

Symbol description:
- • The two oxygen atoms of CO2 are referred to as O1 and O2, respectively. The carbon atom is designated as C.
- • The oxygen atom in the glucose ring is referred to as OS, the hydrogen atoms in the sugar as HS and the hydrogen atom in the methylene as HS2.
- • The oxygen atom adjacent to the carbonyl group is labeled as OE, while the carbonyl oxygen atom is labeled as OAC. The hydrogen atom in methyl group was expressed by HAc.
- • The oxygen atom in OH is referred to as OHy, the hydrogen atom as HHy.
- • The sulfur atom in SH is referred to as SSu, the hydrogen atom as HSu.
- • The sulfur atom in SEt is referred to as SEt, the hydrogen atom as HSu, the hydrogen atom in the methyl group as HEt3, the hydrogen atom as HEt2.
1 Introduction
Carbon dioxide (CO2) has two faces: it is both a greenhouse gas and also a valuable carbon resource and solvent owing to its abundance, low cost and non-toxicity [1–8]. For example, supercritical carbon dioxide (scCO2) can be used as a solvent for extraction, separation, and other chemical processes, including chemical reactions and material handling [9–12]. However, many polar compounds, high molecular-weight substances and organometallic complexes usually exhibit poor solubility in scCO2, which seriously limits its industrial applications [13–16]. Therefore, much effort has been made to look for CO2-philes that can enhance the solubility of insoluble compounds in CO2 or as a potential phase-change physical solvent for CO2 [17]. Compared to the existing CO2-philes, such as fluorinated compounds or silicone polymers, hydrocarbons, especially carbohydrates with carbonyl groups, are one of the most attractive CO2-philic materials owing to their low cost, as well as their easy-modification and environmentally friendly properties [6,17–20]. Therefore, several CO2-phile carbohydrates have been prepared. One of the major classes is peracetylated sugar, such as peracetylated d-glucopyranose (Fig. 1, compound 1). However, peracetylated d-glucopyranose is unstable under acidic conditions, particularly in the presence of sulfur-containing waste gases that released during coal combustion. The exposure to acidic atmosphere leads to 2,3,4,6-tetra-O-acetyl-d-glucopyranose (Fig. 1, compound 2), 1-thiol-d-glucopyranose tetraacetate (Fig. 1, compound 3), and 1-mercaptoethyl-d-glucopyranose tetraacetate (Fig. 1, 4).
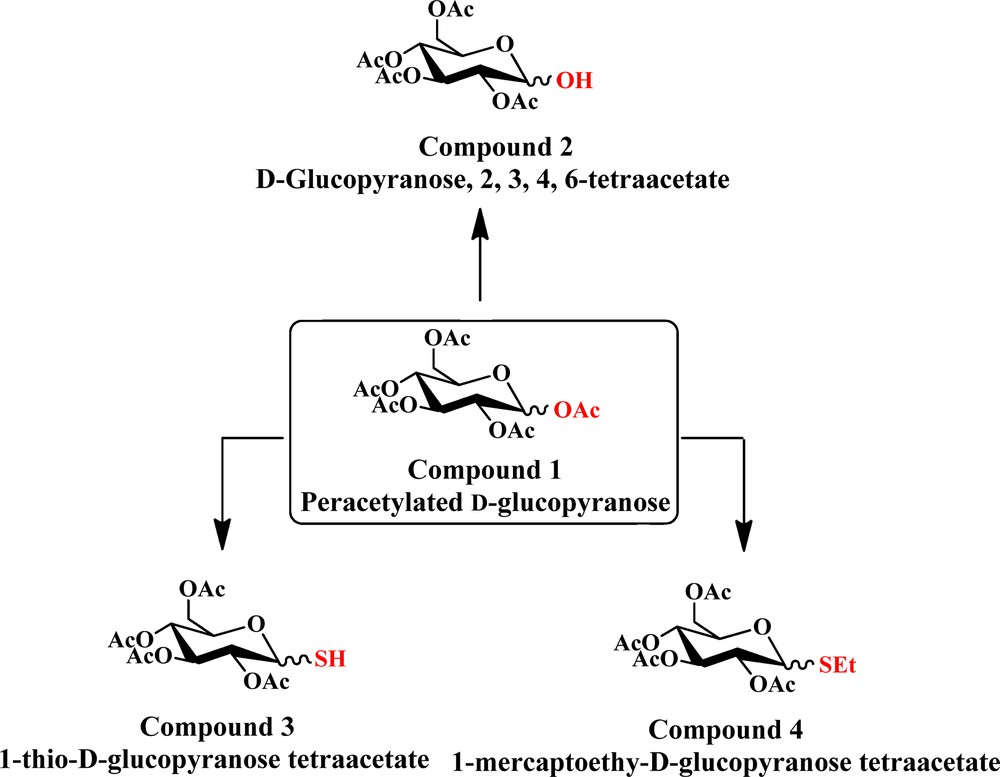
(Color online.) The transformation process from 1–OAc of d-glucose pentaacetate to 1–OH, 1–SH, 1–SEt group.
