

1 Introduction
As an undergraduate student, I tried to synthesize some Ag(II) complexes with N-donor ligands. I found that the unstable Ag(II) complexes reduce to Ag(I) complexes in the presence of water. I suggested that either the water-oxidation or ligand-oxidation reaction is responsible for the reduction of Ag(II) complexes under these conditions. Although ligand oxidation in some cases may occur in the presence of water, I focused on the water-oxidation reaction hypothesis to synthesize stable Ag(II) complexes. Initially I did not think that water oxidation is an important reaction because the product of the reaction is O2, which is present around us! However, on more deep search and the study of water oxidation, I found the importance of the reaction especially by reading the studies by Professor Stenbjörn Styring [1] and Professor Gary W. Brudvig [2]. On the other hand, I read about the role of an Mn compound in an unknown structure in biological water oxidation in a book, Advances in Photosynthesis and Respiration series, in Professor Govindjee series edited by D. R. Ort and C. F. Yocum [3]. After this time, water oxidation and artificial photosynthesis were, and are (!) very interesting for me because they are new, multidisciplinary fields which try to provide a clean environment on our planet.
Artificial photosynthesis is an umbrella term and refers to the processes that mimic natural photosynthesis (Scheme 1) [4–12]. One of the most important goals of the artificial photosynthesis is to use the cheap electrons generated by water oxidation to reduce CO2 or H+ to fuel [4–12]. In contrast to the natural photosynthesis, artificial systems are usually a simplified way to produce clean and renewable fuels (Scheme 2).
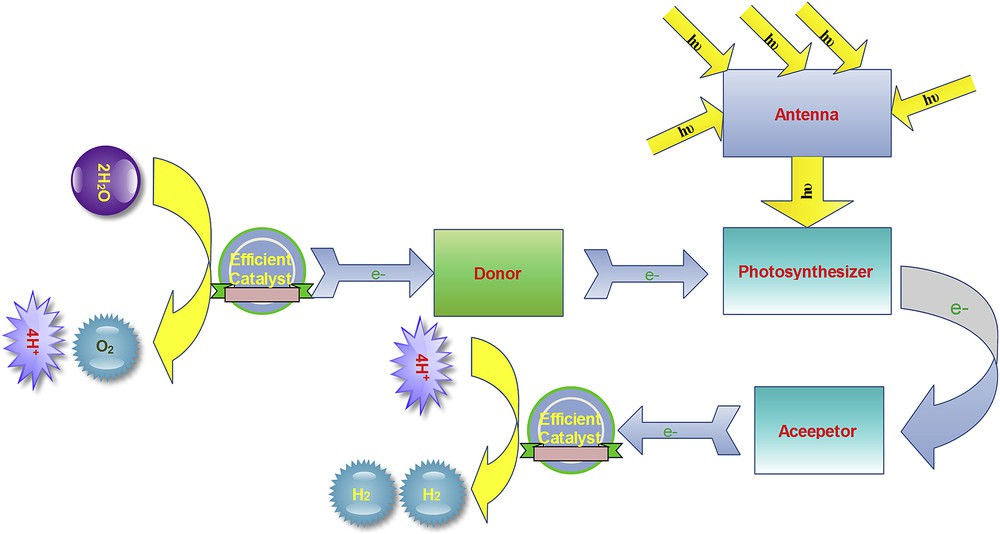
A simple scheme of artificial photosystem to split water into H2 and O2. The following sections are required for both artificial photosynthetic systems: Photosenthisizer, composed of several molecules, electron transfer donor (Donor), electron transfer acceptor (Acceptor), catalyst for chemical H2 reduction, catalyst for chemical water oxidation and light-harvesting molecular construction (Antenna).

During water oxidation oxygen, cheap electron and proton are obtained that could be used for H2 formation.
In an artificial photosynthetic system, we usually need these features [4–12]:
- (1) absorption of a photon;
- (2) a charge-separated state;
- (3) a reducing catalyst:
- (4) accumulation of electrons;
- (5) an oxidizing catalyst;
- (6) substrate(s) to be oxidized and reduced.
For six years, I synthesized and characterized many Mn complexes with different ligands [13–17]. I selected ammonium cerium(IV) nitrate (Ce(IV)), Oxone® (KHSO4, K2SO4 and KHSO5) and photochemically produced to test oxygen evolution catalyzed by these complexes. However, no oxygen evolution was observed in many cases, but Oxone® was promising and a few complexes showed activity toward oxygen evolution [16]. However, the origin of oxygen may not be water under these conditions and the reaction is an oxygen-evolution reaction rather than the water-oxidation reaction [18].
In 2010, in Dr. Philipp Kurz's group, aimed at simulating the water-oxidizing complex (WOC) or oxygen-evolving complex (OEC) in Photosystem II (PSII) (Fig. 1a,b) [19], we showed that the incorporation of Ca ions into Mn oxides can improve the water-oxidation activity of Mn oxides toward water oxidation [20]. The WOC is the only compound to catalyze water oxidation in Nature [21,22] and, as we discussed, it may be considered as an Mn–Ca cluster housed in a protein environment in PSII that controls reaction coordinates, proton movement and water access [23,24]. Shen and his co-workers reported the crystal structure of the Mn–Ca cluster at an atomic resolution (Fig. 1a,b) [21,22]. In the WOC the metal ions, one Ca and four Mn ions, are bridged by five oxygen atoms. Two water molecules are also coordinated directly to both Mn and Ca ions in this structure (Fig. 1a,b) [21,22].
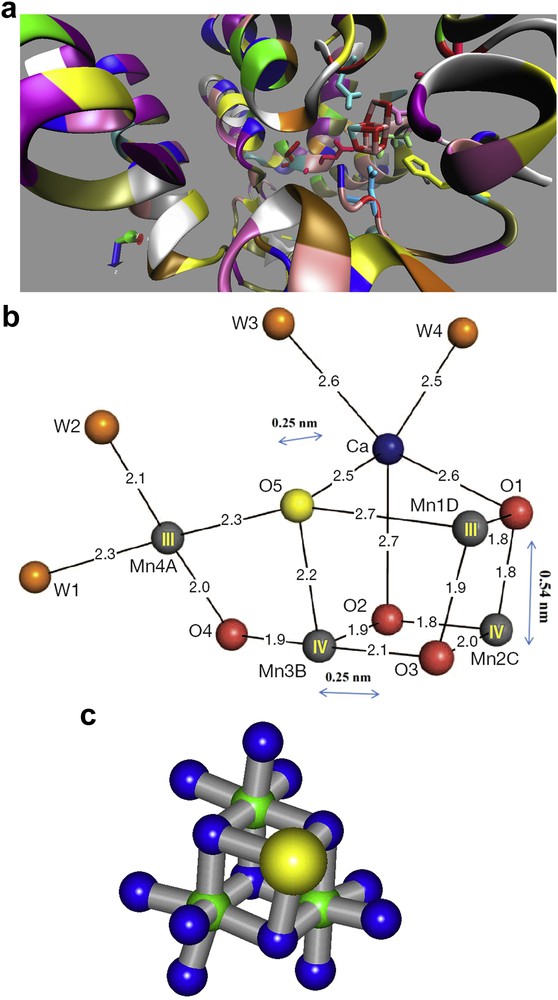
Mn4CaOx and the amino acids around the cluster (a). The structure of the Mn4CaOx cluster, depicted as an asymmetric cubane complex that resembles a distorted chair (b). Images with modification are taken from Ref. [21]. Motif of the edge-sharing MnO6 octahedra (di-μ-oxido bridging) in the manganese–calcium oxides (manganese: green, oxygen: blue, calcium: yellow, nitrogen: red, and carbon: cyan) (c). The image with modification is taken from Ref. [27].
The WOC in PSII, a super-catalyst for water oxidation, is an Mn–Ca cluster [23,24]. There are valuable lessons to learn for the design and synthesis of such catalysts in artificial photosynthetic systems, such as: using the catalyst in the form of a nano-sized oxido cluster embedded in a protein matrix, using channels for O2, water and proton transfer, outer bonds, the spin-flipping rule, redox accumulation in the WOC, regulating the oxidizing power, the photosystem's ability for self-repair, and various strategies used to protect the active site which may all provide valuable insights into the future design of robust artificial water-oxidizing compounds even in a non-biomimetic approach [25,26].
Our biomimetic Mn–Ca oxides were amorphous and therefore their structure could not be identified by the XRD method, but the structures of these amorphous powders were evaluated, using extended-range X-ray absorption spectroscopy (XAS), X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) [27]. These results introduced the compounds as structural and functional analogs to the native WOC in PSII (Fig. 1c) [27,28].
There are two different Ca-containing motifs in these amorphous Mn–Ca oxides. One of them results in the formation of Mn3CaO4 cubes, as also proposed for the WOC in PSII (Fig. 1b) [28]. Other Ca ions are likely to interconnect the oxide-layer fragments [27].
H218O isotope-labeling experiments coupled with membrane inlet mass spectrometry showed the mechanism of oxygen evolution by the Mn–Ca oxides [29]. These results confirmed that in oxygen evolution in the presence of single-electron oxidants such as Ce(IV) or , both oxygen atoms in the dioxygen product originate from water molecules [29]. The results also showed no clear involvement of the surface oxido species in the oxygen evolution reaction from water [29]. These results are important because they show that [29]:
- (7) use of low cost and environmentally friendly ions such as Ca and Mn can form an efficient catalyst for water oxidation;
- (8) Ca as used by Nature improves the water-oxidizing activity of Mn oxido-clusters;
- (9) although Ca is redox-inert, it can improve the catalytic activity;
- (10) in the absence of biomolecules, subunits such as the WOC can be formed.
