were, respectively, 16.1 ± 43.8 and 4.6 ± 7.5 for weddellite versus 300.8 ± 28.3 and 29.3 ± 4.9 for whewellite (p = 0.000053 for Ca/Zn and p = 0.015 for Ca/Sr). It could be of interest to assess the relative ability of Zn and Sr adsorption and/or absorption on weddellite and whewellite crystals in in vitro experiments of synthesis of weddellite or whewellite crystals. As observed in Table 1, the stone morphology compared with the stone composition determined by infrared spectroscopy helps us to identify crystalline conversion and thus stone etiology.
Zn/Ca ratio obtained from calculated areas of the fluorescence peaks of Ca (Kα + Kβ) and Zn (Kα) measured by XRF for different calcium oxalate kidney stones. (Wh: whewellite; Wd: weddellite; CA: calcium phosphate apatite; Prot: proteins; WK: whitlockite). Note that ratio values are not molar quantity.
| Stone reference | Ca/Zn | Ca/Sr | Sex | Morphological type of the stone surface | Morphological type of the stone section | Stone composition (number values are indicated in %) |
| 68,316 | 9.9 | 0.9 | M | IIb | IIb+IVa | Wh 60+CA 21+Wd 15+Prot 4 |
| 56,486 | 14.9 | 9.3 | M | IIa+IVa | IIb+IVa | Wd 44+CA 30+Wh 20+Prot 6 |
| 68,076 | 15.6 | 2.08 | M | IIb | IIb | Wh 52+Wd 40+CA 5+Br 3 |
| 68,285 | 19.2 | 10 | M | IIb | IIb | Wd 56+Wh 35+CA 7+Prot 2 |
| 68,351 | 20.8 | 0.86 | M | IIb+IVa | IIb+IVa | Wd 36+CA 32+Wh 28+Prot 4 |
| 68,114 | 110 | 7.8 | F | IIb+Ia | Ia+IIb | Wh 50+Wd 45+CA 3+Prot 2 |
| 68,097 | 176 | 26 | M | IIb+Id | Ia+IIb | Wh 80+Wd 11+CA 6+Prot 3 |
| 57,503 | 230 | 10.8 | M | Ie | Ie | Wh 100 |
| 68,096 | 240 | 16.6 | M | Ia | Ia | Wh 100 |
| 68,231 | 261 | 30 | F | IIb+IVb+Ia | Ia | Wh 36+Wd 26+CA 25+WK 10+Prot 3 |
| 68,074 | 272 | 23.9 | M | Ia+IIb | Ia | Wh 65+Wd 30+CA 3+Prot 2 |
| 68,214 | 286 | 39 | F | Ia | Ia | Wh 100 |
| 8624 | 328 | 78 | F | Ia | Ia | Wh 100 |
| 68,140 | 380 | 7.9 | M | Ia+IIb | Ia | Wh 64+Wd 28+CA 4+Prot 4 |
| 60,982 | 380 | 38 | M | Ie | Ie | Wh 100 |
| 68,102 | 410 | 30 | F | Ia | Ia | Wh 96+CA 2+Prot 2 |
| 68,258 | 537 | 43 | M | Ia>IIb | Ia | Wh 80+Wd 15+CA 3+Prot 2 |
5 Brushite and X-ray fluorescence
Brushite (dicalcium phosphate dihydrate, CaHPO4·2H2O), named after the American mineralogist George Jarvis Brush (1831–1912), [97] is a chemical compound which has a pivotal role in medicine. Brushite as well as octacalcium phosphate and amorphous calcium phosphate apatite have been proposed as the earliest solid calcium phosphate phase deposited in bone [98]. This is due to the fact that if brushite is more stable than hydroxyl-apatite in sufficiently acid solution, brushite hydrolyzes directly into hydroxyl-apatite at sufficiently basic pH [99]. Brushite is also used in prosthesis to improve the corrosion resistance and biocompatibility of metallic alloys [100,101] or in cement implants for bone regeneration [102,103]. Finally, brushite can be identified in different pathological calcifications [104,105].