As reported, carbohydrate based CO2-philes should have CO2-philic functional groups, such as acetyls, with high free volume and flexibility, displaying weak self-interactions, but relatively strong interactions with CO2. The high solubility of hydrocarbon carbonyl systems in CO2 has been attributed to the Lewis acid (LA)–Lewis base (LB) interactions between CO2 and CO2-philic Lewis base functionalities, such as carbonyl groups [21–23]. Our recent ab initio calculations established that the interactions of compound 1 with CO2 are distance related, and should be of electrostatic origin (point charge effects) and not only LA–LB and hydrogen bond interactions [24]. Accordingly, it is necessary for us to get a deeper insight into the interaction properties of compounds 2, 3 and 4 with CO2 at the molecular level. If the affinity of the thiol (–SH in compound 3) or mercaptoethyl (–SEt in compound 4) group with CO2 were to be clear, the adsorption of CO2 and the conversion of sulfur gas would occur simultaneously and the application of carbohydrates could be widened.
2 Computational models and methods
On the basis of previous work, it was found that one CO2 molecule could only interact with two adjacent Ac groups in peracetylated d-glucopyranose by weak binding and complex formation through the 1,2-di-O-acetyl-d-glucopyranose model [24]. The following substrates were designed: 2-O-acetyl-α-d-glucopyranose (A–OH in Fig. 2), 2-O-acetyl-β-d-glucopyranose (B–OH in Fig. 2), 2-O-acetyl-1-thio-α-d-glucopyranose (A–SH in Fig. 3), 2-O-acetyl-1-thio-β-d-glucopyranose (B–SH in Fig. 3), 1-mercaptoethyl-2-O-acetyl-α-d-glucopyranose (A–SEt in Fig. 4), and 1-mercaptoethyl-2-O-acetyl-β-d-glucopyranose (B–SEt in Fig. 4) instead of their tetraacetated derivatives, as shown in Fig. 1.
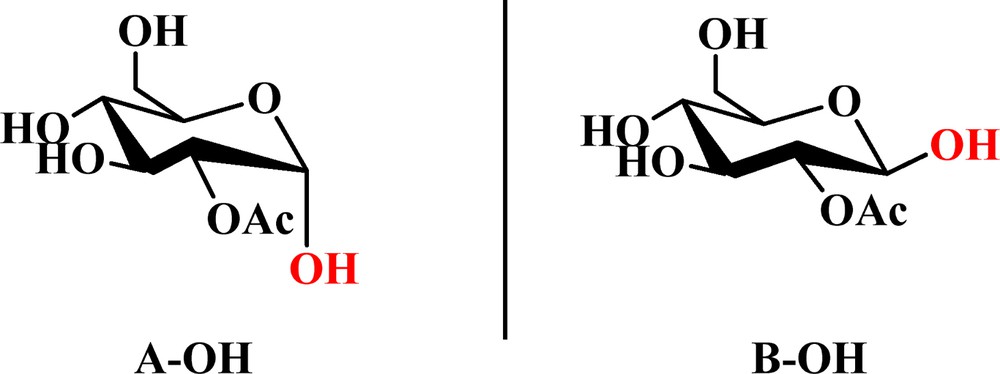
(Color online.) The structures of 2-O-acetyl-α-d-glucopyranose (A–OH) and 2-O-acetyl-β-d-glucopyranose (B–OH).

(Color online.) The structures of 2-O-acetyl-1-thio-α-d-glucopyranose (A–SH) and 2-O-acetyl-1-thio-β-d-glucopyranose (B–SH).

(Color online.) The structures of 1-mercaptoethyl-2-O-acetyl-α-d-glucopyranose (A–SEt) and 1-mercaptoethyl-2-O-acetyl-β-d-glucopyranose (B–SEt).
Ab initio calculations for the interactions of CO2 with the d-glucopyranose derivatives were performed on a 1:1 complex. Preliminary geometry optimizations were carried out at the Hartree–Fock (HF/3-21G) level. In order to determine the configuration property provided within weak interactions, more precise geometry and vibrational frequency calculations were then performed at the B3LYP/6-31+G** level. Single-point energy calculation was carried out at the MP2/aug-cc-pVDZ level, including the zero-point energy (ZPE) to obtain more accurate interaction energies, as shown in our previous work [24]. All the calculations were performed using Gaussian 09. The interaction energies (ΔE) of these complexes were calculated using the “supermolecule” method:
| ΔE = EAB – (EA + EB) |
3 Results and discussion
3.1 The interaction between compound 2 and a CO2 molecule
As mentioned above, the interactions between 2-O-acetyl-α-d-glucopyranose (C1–OH group in axial direction, A–OH in Fig. 2) or its anomer 2-O-acetyl-β-d-glucopyranose (C1–OH group in the equatorial plane, B–OH in Fig. 2) and CO2 are achieved by the formation of a complex.
