

1 Introduction
The modified organic conjugated polymers have been widely used as optical and electronic devices such as organic light-emitting diodes (OLEDs), organic field-effect transistors (OFETs), and organic solar cells (OSCs) [1–5]. These compounds have received a great deal of attention from both academic and industrial research centers [6–9]. During the past few years, the thiophene-based electronic materials such as the oligothiophene derivatives have attracted an extensive interest since they exhibit physical properties that lend them to the development of display devices, transistors, sensors, etc [10–12]. The ease in the chemical and electro-chemical modification of the structures of these materials can potentially allow us to fine-tune their electronic and optoelectronic properties [13–17]. These properties strongly depend upon the degree of electronic delocalization present in such materials and their effective conjugation length; the latter is affected by the introduction of suitable substituents at specific positions in the thiophene ring. However, unsubstituted oligothiophenes show low fluorescence and poor stability compared with the substituted ones [18].
One approach to develop higher performance oligothiophenes is to substitute the thiophene ring. The substituents at the β and β' positions of the five-membered thiophene ring prevent the undesirable α-β couplings that decrease the conjugation length and solubility of the polymers. In fact, the introduction of a proper substituent into the monomer ring plays an important role in the optical and semi-conducting properties of the polymers [19–21]. It has been found that the electronic and structural properties of a functional group contribute to the delocalization of the π-electron conjugated system. In this way, insertion of an electron-donating substituent at the β-position of the thiophene ring might be an interesting strategy to prevent the defects, and withhold or even surpass the desirable properties of polythiophenes [22–24].
An alternative approach is to modify the oligomers by end-capping the α and α' positions with functional groups [25–30]. This approach limits the steric hindrance of the substituents on the electronic and optoelectronic properties of the polymers. The oligothiophenes with end-capped substituents are of growing synthetic interest, and have been shown to act as potential light-emitting materials in OLEDs and as active components in OFETs. However, these features, obtained experimentally, are one of the interesting points in the capped-oligothiophene computational approaches, which have been employed to investigate the different aspects related to the molecular and electronic structures of these materials using the quantum chemical computations [31–38].
In our previous studies [39–41], we investigated theoretically a series of substituted heterocyclic monomers and their related oligomers as compounds designed for novel conductive polymers using the ab initio and DFT methods. In the present study, we extend our research studies to evaluate the effect of the aminoalkyl group as the end-substituent on the structural and conjugational properties of a series of oligothiophenes (repeating units n, from 2 up to 8), in the ground and excited states, and their radical cation (p-type doped) and radical anion (n-type doped) species. A comparative study was used to investigate the effects of the oligomer chain length (dimer up to octamer) and the size of the aminoalkyl group (R) including aminomethyl (AM), aminoethyl (AE), and aminopropyl (AP) on the π-conjugated system and bond length alternation pattern, highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies, electron affinity (EA) values, ionization potential energy (IP) values, Mulliken charges, spin densities, and first excited energies of BRnTs using the DFT calculations. Therefore, quantum chemical investigation of a series of aminoalkyl end-capped oligothiophenes was desirable for designing novel functional materials, and this was the main purpose of this work.
2 Computational methods
Density functional theory (DFT) [42] was employed to optimize the geometry of each titled oligomer in the neutral and singly ionized states by means of the Gaussian 09 program package [43]. Our previous studies [39–41] and the reports given by others [44–50] have shown that the gradient procedures at the hybrid density functional B3LYP [51] have been adapted to describe the oligoheterocycles. A preliminary basis set test performed for the calculations carried out on the electronic ground state of the short oligomer chains showed that 6-31G(d,p) was the best basis set that could be used within our available hardware/software facilities in a reasonable period of time. Thus the DFT-B3LYP functional and the 6-31G(d,p) basis set were selected for the present study of BRnTs, and the optimized structures were calculated for both the neutral and doped states of the oligomers. The nature of the optimized stationary point was characterized by frequency calculations at the same level of theory. The energies of the neutral and doped states in the relevant geometries were obtained for calculating the IP values using the Koopman theorem [52] and the EA values. The absorption spectra of all the oligomers studied were investigated by the time-dependent density functional theory (TD-DFT) [53–55] based on the B3LYP/6-31G(d,p) optimized geometries. The natural bond orbital (NBO) calculations [56,57] were also carried out on the B3LYP/6-31G(d,p) optimized geometries to evaluate the electron conjugation effects.