In urology, the main problem of brushite stones is their resistance to fragmentation through extracorporeal shockwave lithotripsy [106]. Through microcomputerized tomography, it has been shown that resistance roughly correlates with the stone density and increases with the brushite mineral content, which is consistent with clinical experience regarding patients with brushite calculi [107]. The fact that multiple lithotripsy sessions in a patient give rise to the pathogenesis of brushite stones has also been assessed [108,109].
D. Ackermann et al. [110] reported that the state of saturation towards brushite was exclusively determined by urinary calcium and pH, the latter below 5.5 showing a high influence on brushite solubility. As underlined by C.Y. Pak et al. [111], patients with predominantly brushite stones could be distinguished from those with predominantly hydroxyapatite and calcium oxalate stones by higher urinary supersaturation with respect to brushite mainly due to hypercalciuria from absorptive hypercalciuria. Finally, R. Siener et al. [112] noticed that hypercalciuria, a diminished citrate excretion and an elevated pH turned out to be the major urinary determinants of brushite stone formation.
Brushite is the most acidic form of calcium phosphates identified in urine and stones. While carbapatite is a very common component of urinary calculi, identified in various proportions in about 85% of all stones, brushite is much less frequent and is observed in less than 5% of urinary stones. One explanation could be that brushite is considered an instable phase of calcium phosphate which is able to transform with time to carbapatite [113]. However, the occurrence of brushite has been increasing for the past three decades. In addition, the sex ratio (H/F) of patients producing brushite stones is dramatically different from the one obtained for patients producing carbapatite stones (the ratio is 3.08 for brushite and 0.68 for carbapatite stones, p < 0.00001). Finally, the strong relationship between brushite and some pathological states or biochemical disorders suggests that brushite could have a special place in nephrolithiasis. In fact, in contrast to carbapatite, brushite is strongly linked to hypercalciuric states (87% of patients producing brushite stones exhibit hypercalciuria) [114].
Moreover, among well-defined metabolic diseases, primary hyperparathyroidism is especially frequent in the case of brushite stones (close to 20% of cases). Other causes may be considered such as urinary tract infection, medullary sponge kidney, phosphate leak, mellitus diabetes or tubular acidification defect. However, these pathological conditions often induce mixed calcium phosphate stones also containing high proportions of carbapatite. It seems that multiple biochemical disorders such as poorly acidic urine pH, high calcium, high phosphate and low citrate concentration and/or excretion are required for brushite stone formation [114].
Based on the well-established correlation between the stone morphology and pathology [115–119] for COM [115] and for some types of carbapatite stones [65], we examined the brushite stone morphology and the content of the main trace elements by comparison with the calcium content. Four groups of brushite stones were selected according to the pathology: primary hyperparathyroidism (n = 8), hypercalciuria (n = 7), mellitus diabetes (n = 4) and urinary tract infection (n = 8). At the macroscopic and mesoscopic scales, it was not possible to distinguish between all these pathologies from the stone morphology or crystallite morphology assessed by SEM examination. To illustrate this difficulty, the crystallite morphology through a field emission scanning electron microscope (FE-SEM) implemented at the Laboratoire de Physique des Solides (LPS) of brushite kidney stones is shown in Fig. 9.

FESEM photographs of brushite kidney stones. These observations show clearly that brushite kidney stones may display very different morphologies. Unfortunately it was not possible to relate a specific morphology to a particular disease (i.e. primary hyperparathyroidism, hypercalciuria, mellitus diabetes, and urinary tract infection). (f) Phase transition between brushite and apatite observed at a high magnification.