3.1.1 A–OH–CO2
With the anomeric OH group of A–OH in the axial position, and C2–OAc in the equatorial plane, three configurations (Fig. 5) can be formed. These structures are numbered from I to III based on their calculated energies, from the highest to the lowest. If the energy of I is assigned to be zero, the relative energies are –5.6 kJ/mol and –27.9 kJ/mol for II and III, respectively. Even though their energies are different, due to the tiny difference between them (less than 30 kJ/mol), these configurations would all exist in real conditions.

(Color online.) Three optimized structures of 2-O-acetyl-α-d-glucopyranose (A–OH).
The three configurations of A–OH (Fig. 5) can bind with one CO2 molecule to yield five binding models, as shown in Fig. 6. Owing to its quadrupole properties, one CO2 molecule may interact with A–OH through its carbonyl oxygen atom (OAc), its ester oxygen atom (OE), its oxygen atom in the sugar ring (OS), its hydroxyl oxygen atom (OHy), the hydrogen atoms of its Ac group (HAc), the hydrogen atom of its hydroxyl group (HHy), and/or the hydrogen atoms attached on the sugar ring (HS). Each A–OH–CO2 complex structure was optimized and found to have different binding energies (ΔE) from those of the other ones, as detailed in Table 1.

(Color online.) The optimized binding models for A–OH–CO2. (The dashed lines indicate the interaction points between the two molecules. The values represent the distances between two atoms, and the unit is 1 Å).
Interaction energies of CO2 with A–OH.
| Structures | Interaction pairs | Distances of two atoms (Å) | Interaction pairs number | Binding energy (ΔE*, kJ/mol) | Gibbers free energy (ΔG*, kJ/mol) |
| Ia | C–OHy | 3.348 | 3 | –23.4 | 5.4 |
| C–OE | 3.038 | ||||
| C–OAc | 3.276 | ||||
| O1–HS | 3.766 | 2 | |||
| O2–HHy | 3.447 | ||||
| IIa | C–OHy | 3.087 | 2 | –24.3 | 7.3 |
| C–OAc | 2.839 | ||||
| O1–HHy | 3.341 | 3 | |||
| O2–HS | 3.500 | ||||
| O2–HS | 2.839 | ||||
| IIb | C–OHy | 3.195 | 1 | –16.3 | 8.1 |
| O1–HHy | 2.514 | 3 | |||
| O2–HAc | 3.592 | ||||
| O2–HAc | 3.507 | ||||
| IIIa | C–OE | 3.285 | 2 | –22.7 | 9.5 |
| C–OHy | 3.066 | ||||
| O1–HS | 2.709 | 3 | |||
| O2–HAc | 2.797 | ||||
| O2–HHy | 3.631 | ||||
| IIIb | C–OAc | 2.924 | 1 | –17.0 | 9.7 |
| O1–HS | 2.635 | 1 | |||
| Average binding energy (kJ/mol) | –20.7 | 8.0 |
As shown in Fig. 6 and Table 1, structure IIb has one OHy while IIIb has one OAc involved in the interaction, without changing the ΔE. The interaction between CO2 and OHy was as strong as that with OAc, i.e. the chemical environment of the oxygen atom (OHy or OAc) has only minor influence on the ΔE value of the A–OH–CO2 complex. Consequently, the data in Table 1 also revealed that ΔE increased if more oxygen atoms were joined in the binding model (i.e. IIa). Hydrogen atoms could interact with oxygen atoms as presented in all structures in Fig. 6, but this interaction has no saturation and directional features, as expected from a hydrogen bond. Therefore, it should be attributed to the point charge interaction rather than to the hydrogen bond.
3.1.2 B–OH–CO2
B–OH can display three different configurations, as shown in Fig. 7. The energy of structure A was assigned to be zero and the others are –15.2 kJ/mol and –18.9 kJ/mol for structures B and C, respectively (like with 3.1.1).
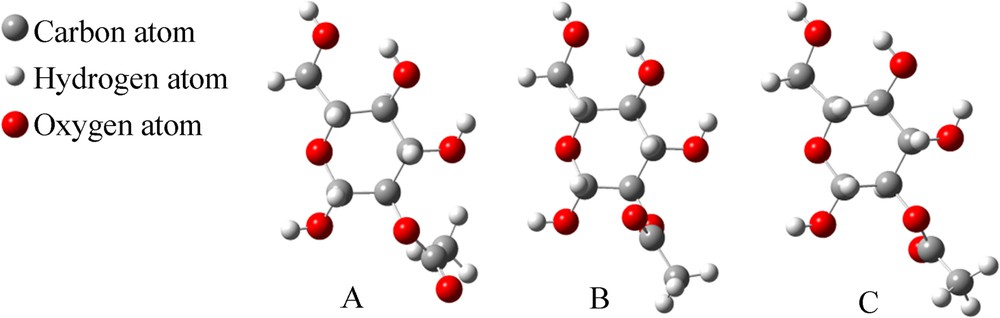
(Color online.) Three optimized structures of 2-O-acetyl-β-d-glucopyranose (B–OH).