Then, I came to the department of chemistry of the Institute for Advanced Studies in Basic Sciences (IASBS), one of the best Iranian centers for basic sciences founded in 1991 by Prof. Yousef Sobouti [30], with an independent postdoctoral position and then became a faculty member.
Fortunately, the field is very interesting for many students, at least in my country, because it is a modern field, which needs electrochemistry, nanotechnology, theoretical calculations, polymer, biochemistry, photochemistry, inorganic/physical/organic/analytical chemistry, physiology, electronics, mathematics and others. On the other hand, the field tries to improve our environment, and thus, is a holy graal, and one of the greatest challenges facing scientists in the twenty-first century.
Moreover, our group studies the metal and especially Mn compounds as water-oxidizing catalysts in artificial photosynthetic systems not only because Mn compounds are usually low cost, environmentally friendly and easy to use in terms of the synthesis and manufacture of the catalyst for water oxidation, but also because such compounds are successfully used by Nature for the same task. However, it is important to note that the conditions of water oxidation in Nature and in industry are clearly different, but the 3 billion year evolutionary experiments by Nature have provided excellent lessons [25,26] in water oxidation, which can be used even in non-biomimetic systems.
Herein, I try to focus more on water oxidation catalyzed by Mn-based catalysts and the related strategies, which have been reported by my group.
There is a growing need to convert sunlight to stored chemical energy in the form of fuels (Scheme 1).
Hydrogen is an attractive energy carrier for energy systems in the future that offers the advantage of clean and complete conversion to water [4–12,31–34]. Hydrogen generation from water splitting is a strategy to store solar, wind, ocean current, tide, wave or other sustainable energies [4–12,31–34]. However, for high-scale hydrogen generation from water splitting in a sustainable manner, first it is necessary to synthesize low cost and efficient, environmentally friendly and easy to use, stable catalysts for water oxidation, which is a more challenging half-reaction of water splitting [8,35]. Two important points in the case of water oxidation are:
- (11) water oxidation and cheap electron production is not only important to reduce proton (hydronium), but also to reduce N2 and CO2 to NH3 and CH4. In other words, a cheap electron is a gold fish for many reduction reactions;
- (12) water oxidation is not only important in artificial photosynthesis, but also it is necessary to apply wind, ocean currents and other intermittent and fluctuating energies for water splitting.
Mn oxides as biomimetic models for the Mn–Ca cluster in PSII were reported many years ago. Historically, for the first time in 1968, Glikman and Shcheglova from Russia reported on the water-oxidizing activity of Mn oxides in the presence of cerium(IV) salts as an oxidant [36]. Then, Morita in 1977 reported on electrochemical water oxidation catalyzed by Mn oxides and on the important role of Mn(III) [37]. Shilov extended the studies of water-oxidizing activity of Mn oxides under different conditions [38]. Harriman's group in 1988 extensively studied cobalt, iridium, Mn(III) and ruthenium oxides as efficient catalysts for water oxidation in the presence of Ce(IV) or as chemical oxidants [39]. The Harriman's group also studied the calcination temperature, different supports and many parameters for water oxidation by metal oxides [39]. After these studies, different Mn oxides were reported as efficient catalysts toward water oxidation by many research groups [40].
2 Nano scale Mn–Ca oxides
In 2011, without hydrothermal conditions and using high concentrations of KOH compared to the previously reported method, we synthesized nano-sized amorphous Mn–Ca oxides as an efficient catalyst with water-oxidation activity in the presence of Ce(IV) [23]. These nano-sized amorphous Mn–Ca oxides (Fig. 2) are stable, low cost, efficient, environmentally friendly and easy to use and could be a good candidate for water-oxidizing catalysts in artificial photosynthesis [23].

TEM images (a and b) and SEM (c) of Mn–Ca oxides.
We could not determine the structure of the compound by XRD, but SEM images show nanoparticles and TEM images (Fig. 2) show the layered structure of these oxides. In the layered Mn–Ca oxides, a long-range order cannot be detected [23]. We concluded that, most probably, the morphology of each nanoparticle is similar to crumpled paper. Such compounds under optimum conditions show more than three times higher water-oxidizing activity than the previously reported Mn–Ca oxides [19]. The next results also showed that small [41], but not very small [42], Mn oxides are very good catalysts for water oxidation at least in the presence of Ce(IV). The order of increasing activity toward water oxidation catalyzed by Mn oxides is as follows: nano-sized > bulk ∼ Ångström-scale, most probably due to the fragile structure at the Ångström-scale [42]. The ångström-scale Mn oxides in the presence of Ce(IV) significantly oxidize to ions [42]. In the biological site of water oxidation, amino acid side chains help to stabilize the very small and fragile Mn cluster [42]. In other words, the Mn-stabilizing protein (PsbO) that is present in all O2-evolving photosynthetic organisms shields the WOC unit from the thylakoid lumen [43].
Electrochemically, we observed that the water oxidation on a Pt electrode modified with Mn–Ca oxides is lower (1.15 V vs. Ag|AgCl) than that on a Pt electrode under acidic conditions [44].
3 Mechanism of water oxidation catalyzed by layered Mn oxides
Eight different mechanisms proposed for water oxidation are shown in Fig. 3 [45]. vi or viii is not the true mechanism for water oxidation in the presence of Ce(IV) or [RuIII(bpy)3]3+ because no O2 evolution in the absence of nano-layered Mn oxides was observed. Previously MIMS was used to detect O2 in the presence of Ce(IV) or [RuIII(bpy)3]3+ in H218O and to calculate the signals for the O2 isotopologues 16O2 (m/z = 32), 16O18O (m/z = 34) and 18O2 (m/z = 36) [29]. The role of nitrate from Ce(IV) in O2 evolution (mechanisms vi, vii and viii in Fig. 3) was denied by MIMS experiments [29].
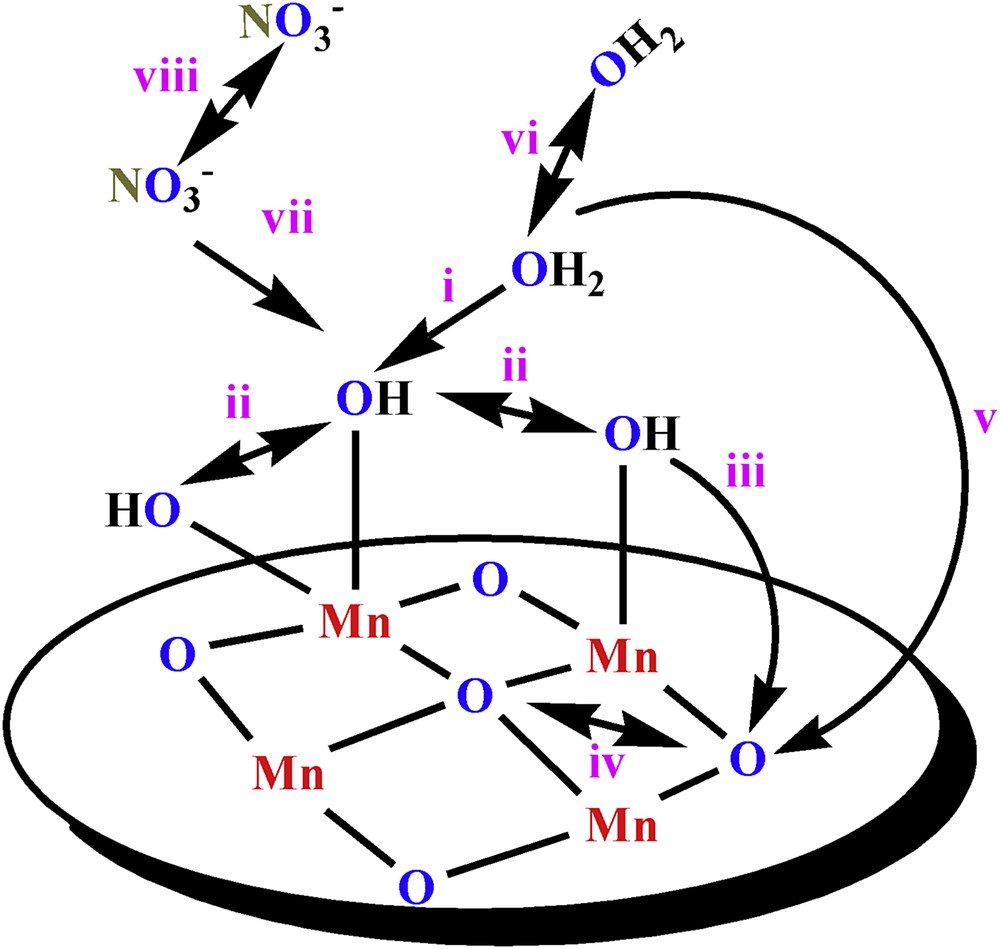
Mechanisms proposed for water oxidation catalyzed by the layered Mn oxides: nucleophilic attack of water molecules on a terminal hydroxide group (i); coupling of the terminal hydroxide ligands (ii), the attack of terminal hydroxide group on a bridging oxido ligand (iii), coupling of the bridging oxido ligands (iv), the attack of water molecules on a bridging oxido ligand (v), and the reaction between two water molecules (vi). The oxygen evolution by the reaction of nitrate ions is also possible (vii and viii). Images and captions are taken from Ref. [45]. Reprinted with permission and modification from Ref. [45], Copyright (2015) by Elsevier.