3 Results and discussion
3.1 Structures of BRnTs
The selected symbols used for representing the chemical structures of the α,α'-bis(aminoalkyl) oligothiophenes (BRnTs) studied in this work, and the sketch map of the structure for the octamer type (n = 8), as an example of the BRnT structures, are illustrated in Scheme 1. It was found that the well-defined oligomer structure contained almost exclusively the head-to-tail (HT) conformation due to the α,α'-couplings between the adjacent heterocyclic monomers in the backbone of the polymer chains [39,40]. Thus geometry optimization of the neutral and ionic species of BRnTs were carried out on the HT conformation at the B3LYP/6-31G(d,p) level of theory. One of the important structural parameters related to the extent of the π-conjugation of the oligomer derivatives is the chain planarity that can be reflected by the torsional angle, D, as defined in Scheme 1. The calculated values for the dihedral angles defining the torsion between two adjacent building units along the oligomer chains for BRnTs and their corresponding ionic species are given in Tables S1–S3 in the Supporting Information section. For a comparative study, the torsion angle values for the neutral, cationic, and anionic species of octathiophenes (8T) and aminomethyl end-capped octathiophenes (BAM8T) are presented in Table 1.

Structure and numbering scheme used for bis(aminoalkyl) end-capped octathiophene BAR8T.
Torsion angle values (deg.) for the unsubstituted (8T) and aminomethyl end-capped octathiophenes (BAM8T) in neutral and ionic states (Di in Scheme 1).
| Species | D1 | D2 | D3 | D4 | D5 | D6 | D7 |
| 8T: | |||||||
| neutral | 15.1 | 13.5 | 11.6 | 11.7 | 11.8 | 13.7 | 15.1 |
| cationic | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| anionic | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| BAM8T: | |||||||
| neutral | 14.8 | 13.5 | 10.8 | 11.5 | 11.0 | 13.3 | 14.6 |
| cationic | 2.0 | 0.2 | 0.1 | 0.0 | 0.1 | 0.2 | 0.8 |
| anionic | 0.4 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.6 |
The results obtained indicate that the structures of all the oligothiophene derivatives in the neutral state have small deviations from the planar structure, in which the aminoalkyl substituent torsions (D1 and D7) are largely twisted from the plane of the backbone structure due to their steric hindrances. According to Table 1, the insertion of two aminoalkyl substituents at the end-sides of the octathiophene chain causes a lower value in their torsion angles with respect to the unsubstituted octamers (8T). It is interesting that the doped oligomers have completely planar structures (all the Di values are near zero, <2°). It is clear that the planarity of the polymer chains has an important effect on their electric conductivity. The calculated results show that with increase in the repeating units, there is a significant reduction in the inter-ring torsion angle at the center of the oligomer, compared to the outside one. It was also found that the size of the aminoalkyl substituents did not have a considerable effect on the torsion angle values of the different BRnTs. In general, crystalline oligothiophenes have been found to be nearly planar, as a result of more favorable crystal packing [58], and, therefore, the DFT-optimized geometries obtained are in agreement with the solid-state structure.
Another structural parameter affecting the π-conjugated system is the bond alternation parameter (δ). It is defined as the bond length difference between two adjacent single and double carbon–carbon bonds. A suitable π-conjugated system corresponds to a lower δ value [59]. Here, the average bond alternation parameter, δave, is defined as the mean value δ along the π-conjugated backbone. The calculated average bond alternation parameters for all the neutral species are tabulated in Table 2. By comparing with the unsubstituted oligothiophenes (R = H), the BRnT compounds, in particular with the small substitutions, have lower alternation bond parameters, which decrease with increase in the oligomer chain length. The end-capped polymers with electron-donating aminoalkyl groups, therefore, showed a better π-electron conjugated structure. It was also found that the size of the aminoalkyl groups did not significantly affect the reduction in the average bond alternation parameter. The insertion of two aminomethyl groups at the end-positions in an octamer chain could decrease the δave values by 0.03 and 0.02 Å for the p- and n-doped states (8T(+) and 8T(−) chains), respectively (rows 9 and 10 in Table 2). In other words, the doped oligomers, especially the p-type ones, show a better π-conjugated system with respect to their neutral states. Therefore, the end-capped oligothiophenes with electron-donor aminoalkyl substituents can enhance both the aromatic and quinoid characteristics of BRnTs with respect to the parent oligothiophenes.
Average bond alternation parameters (Å) for the unsubstituted (R = H) and aminoalkyl end-substituted oligothiophenes.