The three configurations of B–OH (Fig. 7) could bind with one CO2 molecule to yield five binding models (Fig. 8). As shown in Fig. 8 and Table 2, four binding models (A1, B1, B2, C2) display one O atom involved in the interaction. For example, structures A1 or B1 had one OHy, structure B2 had one OAc, while C2 had one OE involved in the interaction, but their ΔEs were all around –19 kJ/mol. The interaction between CO2 and OAc was as strong as that with OE and OHy; the chemical environmental of oxygen atom had no obvious influence on ΔE of B–OH–CO2 complex.

(Color online.) The optimized binding models for B–OH–CO2. (The dashed lines indicate the interaction points between the two molecules. The values represent the distances between two atoms, and the unit is 1 Å).
Interaction energies of CO2 with B–OH.
| Structures | Interaction pairs | Distances of two atoms (Å) | Interaction pairs number | Binding energy (ΔE*, kJ/mol) | Gibbs free energy (ΔG*, kJ/mol) |
| A1 | C–OHy | 2.870 | 1 | –19.5 | 8.7 |
| O1–HHy | 3.050 | 3 | |||
| O2–HS | 2.971 | ||||
| O2–HAc | 2.751 | ||||
| B1 | C–OHy | 2.919 | 1 | –19.0 | 6.8 |
| O1–HHy | 3.039 | 2 | |||
| O2–Hs | 2.862 | ||||
| B2 | C–OAc | 2.936 | 1 | –18.9 | 5.4 |
| O1–HS | 2.646 | 1 | |||
| C1 | C–OAc | 3.020 | 2 | –22.7 | 8.4 |
| C–OHy | 3.140 | ||||
| O1–HS | 2.816 | 1 | |||
| C2 | C–OE | 3.310 | 1 | –18.6 | 10.4 |
| O1–HS | 2.940 | 4 | |||
| O1–HS | 2.800 | ||||
| O2–HAc | 2.890 | ||||
| O2–HAc | 3.420 | ||||
| Average binding energy (kJ/mol) | –19.7 | 8.0 |
From Table 1 and Table 2, the average ΔE of A–OH–CO2 (–20.7 kJ/mol) is similar to that of B–OH–CO2 (–19.7 kJ/mol), which means that the interaction between CO2 and α-d-glucopyranose tetraacetate is almost strong as that with its β-anomer. In addition, because compound 2 has less carbonyl groups than compound 1, the average ΔE values of compound 2–CO2 complexes are lower than those of compound 1–CO2 complexes (–24.2 and–22 kJ/mol). Therefore, the numbers of negative charges are important in such a carbohydrate–CO2 system.
3.2 The interaction between compound 3 and a CO2 molecule
The α- and β-1-thio-d-glucopyranose tetraacetate (compound 3) had one anomeric–SH group and four Ac groups. 2-O-acetyl-1-thio-α-d-glucopyranose (A–SH in Fig. 3) and 2-O-acetyl-1-thio-β-d-glucopyranose (B–SH in Fig. 3) were used as models. One CO2 molecule may interact with A–SH or B–SH through its carbonyl oxygen atom (OAc), an ester oxygen atom (OE), an oxygen atom in the sugar ring (OS), a sulfur atom (SSu) and/or hydrogen atoms, including HSu and HAc.
3.2.1 A–SH–CO2
The anomeric–SH group of A–SH is in axial position, whereas C2–OAc is in the equatorial plane, and these two groups can create some rotation conformers, as shown in Fig. 9. The structures are numbered from I to III based on the calculated energies, from the highest to the lowest. If the energy of I is assigned to be zero, the relative energies are –10.5 kJ/mol, and –31.4 kJ/mol for configurations II and III, respectively (similar to 3.1.1).

(Color online.) Three optimized structures of 2-O-acetyl-1-thio-α-d-glucopyranose (A–SH).
The three configurations of A–SH (Fig. 9) can bind with one CO2 molecule to yield four binding models, as shown in Fig. 10. Compared with others (IIa, IIb and IIIa in Fig. 10), structure Ia had one more sulfur atom besides the oxygen atoms (OAc and OE) that can interact with the CO2 molecule and therefore gives a higher ΔE (–22.4 kJ/mol, Table 3). Moreover, the distance between the CO2 molecule and the –SH group (>S···C, 3.8 Å) is slightly longer than in the case of 1–OH (around 3 Å, Fig. 6); this may be the cause of a weaker electrostatic interaction between A–SH and CO2 and thereby of a lower average ΔE value (18.8 kJ/mol for A–SH in Table 3, –20.7 kJ/mol for A–OH in Table 1).