These results indicate that other scenarios are possible [29]:
- a) μ-O groups on the surface of Mn oxides do not directly participate in the O–O bond formation. Under these conditions, only i or (and) ii in Fig. 3 are the proposed mechanism(s). It is important to note that μ-O groups can participate indirectly in the processes such as deprotonation in water oxidation.
- b) μ-O groups on the surface are oxidized to form O2 (iii and iv in Fig. 3 are possible). Under these conditions the μ-O groups on the surface should exchange with the bulk solution very rapidly because if the exchange is slow more 16O2 and 16O18O should be formed.
- c) μ-O groups are involved in the O–O bond formation, but the number of sites on the oxide's surface is extremely small. However, experiments showed that the number of sites on the oxide's surface is high [29].
Thus, the rate of exchange for the μ-O groups on the surface of Mn oxides is critical to discrimination between the routes a or b. If very fast water exchange, compared to O2 evolution, occurs between the bulk water and μ-O on the surface of these oxides, then μ-O may participate in water oxidation, and i-v pathways are the possible mechanisms (Fig. 3). Usually the time taken for water exchange of μ-O groups in the Mn(III)(μ-O)Mn(IV) or Mn(IV)(μ-O)Mn(IV) complexes is much longer [46], but of course the μ-O groups on the surface of Mn oxides may have different characteristics from the μ-O atoms of the Mn complexes. We used the diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) technique to find out more about the rate of exchange between the bulk water and μ-O on the surface of the nano-sized Mn oxides and found that such exchanges are too slow [47]. But the question is open if the water exchange in the presence of Ce(IV) or [RuIII(bpy)3]3+, not in non-pure water, and under the water-oxidation conditions remains slow or not.
Recently, we performed water oxidation in pure acetonitrile and showed no water oxidation under these conditions. The experiments show that the water molecules from the solvent or OH groups on the surface of the Mn oxides are necessary for water oxidation (Fig. 3) [46]. In other experiments with different ratios of water to acetonitrile, we observed water oxidation is a first order reaction on the concentration of water (Fig. 3). In agreement with the previous results [46], we find Eq. (1) for water oxidation as:
| (1) |
We proposed that for water oxidation catalyzed by layered Mn oxides in the presence of Ce(IV), a mechanism similar to the pathway i in Fig. 3 occurs [46]. In this mechanism, oxygen is formed by the nucleophilic attack of water molecules on an MnO or Mn-OH moiety. Another proposed mechanism is the pathway ii, in which two Mn-OH moieties couple to form oxygen [46]. We hypothesize that there are many Mn–OH fragments on the surface of the Mn oxides, and if the mechanism i is the true mechanism for oxygen evolution, we expect that at least for the first few seconds, and before water exchange by acetonitrile, water oxidation should be observed. Thus, the pathway ii may not be the main mechanism for water oxidation in the presence of Ce(IV).
Generally, two mechanisms for water oxidation were proposed in the presence of layered Mn oxides [47]. The first one is a mechanism with very low overpotential related to the reaction between two OH groups that are coordinated to high-valent Mn ion(s) (pathway ii in Fig. 3), and another mechanism is related to the attack of an outer-sphere water molecule on an OH group that is coordinated to a high-valent Mn ion in the oxide phase (pathway i in Fig. 3) [47,48]. Interestingly, we observed these two ranges for water oxidation for nano-layered Mn oxides, at least, at pH = 6.3 [48]. The first range for water oxidation is near to the peak related to the Mn(III)/Mn(IV) couple with 50–250 mV overpotential, and another one has 600–1000 mV overpotential for water oxidation [48]. We relate these points to pathways ii and i in Fig. 3, respectively. We also suggest that the pathway ii may be related to a four-electron pathway for water oxidation due to a very low overpotential (Fig. 4). As we discussed, the mechanism i is most probably the true mechanism in the presence of Ce(IV) [47].
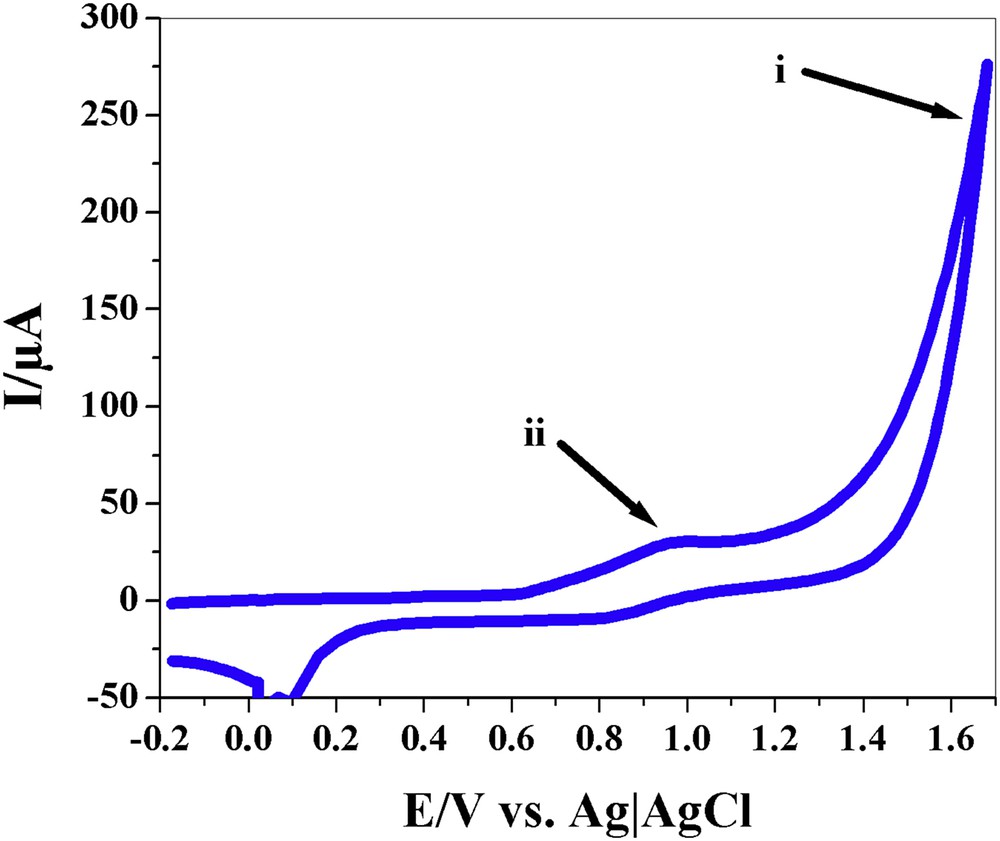
The cyclic voltammogram of a layered Mn oxide. There are two areas for water oxidation for the nano-layered Mn oxides, at least, at pH = 6.3. A range for water oxidation is near the peak related to Mn(III)/Mn(IV) with 100–250 mV overpotential (related to ii pathways in Fig. 3), and another has 600–1000 mV overpotential (related to i pathways in Fig. 3) for water oxidation. Images and captions are taken from Ref. [48]. Reprinted with permission and modification from Ref. [48]. Copyright (2013) by The Royal Society of Chemistry.
In addition to the discussed results, we studied the relation between the Mn content in the MnOx–clay and the rate of water oxidation for different amounts of Mn on the clay [49]. Our experiments showed that at low concentrations of Mn, a polynomial, and at high concentrations of Mn, a linear relation is observed between the Mn content in MnOx–clays and the rate of water oxidation [49]. Based on these data we suggested that cooperation occurs only among the neighboring Mn ions. At low concentrations of Mn on the clay, all Mn ions are significantly used to improve the efficiency of water oxidation [49].
4 Other Mn oxides
We tested many Mn oxides and natural minerals such as Mn-oxide minerals including hollandite, hausmannite and pyrolusite for water oxidation and in the presence of Ce(IV) [23,47–56]. As many minerals possess high crystallinity and low surface area, low water oxidation activities are observed. Mixed-valence Mn–Ca, CaMn4O8 and CaMn3O6 showed promising water-oxidizing activity compared to Mn oxides without Ca ions [53,56]. A soluble form of colloidal Mn(IV) oxide was also tested [55]. However, among all of these compounds, amorphous Mn–Ca oxide is the best catalyst toward water oxidation (see Table 1 for a comparison between different Mn oxides) [40].
The rate of water oxidation catalyzed by various Mn-based catalysts for water oxidation in the presence of a non-oxygen transfer oxidant.
| Compound | Oxidant | TOF mmol O2/(mol Mn·s) | References |
| Nano-scale Mn oxide within NaY zeolite | Ce(IV) | 2.6 | [41] |
| Nanolayered Mn oxide/CNT,G or GO | Ce(IV) | 0.2–2.6 | [78] |
| Nanolayered Mn–Ca oxide | Ce(IV) | 0.5–2.2 | [23] |
| Nanolayered Mn–Al, Zn, K, Cd and Mg oxide | Ce(IV) | 0.8–2.2 | [57,59] |
| Gold deposited on layered Mn oxide | Ce(IV) | ∼2 | [74] |
| Nanolayered Mn oxide/C60 | Ce(IV) | 1.5 | [71] |
| Layered Mn/ND | Ce(IV) | ∼1 | [70] |
| Nanolayered Mn–Ni(II) oxide | Ce(IV) | 0.4–0.6 | [72] |
| CaMn2O4·H2O | Ce(IV) | 0.54 | [19] |
| CaMn2O4·4H2O | Ce(IV) | 0.32 | [19] |
| Mn oxide-coated montmorillonite (low surface) | Ce(IV) | 0.22 | [106] |
| Nanolayered Mn–Cu(II) oxide | Ce(IV) | 0.2–0.35 | [72] |
| CaMn3O6 | Ce(IV) | 0.046 | [53] |
| CaMn4O8 | Ce(IV) | 0.035 | [53] |
| Ca2Mn3O8 | Ce(IV) | 0.016 | [56] |
| CaMnO3 | Ce(IV) | 0.012 | [56] |
| Mn Complexes | Ce(IV) | 0.01–0.6 | [35] |
5 Important factors affecting water oxidation
To answer the question which factors are important in water oxidation catalyzed by layered Mn oxides, different experiments were performed. We briefly discussed each factor separately. Such factors are complicated when considered together (Fig. 5) [57,58].