| Species | H | CH2–NH2 | C2H4–NH2 | C3H6–NH2 |
| 2T | 0.0655 | 0.0635 | 0.0630 | 0.0640 |
| 3T | 0.0590 | 0.0575 | 0.0580 | 0.0585 |
| 4T | 0.0555 | 0.0550 | 0.0550 | 0.0555 |
| 5T | 0.0530 | 0.0525 | 0.0520 | 0.0530 |
| 6T | 0.0515 | 0.0510 | 0.0510 | 0.0515 |
| 7T | 0.0510 | 0.0505 | 0.0490 | 0.0510 |
| 8T | 0.0505 | 0.0495 | 0.0490 | 0.0495 |
| 8T(+) | 0.0377 | 0.0325 | 0.0324 | 0.0334 |
| 8T(−) | 0.0282 | 0.0224 | 0.0222 | 0.0235 |
A structural parameter of the π-conjugated system is the quinoid coefficient, fn [60–62]. This geometry parameter refers to a structure in which the inter-ring bond has a greater double bond character than that in the standard aromatic configuration, and is defined as:
| (1) |
Calculated quinoid coefficients, f8, for the unsubstituted (8T) and aminoalkyl end-capped octathiophenes in neutral and doped states.
| Species | Neutral | p-doped | n-doped |
| 8T | 1.036 | 1.015 | 1.020 |
| BAM8T | 1.035 | 1.013 | 1.019 |
| BAE8T | 1.034 | 1.013 | 1.019 |
| BAP8T | 1.036 | 1.014 | 1.020 |
3.2 Electronic properties
In order to characterize the excitation properties and the ability of electron or hole transport, we calculated the distribution patterns of the frontier molecular orbitals (FMOs) for all the molecules studied based on the current level of theory. For a detailed comparative study, the FMO diagrams of a series of the aminoalkyl-capped oligothiophenes such as BAM2T, BAM4T, BAM6T, and BAM8T are illustrated in Fig. 1. According to this figure, the ground state (S0) → first singlet excited state (S1) excitation process may be mainly assigned to the HOMO → LUMO transition, which corresponds to a π∗–π excited singlet state. As shown in Fig. 1, the FMOs spread over the whole π-conjugated backbone in the oligomers with relatively short conjugation lengths (n < 6), whereas with increase in the thiophene conjugation length (n > 6) and also capping of the end-sides of the oligomeric chains by aminoalkyl groups, the electron clouds on the substituent groups decrease, being localized on the thiophene units. These results can be ascribed to the electron-donating properties of the aminoalkyl groups. The results obtained revealed that the FMO distribution patterns of the oligothiophene derivatives were slightly influenced by the size of the aminoalkyl groups.

Frontier molecular orbitals of BAM2T, BAM4T, BAM6T, and BAM8T calculated at the B3LYP/6-31G(d,p) level of theory.
Furthermore, as shown in Fig. 1, HOMOs are composed of anti-bonding interactions between the joint C–C atoms in the backbone of the oligomer chains. Two electrons occupy each HOMO, so that the twist angles between the rings are large in the neutral species. In the case of the cationic species, one electron is removed from HOMOs, and the repulsive interaction becomes weaker, and so the structure is more close to planar. LUMOs are composed of joint atoms possessing bonding interactions between the rings. The orbital phase of the C–C bonds in the connecting positions is in-phase. Therefore, the structures of the anionic species are purely planar. These results are in line with those obtained in the structural investigation mentioned in the previous section.
For all the unsubstituted and end-capped oligothiophenes studied, the calculated values of the HOMO-LUMO gaps (HLGs) at the B3LYP/6-31G(d,p) level of theory are given in Table 4. Closing the end-sides of oligothiophenes by aminoalkyl groups destabilizes both the HOMO and LUMO levels. It was found that this effect was asymmetric in the sense that the HOMO level was more affected than the LUMO level. Thus the energy gap in the aminoalkyl-capped oligothiophenes decreases with respect to the parent oligothiophenes. It is worth noting that the energy gap decreases with increasing chain length. For the case of the aminomethyl substituent in the ionic states, the HLG values for the unsubstituted and end-capped oligothiophenes were compared, and the results obtained are listed in Table 5. A similar tendency can also be found for ionic states, whereas the HLG values are less than those for the corresponding neutral species (Table 4), which lead to an improved conductivity. It is well-known that the parameters IP and EA are closely related to the degree of electron extraction (hole injection) to form the p-type semi-conductors and electron injection to form the n-type semi-conductors, respectively [48,50].
HLG values (in eV) for unsubstituted and bis(aminoalkyl) end-substituted oligothiophenes.
| n | nT | BAMnT | BAEnT | BAPnT |
| 2 | 4.35 | 4.11 | 4.14 | 4.17 |
| 3 | 3.56 | 3.43 | 3.46 | 3.44 |
| 4 | 3.13 | 3.05 | 3.06 | 3.06 |
| 5 | 2.85 | 2.81 | 2.78 | 2.81 |
| 6 | 2.67 | 2.66 | 2.66 | 2.67 |
| 7 | 2.58 | 2.52 | 2.49 | 2.54 |
| 8 | 2.49 | 2.47 | 2.45 | 2.47 |
HLG values (in eV) for unsubstituted and aminomethyl end-substituted oligothiophenes in ionic states.