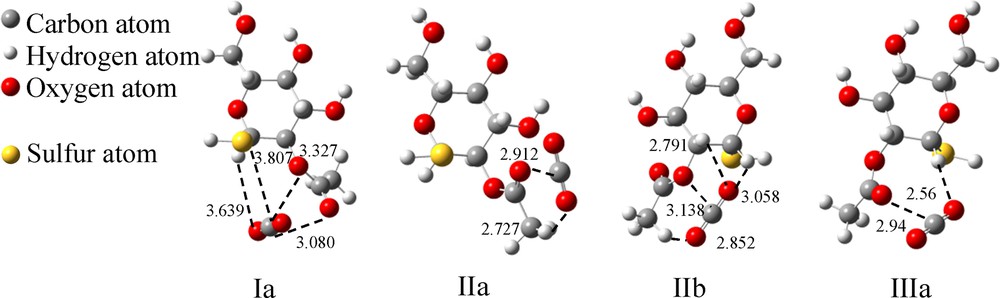
The optimized binding models for A–SH–CO2. (The dashed lines indicate the interaction points between the two molecules. The values represent the distances between two atoms, and the unit is 1 Å).
Interaction energies of CO2 with A–SH.
| Structures | Interaction pairs | Distances of two atoms (Å) | Interaction pairs number | Binding energy (ΔE*, kJ/mol) | Gibbers free energy (ΔG*, kJ/mol) |
| Ia | C–SSu | 3.807 | 1 | –22.4 | 7.4 |
| C–OE | 3.327 | 2 | |||
| C–OAc | 3.080 | ||||
| O1–HS | 3.639 | 1 | |||
| IIa | C–OAc | 2.912 | 1 | –16.5 | 10.4 |
| O1–HAc | 2.727 | 1 | |||
| IIb | C–OE | 3.138 | 1 | –18.8 | 9.6 |
| O1–HS | 3.058 | 3 | |||
| O1–HS | 2.791 | ||||
| O2–HAc | 2.852 | ||||
| IIIa | C–OAc | 2.940 | 1 | –17.5 | 9.0 |
| O1–HS | 2.560 | 1 | |||
| Average binding energy (kJ/mol) | –18.8 | 9.1 |
3.2.2 B–SH–CO2
B–SH has one equatorial anomeric–SH group and one C2–OAc group similar to that of A–SH, and can create three rotation conformers, as shown in Fig. 11. The structures are numbered from A to C based on the calculated energies from the highest to the lowest, i.e. the energy of A was assigned to be zero, which gives –19.0 kJ/mol for B and –22.8 kJ/mol for C. There are four binding models with three configurations of B–SH binding with one CO2 molecule (Fig. 12), and the ΔEs of these models are listed in Table 4.

Three optimized structures of the 2-O-acetyl-1-thio-β-d-glucopyranose (B–SH).
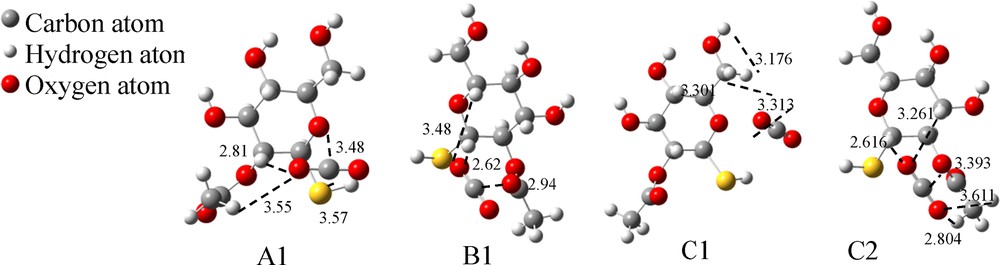
The optimized binding models for B–SH–CO2. (The dashed lines indicate the interaction points between two molecules. The values represent the distances between two atoms, and the unit is 1 Å).
Interaction energies of CO2 with B–SH.
| Structures | Interaction pairs | Distances of two atoms (Å) | Interaction pairs number | Binding energy (ΔE*, kJ/mol) | Gibbs free energy (ΔG*, kJ/mol) |
| A1 | C–SSu | 3.570 | 1 | –21.1 | 7.8 |
| C–Os | 3.480 | 1 | |||
| O1–HS | 2.810 | 2 | |||
| O1–HAc | 3.550 | ||||
| B1 | C–OAc | 2.940 | 1 | –19.1 | 8.2 |
| O1–HS | 2.620 | 2 | |||
| O1–HS | 3.480 | ||||
| C1 | C–OS | 3.301 | 1 | –18.2 | 7.1 |
| O1–HSu | 3.313 | 2 | |||
| O1–HS2 | 3.176 | ||||
| C2 | C–OE | 3.393 | 1 | –18.8 | 6.6 |
| O1–HAc | 3.611 | 4 | |||
| O1–HAc | 2.804 | ||||
| O2–HS | 3.261 | ||||
| O2–HS | 2.616 | ||||
| Average binding energy (kJ/mol) | –19.3 | 7.4 |
Except A1, which has one more S atom, all the other structures have only one O atom interacting with CO2. Correspondingly, the ΔE value of A1 is higher (–21.1 kJ/mol, Table 4) than that of the others, which indicates that ΔE increases when a more negative charge is involved in the complex. In addition, the ΔE values of B1, C1 and C2 are similar, even though there are four O···H interactions existing in the C2 complex (two in B1 and C1), i.e. the number of O···H bonds has no significant influence on ΔE. The average binding energy of CO2 with B–SH is the same as that with B–OH, but the latter has more binding modes with CO2.