The most important factors in the water oxidation reaction catalyzed by layered Mn oxides. Blue lines attach the related parameters together. The image is taken from Ref. [57]. Reprinted with permission and modification from Ref. [57]. Copyright (2013) by The Royal Society of Chemistry.
6 Surface
The surface of the layered Mn oxides can affect the activity because the active sites are on the surface of the compound and usually increasing the surface means increasing the number of active sites. However, in comparison to other factors, this factor is not very important.
7 Water content
We observed that the best catalysts need the removal of water from their structures. In other words, wet Mn oxides usually are not very efficient catalysts toward water oxidation.
8 Oxidation state
Oxidation states could be a significant factor in water oxidation catalyzed by Mn oxides and in the presence of Ce(IV); the following activity order is observed toward water oxidation:
9 Crystalline Mn oxides
Crystalline layered Mn oxides show low activity toward water oxidation.
10 Calcination temperature
The best catalysts are obtained by calcination in temperatures of more than 200 °C. At temperatures higher than 550 °C, efficient catalysts are not obtained.
11 Mn content
When we consider the procedure to synthesize layered Mn oxides, the Mn content may be a factor affecting water-oxidizing activity. At low Mn contents and high contents of redox-inert ions, Mn2O3 or MnO2 is the major phase. Thus, the layered Mn oxide phase as an efficient catalyst for water oxidation is not formed under these conditions.
12 The role of redox-inert ions in the layered Mn oxides
Different layered Mn oxides with a few redox-inert ions, such as Ca(II), Ce(III), La(III) Zn(II), Al(III), Cd(II), K(I) or Mg(II), were synthesized; they showed very similar water-oxidation activity [57,59–61].
We observed no special effect for the redox-inert ions but generally a redox-inert ion can increase or decrease the water-oxidation activity of Mn oxides toward water oxidation [57,59–61]. However, layered Mn oxides with different ions display different properties. For example, Mn–Ca oxides are very amorphous until ∼500 °C, but a similar compound with Cd(II) shows a crystalline phase even at ∼300 °C [81,134]. In other words, we related the few different water oxidation activities of these compounds to the physical characteristics of these compounds (size and shape of particles) rather than redox-inert ions and we believed that we should first optimize each ion between the layers of Mn oxides and then compare the water-oxidizing activities of these compounds. However, the role of redox-inert ions in layered Mn oxides is a challenging issue [62].
Very complicated relations occur among the factors affecting water oxidation. For example, as shown in Fig. 5, calcination temperature affects the water content, oxidation state, phase, crystallinity and surface. Thus, to compare the water-oxidizing activities of different Mn oxides all these factors should be carefully analyzed. Recently, we used a new strategy to find the effect of different ions between the layers of Mn oxides on water oxidation [61]. First we synthesized layered Mn oxides with K(I) between the layers and then replaced them by Ca(II), Mg(II), La(III) or Ni(II). Using the strategy, very similar oxides only with different ions between the layers could be obtained [61]. We found that in the presence of cations such as Ca(II), K(I), Mg(II), La(III) and Ni(II) between the layers very similar activity to Mn oxides could be obtained, but no ion is specific for water oxidation at least in the presence of Ce(IV) as an oxidant [61]. However, Cu(II) ions decrease the water-oxidizing activity of layered Mn oxides [61].
As we discussed, different factors are important for Mn oxides in water oxidation. Thus, a quantitative structure-activity relationship (QSAR) model was constructed to predict the catalytic activities of twenty Mn–Ca oxides toward water oxidation using multiple linear regression (MLR) and genetic algorithm (GA) for multivariate calibration and feature selection, respectively [63]. The potassium content, the ratio of Ca:Mn and the average Mn oxidation state seems more important among the eight controlled parameters (ripening time, temperature, Mn content, Ca content, potassium content, the ratio of Ca:Mn, the average Mn oxidation state and the surface of catalyst) during the synthesis of the desired catalysts [64]. The model's accuracy criteria R2 and Q2 for predicting the catalytic rate for the external test set experiments were 0.941 and 0.906, respectively. Therefore, the model reveals acceptable capability to anticipate the catalytic activity. Such theoretical methods are promising to predict efficient catalysts for different reactions [63].
13 Self-healing
In addition to efficiency, the stability of the water-oxidizing compounds is very important. In particular, the compounds which catalyze multi-electron reactions are prone to structural rearrangement, and instability during their turnovers [65]. PSII uses a self-healing process for the WOC as an enzyme that performs a four-electron reaction [66]. In 2011, Hocking et al. [67] proposed a self-healing mechanism for water oxidation by a tetranuclear Mn cluster with a diarylphosphinate ligand. The group showed that in situ cycling between the Mn(II) photoreduced product and an oxidized, disordered Mn(III/IV) oxide phase most likely forms the basis of the observed water-oxidation catalysis (Fig. 6).
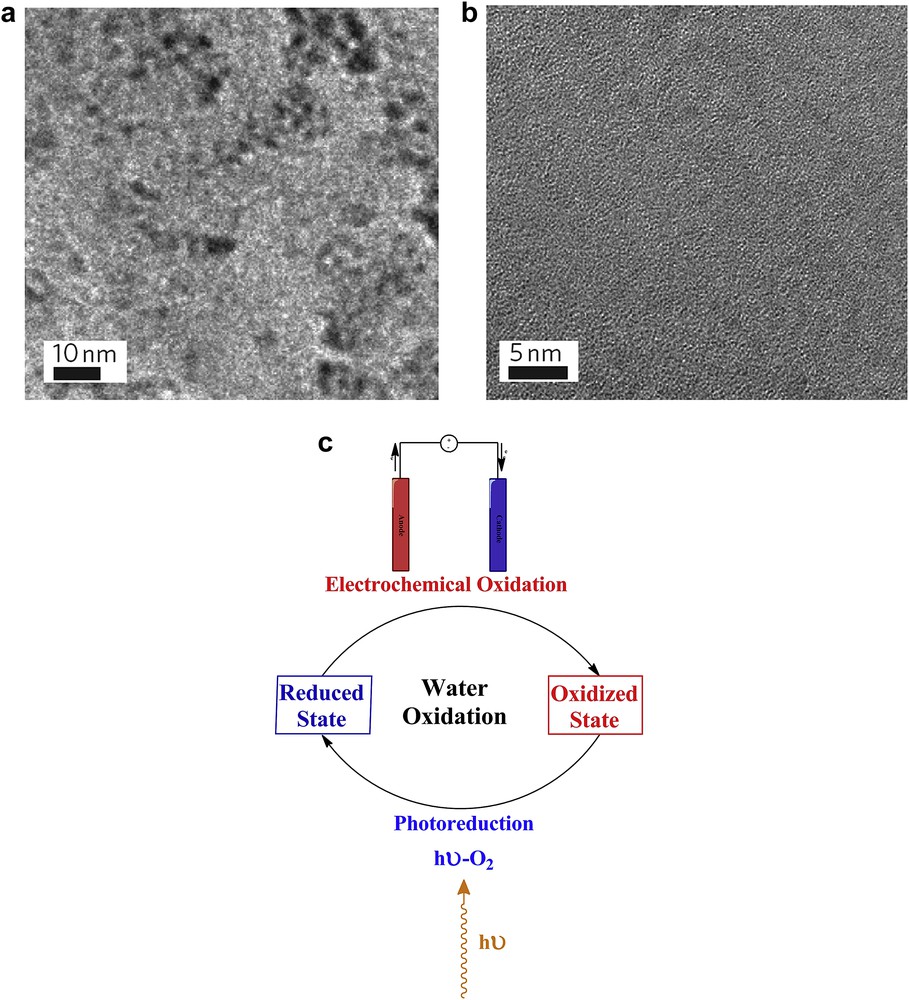
Self-healing cycle for Mn oxides proposed by Spiccia and co-workers. High-resolution transmission electron microscopy images: Nafion film showing the presence of manganese-oxide nanoparticles formed by the electrooxidation of [Mn4O4L6]+ at 1.0 V (versus Ag/AgCl) (a). Nafion film examined after the introduction of [Mn4O4L6]+ into the film (b). Images and captions are from Ref. [12]. Reprinted with permission and modification from Ref. [67]. Copyright (2011) by the Nature Publishing Group. A mechanism proposed for self-healing in the system (c).
To prove the cycling, K-edge X-ray absorption spectroscopy (XAS) of the complex both in acetonitrile and embedded in a Nafion film coated on an electrode was used and the results were consistent with an Mn(II) inner sphere occupied by six oxygen donors with no ordered second sphere (that is, not part of the crystal lattice) [67]. The Spiccia's group concluded that the [Mn4O4L6]+ ion (L: diarylphosphinate) is not the true catalyst for the prolonged water-oxidation catalysis. High-resolution TEM studies of both the oxidized and reduced states of Mn in the Mn compound show that nanoparticles (1–2 nm diameter) can be identified in the oxidized films from both Mn(II) and [Mn4O4L6]+, whereas no nanoparticles were found in the reduced state (Fig. 6). This is consistent with other methods which indicate that in situ reduction occurs to form [Mn(OH2)6]2+ with no higher-order structure [67]. The proposed self-healing process for the system is shown in Fig. 6.