Trace elements identified by X-ray fluorescence (Figs. 10 and 11) were Br, Pb, Sr, Zn and Fe. The results were expressed as the ratio of each element to the calcium content (Table 2). Our data failed to find any difference between groups concerning the ratio Sr/Ca or Br/Ca. In contrast, we observed that the Zn/Ca ratio was significantly higher in stones from diabetic patients in comparison to hypercalciuria (0.0704 ± 0.0108 vs 0.0253 ± 0.008 (p < 0.01). These data suggest that other factors than hypercalciuria (often present also in diabetic patients because of a defect in calcium reabsorption) could explain the high content in Zn. Especially, the inflammatory process could be involved in those patients [120,121]. Of note, the presence and sometimes the relatively high content of Br in several brushite stones remains unexplained and deserves further investigation.
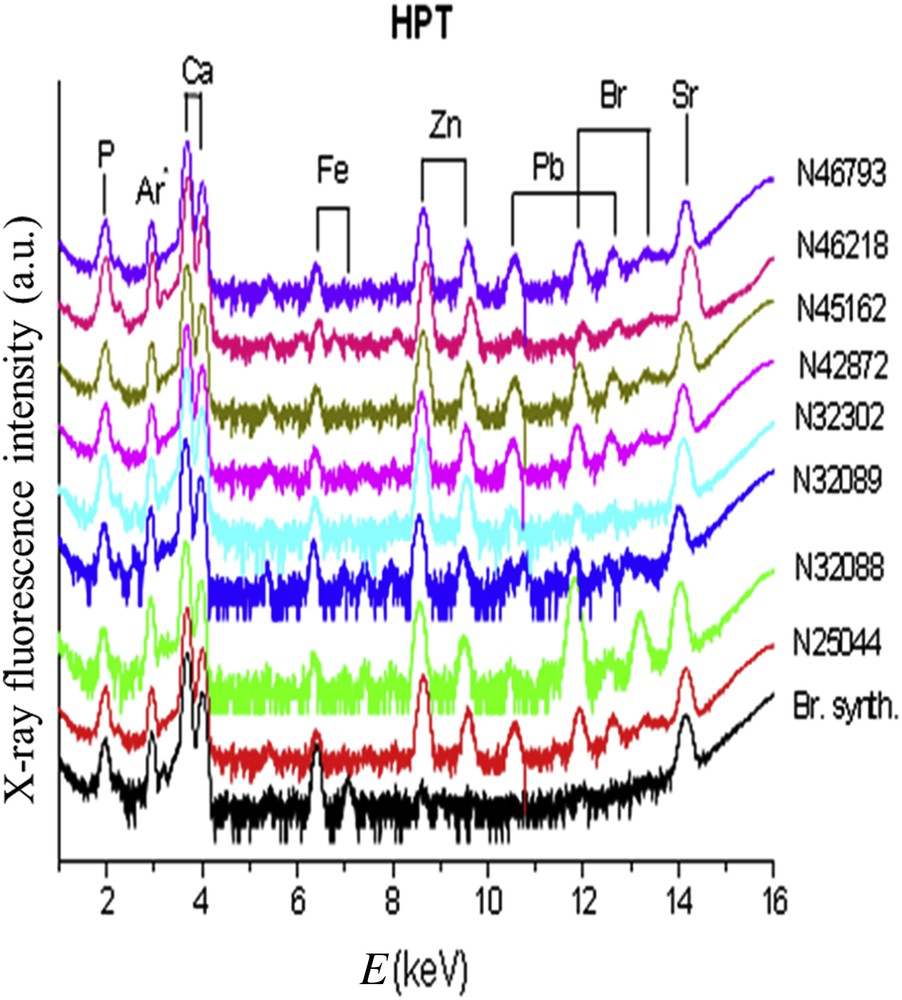
X-ray fluorescence spectra collected for brushite kidney stones related to primary hyperparathyroidism (HPT). The X-ray fluorescence coming from Ar is due to the fact that measurements are performed under ambient pressure.