3.3 The interaction between compound 4 and the CO2 molecule
1-Mercaptoethy-d-glucopyranose tetraacetate has one anomeric SEt group and four Ac groups. 1-Mercaptoethyl-2-O-acetyl-α-d-glucopyranose (A–SEt) and 1-mercaptoethyl-2-O-acetyl-β-d-glucopyranose (B–SEt) are preferred as the final models, as shown in Fig. 4.
3.3.1 A–SEt–CO2
The anomeric SEt group of A–SEt is in the axial position, whereas C2–OAc is in the equatorial plane, and these two groups can create three rotational conformers, as shown in Fig. 13. The structures are numbered from I to III based on the calculated energies, from the highest to the lowest. If the energy of I was assigned to be zero, the relative energies were –10.1 kJ/mol and –32.0 kJ/mol for configurations II and III, respectively (similar with 3.1.1). There are five binding models by three configurations of A–SEt binding with one CO2 molecule (Fig. 14). The ΔE values of the binding models are listed in Table 5.

Three optimized structures of 1-mercaptoethyl-2-O-acetyl-α-d-glucopyranose (A–SEt).
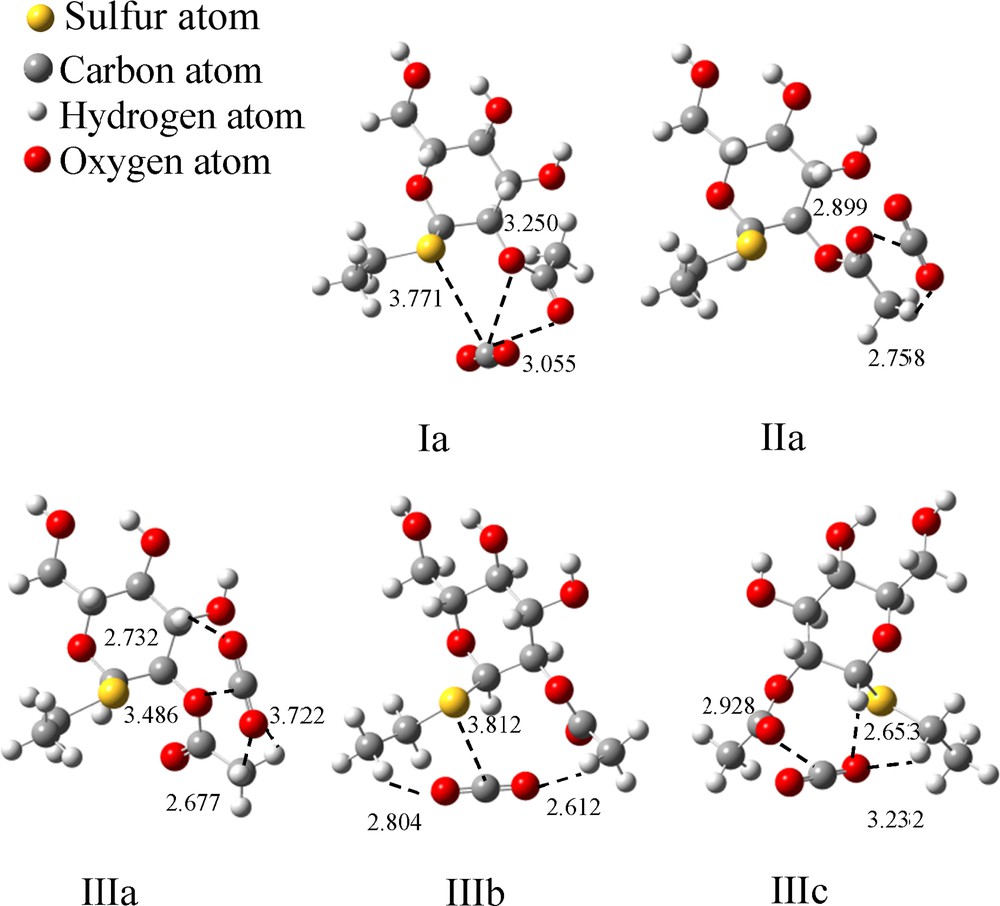
The optimized binding models for A–SEt–CO2. (The dashed lines indicate the interaction points between the two molecules. The values represent the distances between two atoms, and the unit is 1 Å).