Our group also introduced a new mechanism for self-healing of the Mn oxide phase in the presence of Ce (IV) (Fig. 7a) [41,52,68,69]. From the Pourbaix diagram (Fig. 7b) we understand that in the absence of any higher activation energy all species that occur above the water oxidation line, such as , should cause water oxidation.
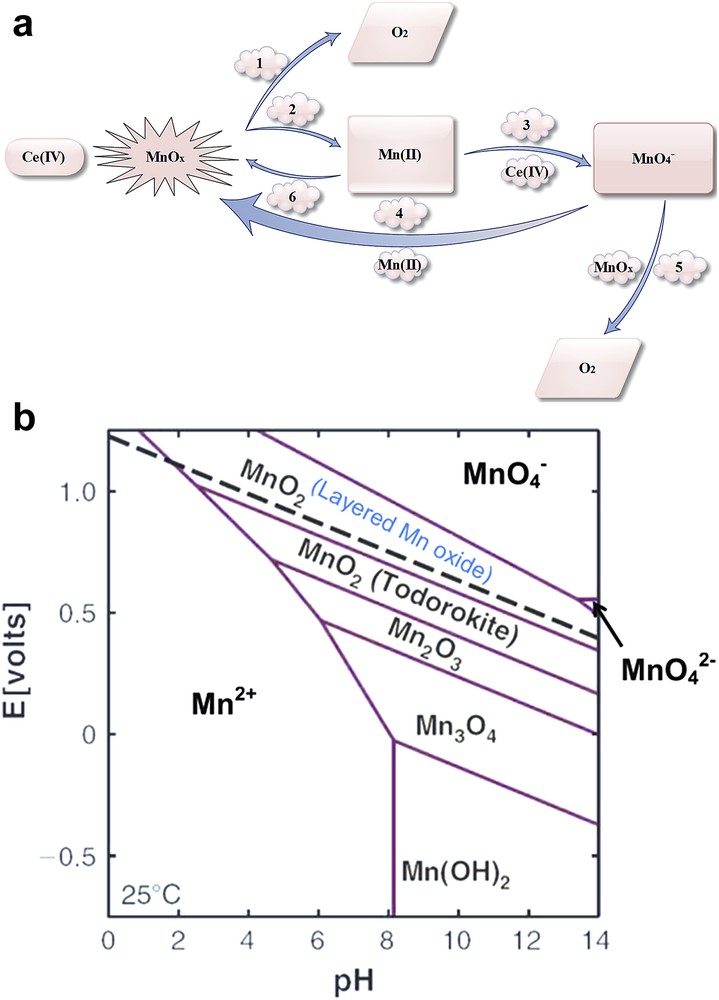
Self-healing in water oxidation catalyzed by Mn oxides in the presence of Ce(IV). 1: Oxygen evolution was detected by an oxygen meter. The origin of oxygen is water. 2: Mn(II) was detected by EPR (see text). 3: formation could be detected by UV–Vis spectrophotometry in the reaction of Mn(II) and Ce(IV). 4: It is known that in the reaction of Mn(II) and at different pH values Mn oxides are produced. 5: ions in the presence of Mn oxides oxidize water. In the reaction, ions are reduced to Mn oxides. 6: Mn(II) in the presence of Ce(IV) forms Mn oxides. In a typical experiment, the reaction of MnSO4 in the presence of Ce(IV) (1.0 M) forms MnO2 that can be detected by XRD. Images and captions are taken from Ref. [26] (a). Reprinted with permission and modification from Ref. [68]. Copyright (2013) by The Royal Society of Chemistry. The Pourbaix diagram for Mn ions (b).
However, water-oxidation reactions are kinetically usually slow by the compound. In high enough potential both water oxidation and reactions take place. ions and similar species cause a long-term stability problem, separate Mn ions from the catalysts and decrease the activity of these compounds toward water oxidation [41,52,68,69].
Our group reported a new self-healing process of Mn oxides in the presence of Ce(IV). We found that in the reaction of these Mn oxides with Ce(IV), ions are produced that change their concentration with time [69]. At the end of this reaction, all ions are consumed, suggesting the operation of a self-healing mechanism according to these experiments:
- a) An solution in the presence of K-layered oxides showed a linear reduction in the concentration of ions.
- b) formation was observed in the reaction of Mn(II) nitrate (0.5 mM) and Ce(IV) (0.2–2 M).
- c) Without any Mn oxide, the amount of showed no change for 100 h of the experiment indicating that Mn oxides are necessary for the reduction of .
Using the multivariate curve resolution-alternative least squares, we found that the concentration profile of Ce(IV) (red line) is in a descending manner and ions are produced and consumed during the reaction time. The concentration profile of the ions (the blue line) is similar to an intermediate during water oxidation.
Regarding these experiments, we proposed a self-healing reaction. In the mechanism [66]:
- d) ions may be formed by oxidizing the leaked Mn(II) from Mn oxides by Ce(IV).
- e) Mn oxides are responsible for the reduction of . At least two reactions are possible for the reduction of in the presence of Mn oxides: i) The reaction of ions with Mn(II) ions on the surface of Mn oxides (Fig. 7a). ii) It is also possible that , as an oxidant, and Mn oxides, as water-oxidizing catalysts, react to oxidize water (Fig. 7a).
Self-healing mechanisms in biological systems are very complicated. However, a comparison between the biological and artificial mechanisms toward self-healing in water-oxidizing catalysts was reported, which shows that there are a few similarities [69]. Such systems can be considered to be composed of a few simple “building blocks” [66]:
- The Mn–Ca cluster in Photosystem II: Mn, Ca and O
- Mn oxides: primarily Mn and O
- Co oxides: primarily Co and O
Both decomposition (red arrows in Fig. 8) and self-healing (green arrows in Fig. 8) reactions can occur for these mechanisms. The detachment and reattachment of these components is related to the decomposition and self-healing processes, respectively [66].
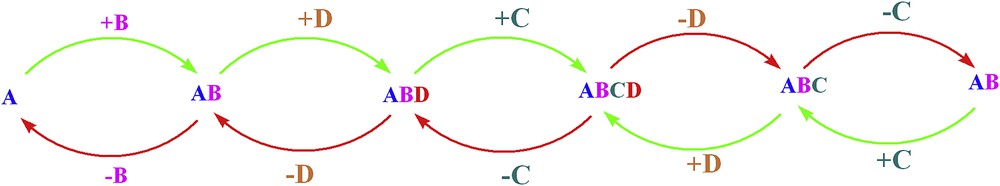
Decomposition and self-healing of the PSII-WOC (during photodeactivation) or metal oxides (during catalytic operation) could be summarized by this simple schematic image. In this image, each structure contains only a few components. The attachment or removal of these components results in self-healing (green arrow) or decomposition (red arrows). Reprinted with permission and modification from Ref. [66]. Copyright (2015) by the American Chemical Society.
14 Supports
To obtain a good water-oxidizing catalyst in the form of Mn oxides, we used many Mn oxides on supports or Mn oxide composites [70–78]. The details are not discussed here but for water oxidation and in the presence of Ce(IV):
- i) Although, carbon nano-structures as supports are conductive and very promising toward water oxidation, these carbon-based supports are not stable under the conditions of water oxidation.
- ii) Compared with other Mn oxides, gold or platinum as a support has no significant effect on the water-oxidizing activity of the Mn oxides, at least in the presence of Ce(IV). The results show that the previous reports on the effect of inert metals may depend on the synthetic procedure or water-oxidation conditions. Thus, the role of the redox-inert metals under these conditions should be carefully reconsidered.
- iii) The effect of the support depends on the catalysts and the conditions of the water-oxidation reaction (pH, etc.).
- iv) An unstable support decreases the water oxidation rate of these catalysts. As an example, MgO is not stable in the presence of Ce(IV) and decomposes after a few minutes. Under these conditions, the very fragile nano-layered Mn oxides decompose to immediately.
- v) Redox-active supports show complicated effect on water-oxidizing catalysts. Most probably, redox reactions between the supports and the Mn oxide phase are important for these effects.
- vi) The redox-inert supports such as SiO2 or ZrO2 show no specific effect and it seems that these supports are important for controlling size, crystallinity and morphology. We suggested that the crystallinity and morphology of Mn oxides depend on the sort, crystallinity and morphology of the support.
Some reports show that gold or platinum has an important effect on the water-oxidizing activity of Mn oxides, but we indicated that such results should be carefully reconsidered because the effect may be related to the reductants and the synthetic methods.
The water oxidation catalyzed by such Mn oxide composites is usually faster than that for many pure Mn oxide phases [79,80], but similar to or lower than that for layered Mn oxides [81,82].
15 An approach for catalyst design in artificial photosynthetic systems: focus on nano-sized inorganic core
In addition to Mn–Ca oxides in PSII, some enzymes can be considered as catalysts having nano-sized inorganic cores in protein matrices (Fig. 9) [83,84]. The metal-oxido or sulfide-clusters [85], which can be considered as cofactors in enzymes, may be recruited for use in other related reactions. The hypothesis is an extended form of the idea of Mn–Ca oxides for water oxidation.