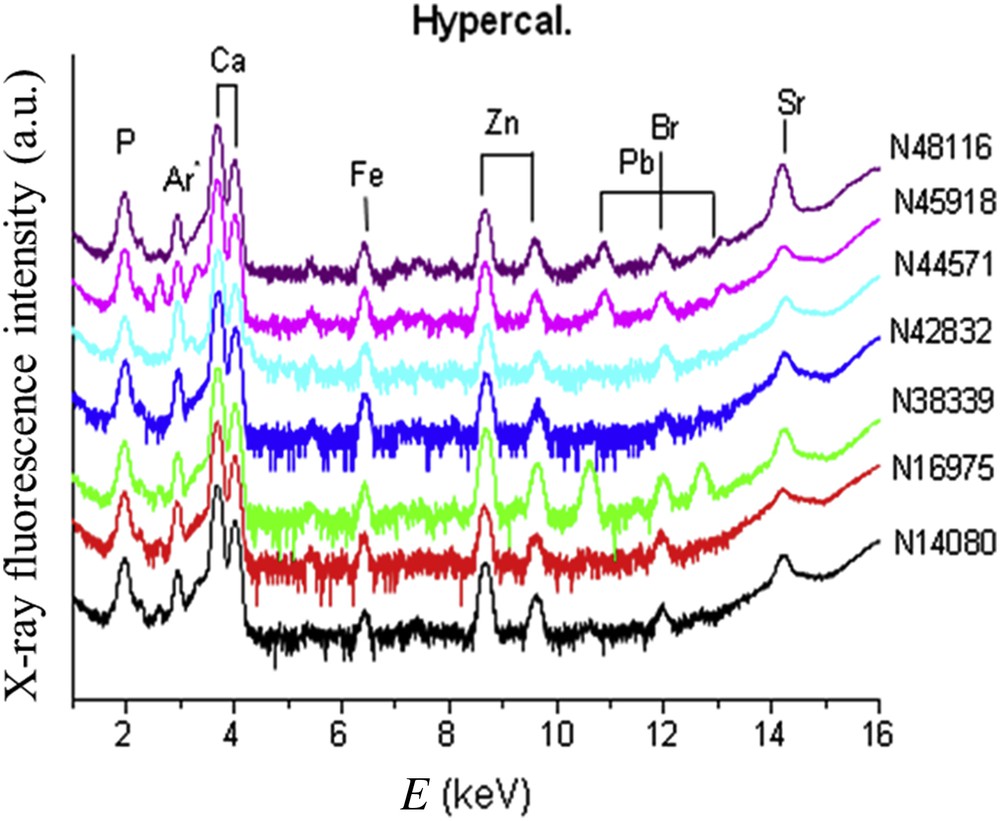
X-ray fluorescence spectra collected for brushite kidney stones related to hypercalciuria.
Zn/Ca ratio obtained from the fluorescence of Ca (Kα + Kβ) and Zn (Kα) measured by XRF (counting acquisition of 20 min for each spectrum) for different brushite kidney stones related to hyperparathyroidism, hypercalciuria, diabetes and urinary tract infections.
| Disease | Zn/Ca ratio |
| Primary hyperparathyroidism | 0.0417 ± 0.0082 |
| Hypercalciuria | 0.0253 ± 0.008 |
| Diabetes | 0.0704 ± 0.0108 |
| Infections | 0.0423 ± 0.0078 |
6 Toward in vivo XRF at the hospital
In the literature, numerous studies (Table 3) have investigated the relationship between the content of trace elements and different diseases [12,122–186] for different kinds of biological samples. For example, L.M. Marco et al. [130] and C.G.L. Canellas et al. [149] have determined the Zn/Cu ratio in serum samples. D. von Czarnowski et al. [127] have evaluated through total reflection X-ray fluorescence analysis the distribution of trace elements in carcinomas of the digestive tract and in normal tissues of human stomach, colon and rectum in correlation. Finally, XRF offers the possibilities to determine the distribution of Pt based anticancer drugs within tumor and in its proximity [126].