Interaction energies of CO2 with A–SEt.
| Structures | Interaction pairs | Distances of two atoms (Å) | Interaction pairs number | Binding energy (ΔE*, kJ/mol) | Gibbs free energy (ΔG*, kJ/mol) |
| Ia | C–SSu | 3771 | 1 | –25.2 | 2.0 |
| C–OE | 3.250 | 2 | |||
| C–OAc | 3.055 | ||||
| IIa | C–OAc | 2.899 | 1 | –16.9 | 9.2 |
| O1–HAc | 2.758 | 1 | |||
| IIIa | C–OE | 3.486 | 1 | –19.1 | 11.1 |
| O1–HS | 2.732 | 3 | |||
| O2–HAc | 2.677 | ||||
| O2–HAc | 3.722 | ||||
| IIIb | C–SSu | 3.812 | 1 | –14.5 | 13.4 |
| O1–HEt3 | 2.804 | 2 | |||
| O2–HAc | 2.612 | ||||
| IIIc | C–OAc | 2.928 | 1 | –19.1 | 7.3 |
| O1–HS | 2.653 | 2 | |||
| O1–HEt2 | 3.232 | ||||
| Average binding energy (kJ/mol) | –18.9 | 8.6 |
As shown in Fig. 14, one CO2 molecule may interact with A–SEt through its carbonyl oxygen atom (OAc), ester oxygen atom (OE), oxygen atom in the sugar ring (OS), sulfur atom of SEt group (SEt) and/or hydrogen atoms. Both Ia and IIIb have SE in the complex, but two additional O atoms in Ia instead of two H atoms in IIIb interacted with CO2. Meanwhile, the other three models (Fig. 14) have only one O atom and no S atom to interact with CO2, and Ia has the highest ΔE value (–25.2 kJ/mol), as shown in Table 5. So it can be deduced that ΔE will increase if a more negative point charge induced by oxygen or sulfur atoms is added in the binding model (three negative charges in Ia), i.e. if the interaction between A–SEt and CO2 is electrostatic.
All the structures, IIa, IIIa and IIIc, have only one O atom, but different numbers of H atoms to interact with CO2. IIa has only hydrogen atom interacting with the CO2 molecule, and its ΔE is –16.9 kJ/mol, three hydrogen atoms of IIIa and two hydrogen atoms of IIIc form a H···OCO bond and give a ΔE around –19.1 kJ/mol. The interaction of H with CO2 is complicated. It could be a hydrogen bond (interaction distance is around 2 Å) or a point charge interaction, which could be influenced by distance, charge density, and so on. From the distance of H···OCO, it appeared that the contribution of H···O interactions to ΔE is larger if the distance is shorter (i.e. 2.6 and 3.2 Å in IIIc) but it could be negligible if the distance is too long (i.e. 3.7 Å in IIIc).
It is notable that the ΔE of IIIb is the lowest (–14.5 kJ/mol, Table 5), and this might be caused by a difference in electronegativity between O and S atom (compared with IIIc) and a longer distance from sulfur to CO2 (about 3.8 Å).
3.3.2 B–SEt–CO2
There are three rotation conformers of B–SEt, as shown in Fig. 15. The structures are numbered from A to C based on the calculated energies, from the highest to the lowest. If the energy of A is assigned to be zero, the relative energies are –19.1 kJ/mol and –22.4 kJ/mol for configurations B and C, respectively. There three configurations of B–SEt could bind with a CO2 molecule through four binding modes (Fig. 16). The ΔE values of the above binding models are listed in Table 6.

Three optimized structures of ethyl 2-O-acetyl-1-thio-β-d-glucopyranose (B–SEt).
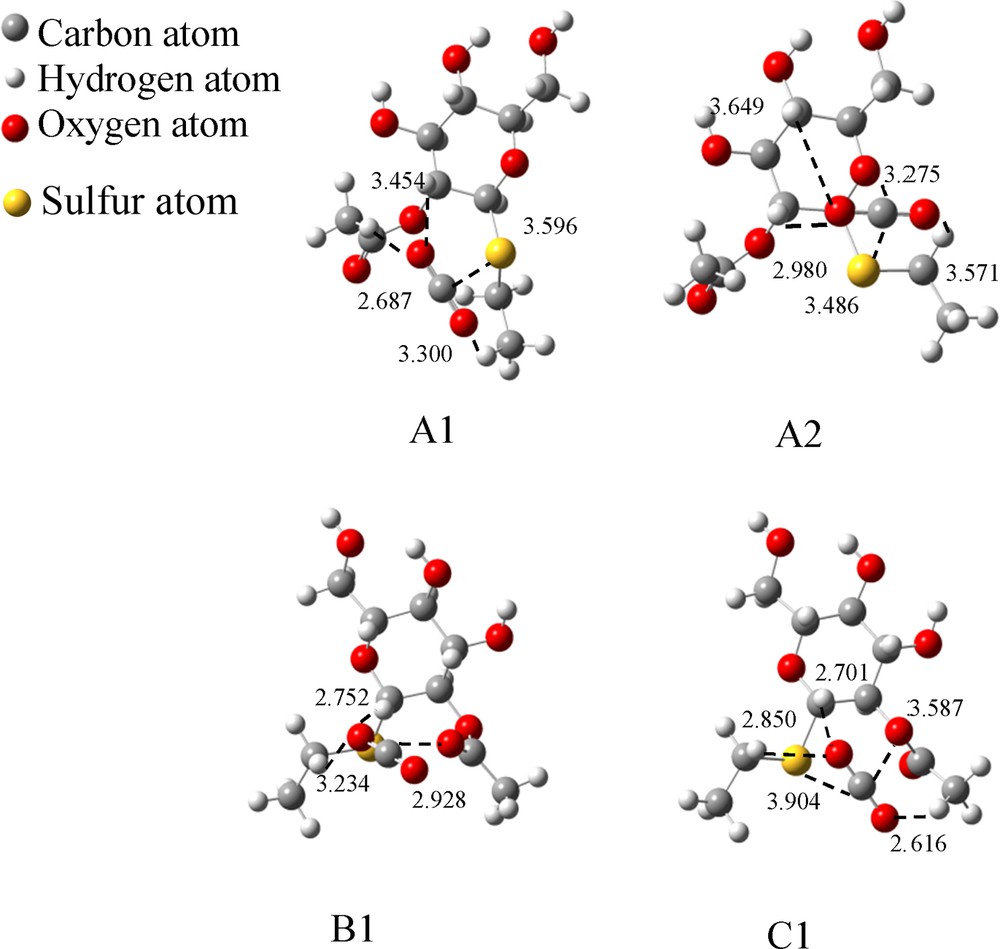
The optimized binding models for B–SEt–CO2. (The dashed lines indicate the interaction points between the two molecules. The values represent the distances between two atoms, and the unit is 1 Å).