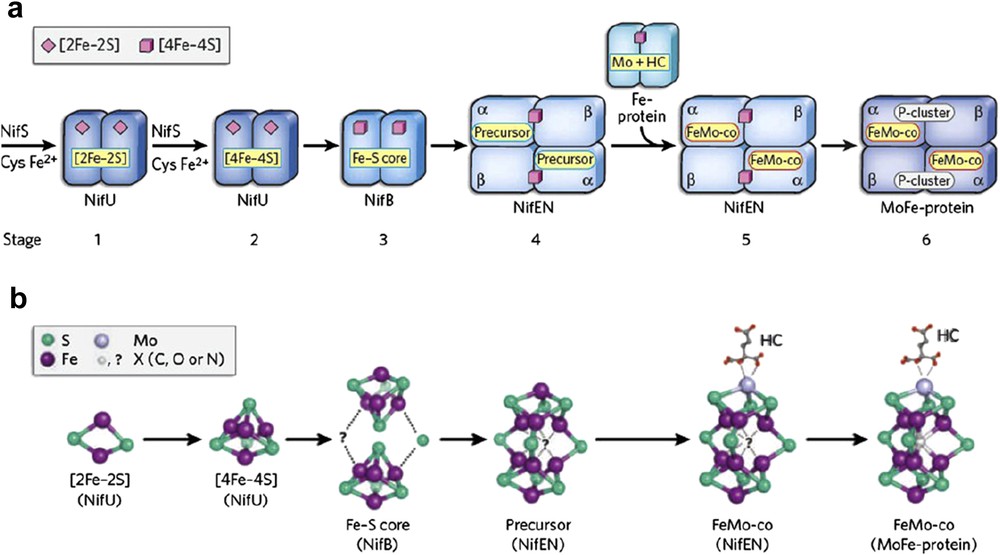
Sequence of events during the FeMoco assembly. The biosynthetic flow of FeMoco is as follows: NifU → NifS → NifB → NifEN → MoFe-protein. The permanent metal centres of the scaffold proteins are colored in pink; the transient cluster intermediates are colored in yellow. HC, homocitrate. Structures of intermediates during FeMoco assembly. The cluster types that were identified (on NifU, NifEN, and MoFe-protein) or proposed (for NifB) are shown. Hypothetically, NifB could bridge two [4Fe-4S] clusters by inserting a sulfur atom along with the central atom, X, thereby generating a FeeS scaffold that could be rearranged into a precursor closely resembling the core structure of the mature FeMoco. In the case of the NifEN-associated precursor, only the 8Fe model is shown. The potential presence of X in the intermediates of FeMoco biosynthesis is indicated by a question mark. Reprinted with permission and modification from Ref. [85]. Copyright (2009) Nature Publishing Group.
16 Mn oxides with organic compounds
There are many amino acids in PSII, but there are only a small fraction of the residues that come in direct contact with the Ca and Mn ions in the Mn–Ca cluster [21,22]. Such amino acids in PSII are involved in proton, water and oxygen transfer and we may note the structure as a nano-sized Mn–Ca oxide in a protein matrix [21,22]. We introduced the Mn–Ca oxides as structural and functional analogs to the native WOC in PSII, but these models contain no groups such as guanidinium or imidazole to stabilize Mn oxides or (and) decrease the activation energy for water oxidation as there are in PSII. In the synthetic model metal complexes, for example in hydrolytic enzymes, the positioning of groups similar to guanidinium could lead to more than 1000-fold increase in reactivity [86]. To arrange guanidinium, imidazole or other groups around Mn ions of Mn oxides, we reported a strategy using Mn oxide monosheets (Fig. 10a) [87]. The Mn(III, IV) oxide monosheets, similar to other cationic monosheets, could aggregate to form layered structures with cationic ions such as guanidinium (Fig. 10b) and imidazolium [87]. The hybrid of guanidinium or imidazole and Mn-based water-oxidizing catalysts are interesting for water oxidation because such structures are found in the WOC in PSII.

TEM image of a monosheet of Mn oxides (a). Mn oxide monosheets aggregate to form layered structures with cationic ions such as guanidinium between the layers (b). Reprinted with permission and modification from Ref. [87]. Copyright (2013) by Springer.
In photosynthesis, first, photons are absorbed by PSII antenna molecules, primary charge separation occurs and a special chlorophyll is oxidized (P680+) [88]. Then, P680+ is reduced by electron transfer from a tyrosine (Tyrosine 161) residue (YZ), located on the D1 protein of PSII, and a tyrosine radical (YZ*) is formed. Electrons for the reduction of YZ* are extracted from the WOC and it finally oxidizes water to oxygen [88]. We reported on a self-assembled layered hybrid of phenol-Mn ions similar to the one in the WOC of PSII as a model to Yz near the WOC (Fig. 11) [89].
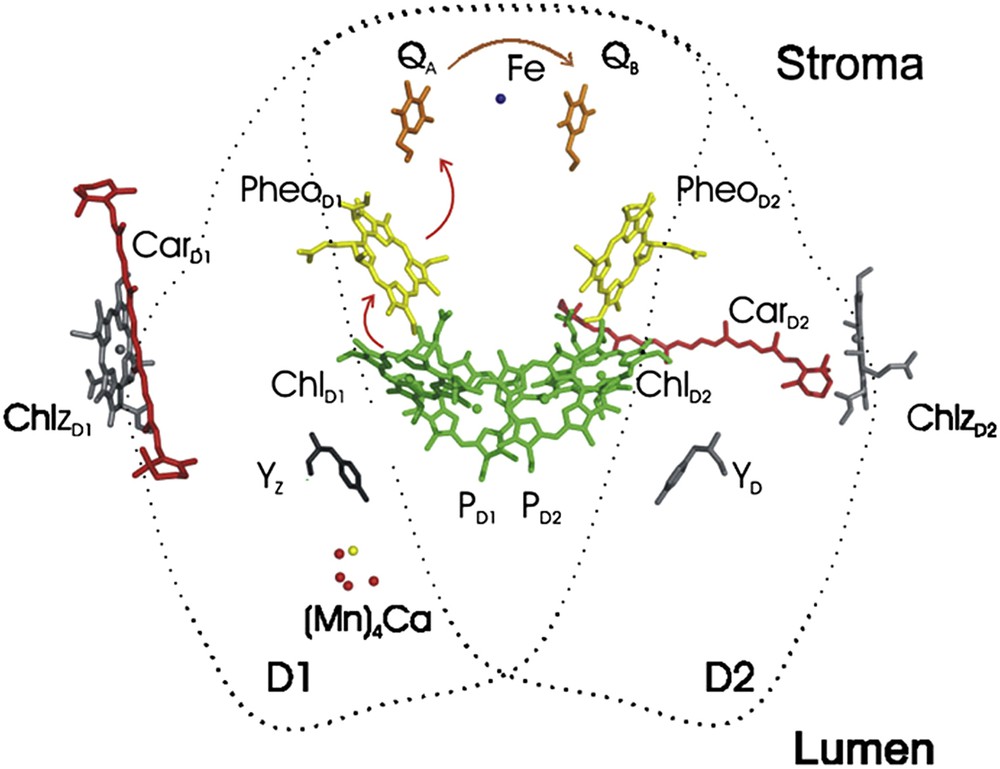
The cofactor arrangement in the core of the reaction center (the view is along the membrane plane). Organic cofactors (for the sake of simplicity, the heme group of cytochrome b559 is omitted) are colored in green (Chl), yellow (Pheo), magenta (plastoquinones QA and QB) and red (carotenoids). Ca (yellow), Fe (blue), and Mn (red) are shown as spheres; the figure was generated using PyMOL. The coordinating protein subunits D1 and D2 are indicated by dotted lines. Reprinted with permission and modification from Ref. [88]. Copyright (2012) by Elsevier.
The compound could be the first step to synthesize a self-assembled layered hybrid of phenol – Mn ions, similar to the WOC of PSII.
As shown in Fig. 12a, the proton acceptor from YZ in PSII is a histidine residue, histidine 190 (His 190), which is hydrogen-bonded to the phenolic proton [88].
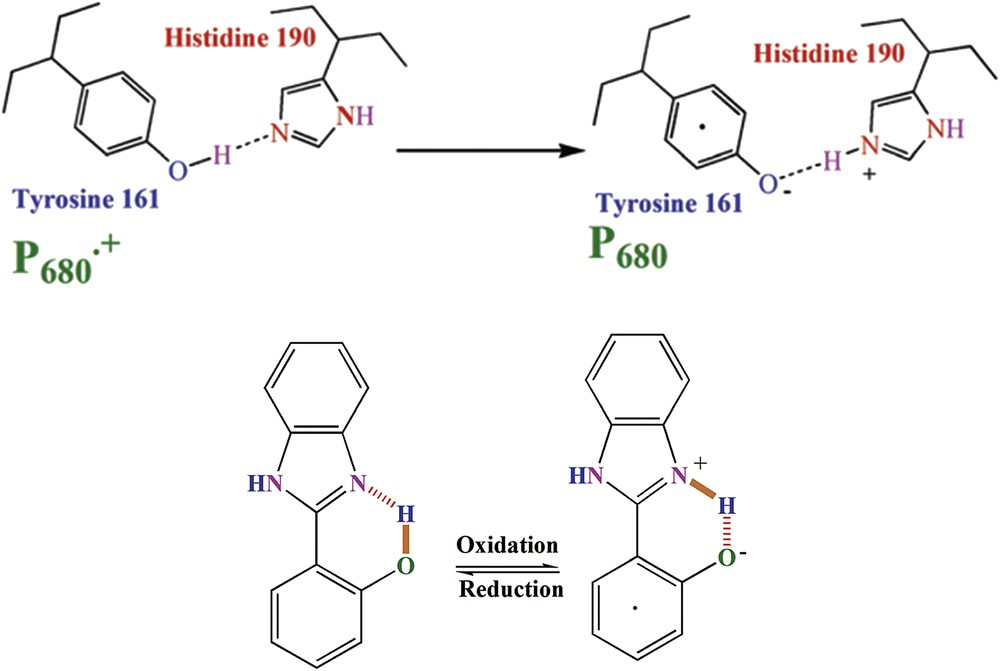
2-(2-Hydroxyphenyl)-1H-benzimidazole phenol (IP) used as a model for Yz/His 190 in PSII. As shown in the scheme, oxidation of IP to oxidized IP (IP) could change the hydrogen bond pattern as observed in PSII. The dot shows an unpaired electron in IP. Reprinted with permission and modification from Ref. [94]. Copyright (2013) by The Royal Society of Chemistry.