Biological tissues and diseases studied by XRF.
| Biological tissues | Etiology |
| Hair | Environmental disease [129,151] |
| Brain | Neurodegenerative diseases [131,134,142–144] |
| Breast | Cancer [138] |
| Bone | Relationship between Pb exposure and memory impairment [124,141] |
| Cartilage | Environmental disease [136], and Arthritis [137] |
| Gallstones | Environmental [123] |
| Kidney stones | Environmental [125] |
| Nails | Environmental [129,145] |
| Prostate | Cancer [135,139] |
| Serums | Sickle cell anemia [149] |
| Skin | Body iron burden [131] and Environmental [129] |
| Stomach | Cancer [127] |
| Tooth enamel | Environmental [129,142] |
With the development of X-ray experimental setups [12,124,133,135,165–186], some investigations have tried to perform in vivo measurements of trace elements such as Fe [185], Au [167], Zn [133,135], Cd [166,170], I [182], Hg [171,179], Pu [186], U[174], Sr [178] and Pb [124,173]. As underlined by several authors, such technique constitutes a safe, rapid, and non-invasive approach. Some of these trace elements namely Au [167] or I [182] are part of drugs or biomedical markers. Gold salts are used in the treatment of rheumatoid arthritis and such XRF experiments allow the clinician to gather information on their mode of action [167]. H. Matsukiyo et al. [182] have developed an XRF camera for carrying out mapping of iodine-based contrast media used in medical angiography. In the case of excessive body toxic element burdens, such as Pb [180], Hg [179] or Cd [166,170], XRF measurements are a practical method for monitoring the efficacy of a therapy as well as for establishing the diagnosis. In vivo XRF experiments have also been collected on kidney [170]. As underlined by U. Nilsson et al. [170], except in the presence of very deeply situated kidneys, where the minimum detectable concentration is high, non-invasive in vivo XRF analysis of kidney cadmium should be a useful tool for evaluating the effects of long-term low-level exposure to cadmium and the risk of kidney damage.
Quite recently, S.S. Shilstein et al. [133,135] described a phantom-based feasibility investigation for a potential in vivo determination of zinc in prostate. Such an approach is based on several publications which have detailed the compelling evidence and basis for the concept that zinc is involved in the pathogenesis of prostate cancer and that zinc could be efficacious in the prevention and treatment of this cancer [157–160]. Such measurements could bring about improved diagnosis of prostate cancer which is the most common malignancy affecting men and is the second-leading cause of cancer death [161].
Finally, L.H. Nie et al. [184] investigated the methodology and feasibility of developing a portable X-ray fluorescence (XRF) technology to quantify Pb in bone in vivo. The authors report that with such a portable XRF device, the detection limit was about 8.4 ppm with 2 mm soft tissue thickness. Also, the entrance skin dose delivered to the human subject was about 13 mSv and the total body effective dose was about 1.5 μSv and that should pose minimal radiation risk. They conclude that portable XRF technology can be used for in vivo bone lead measurements with sensitivity comparable to a standard technology. Similar conclusions have been made by M. Estevam and C.R. Appoloni [185] for the assessment of iron levels in patients with thalassemia and hemochromatosis.
7 Conclusion and perspectives
Through these different examples, some of those being already published, we have clearly demonstrated that in-lab facilities bring significant information to clinicians. Such experimental devices allow the detection of trace elements such Mg, Zn, Fe, Sr or Pb which play a major role in medicine [187,188]. Their place in medical research will increase drastically due to major public health problems related to infection and environmental diseases. In the field of kidney stones, a new set of experiments will be dedicated to assess through XRF the relationship between infection and the content of Zn in the stones.
Acknowledgments
This research has been performed through scientific discussions with Dr. P.-A. Albouy (LPS) and Dr. E. Foy (Iramis-CEA). This work was supported by the Physics and Chemistry Institutes of CNRS and by contract ANR-09-BLAN-0120-02.