Interaction energies of CO2 with B–SEt.
| Structures | Interaction pairs | Distances of two atoms (Å) | Interaction pairs number | Binding energy (ΔE*, kJ/mol) | Gibbs free energy (ΔG*, kJ/mol) |
| A1 | C–SSu | 3.596 | 1 | –16.1 | 20.9 |
| O1–HEt3 | 3.300 | 3 | |||
| O2–HS | 3.454 | ||||
| O2–HAc | 2.687 | ||||
| A2 | C–SSu | 3.486 | 1 | –26.5 | 3.1 |
| C–OS | 3.275 | 1 | |||
| O1–HEt2 | 3.571 | 3 | |||
| O2–HS | 3.649 | ||||
| O2–HS | 2.980 | ||||
| B1 | C–OAc | 2.928 | 1 | –20.9 | 7.3 |
| O1–HEt2 | 3.234 | 2 | |||
| O1–HS | 2.752 | ||||
| C1 | C–SSu | 3.904 | 1 | –22.0 | 7.4 |
| C–OE | 3.587 | 1 | |||
| O1–HAc | 2.616 | 3 | |||
| O2–HEt2 | 2.850 | ||||
| O2–HS | 2.701 | ||||
| Average binding energy (kJ/mol) | –21.4 | 9.7 |
A1 shows the lowest binding energy (–16.1 kJ/mol) because it has only one S atom, which interacts with CO2, and ΔE is increased to –20.9 kJ/mol if the S atom is replaced by an O atom (B1) because the O atom could donate a more negative charge. ΔE will be even higher if one S plus an O atom can interact with CO2 (–26.5 kJ/mol for A2, –22 kJ/mol for C1). Moreover, the ΔE value of C1 is lower than A2 because the distance of S and O with CO2 is shorter (i.e. 3.275 Å for S–CO2 and 3.486 Å for O–CO2 in A2; 3.587 Å for S–CO2 and 3.904 Å of O–CO2 in C1).
From Tables 5 and 6, the average binding energy of A–SEt is slightly lower than B–SEt, but A–SEt had one more binding mode than B–SEt. Compared with compound 3, the ethyl group in compound 4 makes the S atom more negative and thereby there are more H atoms in the binding site, but these factors did not affect the ΔE value of compound 4 (data shown in Tables 3–6).
4 Conclusion
In order to study the interaction properties of d-glucopyranose-2,3,4,6-tetraacetate, 1-thio-d-glucopyranose tetraacetate, 1-mercaptoethyl-d-glucopyranose tetraacetate with CO2, their analogous models (2-O-acetyl derivatives) were selected as substrates, and the complex model was one CO2 molecule combined with one sugar substrate (1:1). Ab initio calculations of these six systems were accomplished including geometry optimizations with HF/3-21G, B3LYP/6-31+G**, and single-point energy calibration with MP2/aug-cc-pVDZ. The results indicated that the electrostatic interactions between substrates with CO2 are mainly influenced by interaction distance, numbers of donors of negative charge, or interacting pairs involved in the complex. ΔE increased greatly if S and O atoms could interact with CO2 simultaneously. The binding was to be strengthened if CO2 could interact with S, O, and H atoms at shorter distance. The binding energy is not highly related to the chemical environment of the oxygen atom (i.e. OAc, OE, or OS) or sulfur atom (i.e. SEt or SH), and to the differences in electronegativity between S and O atoms. The average ΔE value of these three substrates were all around –20 kJ/mol, even though their C-1 (anomeric) functional groups were different (i.e. 1–OH, 1–SH and 1–SEt), but this energy is slightly lower than that of peracetylated d-glucose (–22 kJ/mol), which has one more oxygen atom that could interact with CO2.
Acknowledgements
The authors would like to acknowledge the financial support from the Natural Science Foundation of China (No. 21106172), and the Natural Science Foundation for Youths of Shanxi (2013021008-7). This work was also supported by grants from the Chinese Academy of Sciences (grant 2013YC002).