Tyrosine 161 is oxidized by P680+ (the dot within tyrosine shows that it is in its oxidized form). There is a hydrogen bond between YZ (tyrosine161) and the ε-nitrogen of a histidine (D1-His 190), which is important in proton-coupled electron transfer [88].
2-(2-Hydroxyphenyl)-1H-benzimidazole phenol (IP) was used as a model for YZ/His 190 in PSII by the research groups of Ally Aukauloo and Thomas and Ana Moore [90–93].
We have used Mn oxide monosheets to arrange IP between the Mn layers in Mn(III, IV) oxides with birnessite structures to synthesize 2-(2-hydroxyphenyl)-1H-benzimidazole–Mn oxides, as a model for YZ and His 190 near the Mn cluster in PSII (Fig. 12) [94].
In 2012, Nakamura et al. reported that amines, pyridine and its derivatives significantly decrease the overpotential of water oxidation at neutral pH catalyzed by Mn oxides [95]. The research group related the effect to Mn–N bonds stabilizing Mn(III) or proton transfer by amine groups [96]. The role of Mn(III) in water oxidation is not known, but it was proposed that the electronically degenerated Mn(III), having an eg1 configuration and displaying the Jahn–Teller effect, contributes to the structural flexibility for catalytic turnover in water oxidation at the surface [97]. Our group used the strategy of inserting nano-sized Mn oxides into organic polymer/poly peptides [48,98–100].
We reported on a nanolayered Mn oxide/poly(4-vinylpyridine) as a model for an Mn cluster in PSII [48].
The compound is a very efficient water-oxidizing catalyst and sizable oxygen evolution was detected at 50 mV overpotential at near neutral pH [48]. However, the amount of oxygen is much lower than that of the oxygen produced by the WOC. The number is as low as the overpotential used by nature in cyanobacteria, algae and green plants for similar reactions. We considered poly(4-vinylpyridine) because of the pyridine groups that are most probably important as proton acceptors, in the stabilization of Mn(III) and reduction of overpotential for water oxidation, to attach Mn oxides to electrodes. Other reasons to select the polymer are:
- - the polymer is stable in the presence of powerful oxidants;
- - the polymer and its salts are conductive and polyelectrolytes;
- - the polymer can be considered as a Mn-stabilizing protein;
- - the polymer with many pyridine groups can inhibit acidic conditions and provide a buffering environment for Mn oxides.
Interestingly, we used the catalyst for seawater electrolysis at low overpotentials. At such low overpotentials decomposition to was not observed [48].
As we discussed, there are many carboxylate groups around the Mn–Ca cluster in PSII. We synthesized an Mn–Ca cluster in a bovine serum albumin (Fig. 13a,b) [98] or polyglutamic matrix (Fig. 13c,d) [99].
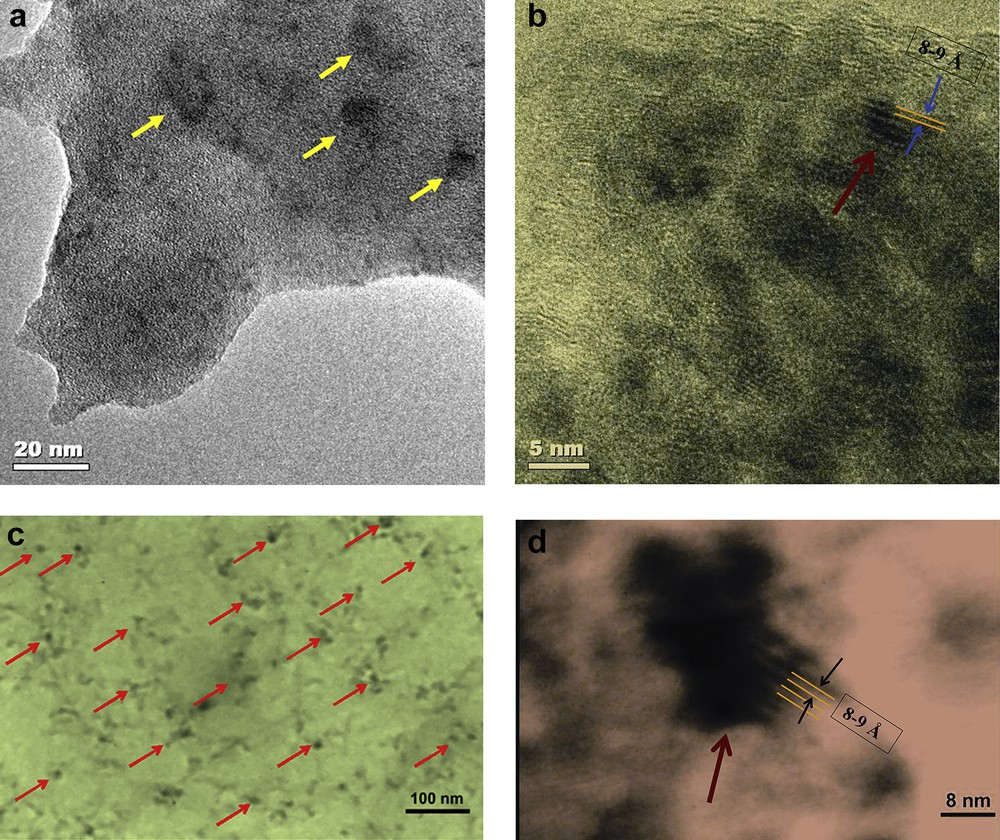
TEM and HRTEM images of BSA (a and b) and MnCaOx-poly-l-glutamic acid (c and d) in water. Black particles are Mn oxide nanoparticles. The crystalline particles are layered Mn oxides with a distance of 8–9 Å between layers.
The residues participate in proton transfer and management, stabilization of Mn(III) or Mn(IV), decreasing the overpotential for water oxidation and core nucleation of Mn–Ca oxides. Although the compounds showed no water oxidation activity, they are interesting structural models for the WOC [98,99]. Recently, we introduced a novel engineered polypeptide containing tyrosine around a nano-sized Mn–Ca oxide, which oxidizes water at a low overpotential (240 mV) (Fig. 14), with a high turnover frequency of 1.5 × 10−2 s−1 at pH = 6.3 and interestingly in the Mn(III)/Mn(IV) oxidation-range [100]. The catalyst is a new model for the WOC in PSII [100]. According to the related peaks of Mn(II)/Mn(III), Mn(III)/Mn(IV) and tyrosine oxidation, it was proposed that tyrosine could reduce Mn(IV) ions to Mn(III) ions and also cause Mn oxidation from II to III [100]. Thus, the increasing amounts of Mn(III) ions because of the Mn(II) oxidation and (or) Mn(IV) reduction by tyrosine may be important for the efficiency of the catalyst [100]. On the other hand, Mn(II) is labile and the oxidation of this ion to Mn(III) also inhibits Mn leaching to the solution [100].
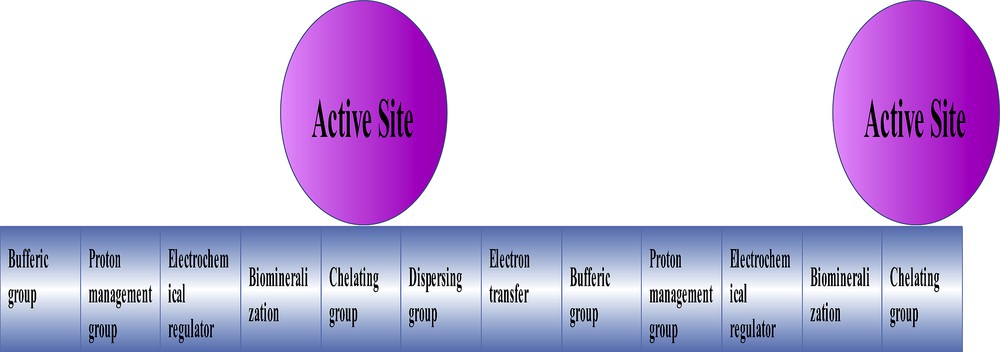
A schematic image of the engineered groups around the catalyst particles. Such groups as present in enzymes can efficiently increase the catalytic activity of active sites. Reprinted with permission and modification from Ref. [100]. Copyright (2015) by The Royal Society of Chemistry.
The observation of oxygen evolution in a potential near the Mn(III)/(IV) oxidation-range shows a relationship between the capacitor behavior and water oxidation. In other words, a charge accumulation related to the Mn(III) to Mn(IV) oxidation occurs before the water oxidation. Such charge accumulation was known for the WOC of PSII in the Kok cycle [100]. Thus, a clever strategy used by Nature just before water oxidation is the charge accumulation with substantial potential to oxidize Mn(III) to Mn(IV) ions [100]. I believe that the engineered organic or inorganic compound around a nano-sized metal oxide is a good strategy to synthesize efficient catalysts with high active-site densities, compared to natural enzymes, with promising characteristics such as substrate transfer and access to active groups, as well as sections with buffering, proton management, electrochemical, biomineralization, chelating, dispersing and electron transfer effects on not only water oxidation, but also on many different reactions. The engineered sections can also control electronic, magnetic properties or solubility of the inorganic cores.
Tris(2,2′-bipyridyl)ruthenium (II) ([Ru(bpy)3]2+) is an important sensitizer for visible-light water oxidation in the presence of [Co (NH3)5Cl]2+ or as an electron acceptor and visible-light (λ > 400 nm) with the aim of studying a model for PSII [101]. By using monosheets, we employed a new strategy to arrange [Ru(bpy)3]2+ ions as photosensitizers and Mn (III, IV) oxides as water-oxidizing catalysts in a self-assembled layered hybrid of [Ru(bpy)3]+2/Mn oxide [102]. Our strategy was the aggregation of Mn(III, IV) oxide monosheets in the presence of [Ru(bpy)3]+2 [102]. The strategy increases the rate and the yield of oxygen evolution because of the decreasing intermolecular interactions between the [Ru(bpy)3]3+ ions. The negative electrostatic charge of the Mn(III, IV) oxides may also have a positive effect on binding and reaction of the oxide with [Ru(bpy)3]3+ [102]. The rate of oxygen evolution catalyzed by this compound, without any additional [Ru(bpy)3]2+ ions in solution with a 250 W tungsten–mercury lamp in the presence of [Co(NH3)5Cl]2+ (10.0 mM) as the sacrificial electron acceptor, is 7.0 μmolO2molMn−1 s−1 [102].
The organic/inorganic engineered compound around a nano-sized metal oxide is a promising way to synthesize a super-catalyst (Fig. 14).
17 Mn oxides as true catalysts for water oxidation
Previously, we reported on an oxygen-evolution reaction upon mixing solid Mn(III) bidentate Schiff base complexes with aqueous solutions of Ce(IV) [13]. We reported on Mn(II), and insoluble Mn products which show weak absorption bands characteristic of an {Mn2O2} core in the region of 600–700 cm−1 [13]. Using ESI(+)–MS/MS at pH∼4.5 we also showed that the mononuclear complex [Mn(tptz)(CH3COO)(OH2)2]NO3 is not fully preserved and is in equilibrium with some cationic species [15]. In this case, a dissociative Mn-ligand process occurs in the water/acetonitrile solution [15]. The experiment also showed that the most important reaction in the presence of Oxone® is a “decoordination”.
Metal ions can catalyze such decoordination reactions [103,104]. In 2011, we reported a pentanuclear heterobimetallic cation with Ce(III), Ce(IV) and Mn(II) obtained in the reaction of [Mn2(μ-dipic)2(H2O)6]·2H2dipic and Ce(IV) [105]. The heterobimetallic complex, again, shows that Mn complexes are not very stable in the presence of the oxidant [105]. In 2012, based on UV–Vis spectroscopy, X-ray diffraction spectrometry, dynamic light scattering, Fourier transform infrared spectroscopy, electron paramagnetic resonance spectroscopy, transmission electron microscopy, scanning electron microscopy and X-ray photoelectron spectroscopy, we proposed nano-sized Mn oxides as the true catalysts for water oxidation in the reaction of some Mn complexes and Ce(IV) [106]. In 2014, the reaction of [(OH2)(terpy)Mn(μ-O)2Mn(terpy)(OH2)]3+ on clay as a water-oxidizing catalyst in the presence of Ce(IV) was considered. It was found that under the chosen reaction conditions, this complex [107] and other similar complexes underwent transformation into a layered Mn oxide phase, which was the true catalyst for water oxidation (Fig. 15) [108,109].

In many oxygen evolution reactions catalyzed by Mn-based compounds, a layered Mn oxide is the true catalyst. *In the case of Oxone® as an oxidant, other intermediates than Mn oxides are also present. In the case of this oxidant in the presence of Mn oxides oxygen evolution and not water oxidation occurs. Reprinted with permission and modification from Ref. [112]. Copyright (2014) by The Royal Society of Chemistry.
I suggest that the Mn oxide and even many other transition metal oxides are at least one of the true catalysts in many oxidation reactions. Our group showed such results for many Mn complexes in water-oxidation reactions. Recently, our group considered the water-oxidizing activity of a copper complex with (E)-3-(pyridin-2-yldiazenyl)naphthalen-2-ol as a ligand [110]. No oxygen evolution was observed for this complex in the presence of Ce(IV) at pH = 1 and 11, but water electrooxidation at pH = 11 is observed, which is related to a Cu oxide formation [110]. In 2015, we detected Mn oxides as the true catalyst in an alcohol oxidation reaction [111].
All these experiments indicate that [106–112]:
- - in the reaction of metal complexes with powerful oxidants the true catalyst should be carefully checked because usually such oxidizing conditions decompose the ligands in the complexes;
- - for oxidation reactions catalyzed by metal complexes under electrochemical conditions, the precipitation of metal oxides should be carefully checked because such metal oxides can form films on the surface of the electrode even without the decomposition of the ligand. Such films are active in water oxidation,
| ML ↔ M+ + L− |
| M+ → M oxide (true catalyst for the oxidation reaction); |
- - organic ligands in the presence of strong reductants and oxidants were extensively used as catalysts toward different oxidation and reduction reactions, careful analyses should be considered to detect the true catalysts.
I personally think that metal oxides are at least one of the true catalysts in many oxidation reactions where strong or even moderate oxidants are used.
More interestingly, even other phases of Mn oxides convert into the layered Mn oxide phase with no long-range order in both electrochemical and chemical water oxidation reactions [113,114]. This finding can be predicted by the Pourbaix diagram and it shows that the true catalyst should be considered very carefully even for solid-state compounds. After the conversion of many Mn oxides to the layered Mn oxide phase with no long-range order, the obtained solid is even a better catalyst compared to fresh catalysts [113,114]. It is very interesting and is an ‘evolution-like’ phenomenon in chemistry [113,114].
18 The effect of UV radiation on the decomposition of the layered Mn oxides
Light absorption by plastosemiquinone or Mn ions in PSII has been proposed to account for photoinhibition under UV or near-UV illumination. The effect of UV radiation on the decomposition of the Mn–Ca oxides in the presence of organic groups was studied [115]. Our results showed that there are similarities between the reactions of the Mn–Ca oxides and the WOC of PSII in the presence of UV radiation [115].
No effect on releasing of Mn(II) to solution was observed without any organic compounds in the presence of UV radiation. However, high amounts of Mn(II) released in the presence of aspartic acid and pyrazine-2,3-dicarboxylate were observed in the presence of UV radiation [115]. Our results showed that the carboxylate groups, as hard and good ligands for Mn ions, accelerate decomposition of the oxide phase. Imidazole and phenol ligands have little effect on the release of Mn ions to solution. Most probably, these groups reduced Mn(IV) on the surface of oxides and released it from the solid phase [115]. Interestingly, Bovine Serum Albumin (BSA) as a protein could separate nanoparticles from an aggregated Mn–Ca oxide and also release Mn(II) ions even in the absence of UV radiation [115].
19 A Swiss army knife: Mn oxides
In addition to water oxidation, a variety of important reactions can be catalyzed by Mn oxides. With this respect Mn oxides truly deserve to be called a “catalytic Swiss army knife” [116–122]. Using Mn oxides as catalysts, our group reported oxidation of alcohols, and sulfides and epoxidation of olefins in the presence of different oxidants [116–122]. They are not discussed here, but such systems could be coupled to artificial photosynthetic systems to produce important compounds. Layered Mn oxides can be used for water treatment to remove toxic ions. Interestingly, Nature uses Mn oxides to control heavy metal concentrations in soils and in aquatic sediments [123,124]. We synthesized nano-layered Mn oxides and deposited Ag on it [125]. The Ag deposited on nano-layered Mn oxides shows antibacterial activities toward Escherichia coli and Staphylococcus aureus [125]. On the other hand, layered Mn oxides can be used to remove heavy metal ions (Pb(II), Cd(II), As(III), Cr(III), etc.) from water [126]. Thus, the system combines both heavy-metal ion removing activity and antibacterial effect in one compound [125].
We reported Mn oxides as, at least, a true catalyst for many oxidation reactions catalyzed by Mn and many metal complexes [127–133]. However, we could not detect any nanoparticles in some cases [134,135].
I believe that with learning strategies from natural systems [136–143] and extending these strategies to other reactions, design of modern catalysts for all reactions using only earth-abundant, low cost and environmentally friendly metal ions is possible.
Acknowledgment
I am grateful to the Institute for Advanced Studies in Basic Sciences and the National Elite Foundation for financial support. In 2009, I had a chance to join the group of Professor Philipp Kurz as a visiting scientist. He is not only a very kind person, but also a nice teacher, mentor and supervisor. I learned from him and his kind PhD students, now Dr. Anne Westphal and Dr. Hans-Martin Berends, a lot about metal complexes and metal oxides. I do thank them for all their help, considerations and kindness. I also would like to acknowledge the members of my research group and collaborators who have contributed to the work described herein. I thank Dr. Małgorzata Hołyńska for comments and helpful discussions.