| n | nT | BAMnT | ||
| Cationic | Anionic | Cationic | Anionic | |
| 2 | 3.90 | 2.92 | 3.66 | 2.81 |
| 3 | 3.12 | 2.16 | 3.00 | 2.08 |
| 4 | 2.71 | 1.70 | 2.64 | 1.65 |
| 5 | 2.46 | 1.39 | 2.41 | 1.36 |
| 6 | 2.30 | 1.16 | 2.26 | 1.14 |
| 7 | 2.19 | 0.98 | 2.16 | 0.97 |
| 8 | 2.12 | 0.83 | 2.10 | 0.83 |
As it can be seen in Table 6, the IP and EA values show that the BRnT oligomers have higher IP values (corresponding to a less energy barrier for the hole injection) and lower EA values (corresponding to the electron injection facility) compared with those corresponding to the unsubstituted ones, which are in line with the corresponding orders of their HLG values. It was also found that as the chain length of an oligomer increases, its IP value decreases, which corresponds to the improvement in the hole injection properties. This is consistent with the observed trend in the case of HLGs. It is clear that the first step in the electro-polymerization mechanism of conductive polythiophenes is the formation of the intermediate radical cations by the electro-oxidation of monomers. This oxidation reaction can be considered as an ionization process [63–65]. It may be concluded that the initiation step in the electro-polymerization of the BRnT species requires a lower applied potential. This potential is reduced when the electron conjugation of molecules increases, which is in agreement with the lower oxidation potential measured experimentally [63].
Ionization potential (and electron affinity) values for BRnT oligomers (in eV).
| Species | H | CH2–NH2 | C2H4–NH2 | C3H6–NH2 |
| 2T | 7.11 (0.34) | 6.50 (−0.04) | 6.56 (0.43) | 6.43 (0.36) |
| 3T | 6.49 (−0.36) | 6.08 (−0.33) | 6.07 (−0.38) | 5.98 (−0.23) |
| 4T | 6.14 (−0.78) | 5.69 (−0.78) | 5.81 (−0.77) | 5.70 (−0.70) |
| 5T | 5.90 (−1.05) | 5.54 (−1.05) | 5.61 (−0.92) | 5.59 (−1.13) |
| 6T | 5.74 (−1.25) | 5.43 (−1.24) | 5.55 (−1.07) | 5.56 (−1.18) |
| 7T | 5.62 (−1.39) | 5.35 (−1.39) | 5.36 (−1.24) | 5.41 (−1.27) |
| 8T | 5.53 (−1.50) | 5.29 (−1.44) | 5.43 (−1.68) | 5.53 (−1.44) |
The calculated Mulliken charges on each thiophene ring (Ti) in the conjugated chains of the neutral species are given in Table S5. In the case of the end-capped and parent octathiophenes, the positive charge is locally generated in the outermost thiophene rings, while the central thiophene rings have a negative charge (see Table 7). However, in the BR8T species, the total negative charge was distributed over the thiophene rings in the oligomer backbone, while the aminoalkyl groups bear positive charges. This indicates that electrons are slightly transferred from the aminoalkyl groups to the π-conjugated carbon backbone. One can expect a larger dipole moment in the end-substituted oligomers, and an improvement in charge transition along the conjugated system, in comparison with the parent oligomers.
Mulliken charge values on thiophene rings (Ti in Scheme 1) of unsubstituted and aminomethyl end-substituted octamers.
| Species | T1 | T2 | T3 | T4 | T5 | T6 | T7 | T8 |
| 8T | 0.000 | 0.005 | −0.002 | −0.002 | −0.002 | −0.002 | 0.005 | 0.000 |
| BAM8T | −0.017 | −0.007 | −0.007 | −0.005 | −0.005 | −0.006 | −0.007 | −0.017 |
| BAE8T | −0.042 | −0.008 | −0.008 | −0.005 | −0.006 | −0.007 | −0.009 | −0.042 |
| BAP8T | −0.032 | −0.009 | −0.006 | −0.005 | −0.005 | −0.007 | −0.008 | −0.032 |
It is well-known that the uniform distribution of the spin density in the π-conjugated system results in a better delocalization of charge carriers in the oligomeric chain length, and it improves the conductivity [65,66]. In the case of the p-doped state, the diagrams of the spin density distribution on the carbon atoms of the aminomethyl end-capped di-, penta-, and octa-thiophenes are shown in Fig. 2. The results obtained show that the spin density is symmetrically divided on the carbon atoms of the conjugated chains, and that an increase in the oligomer chain length causes a more uniform distribution of spin density along the oligomer backbone. Delocalization of the unpaired spins onto the oligomer chains leads to satisfactory resonance structures, which reflects an improvement in the charge transfer of the p-doped oligomers. The results obtained are in-line with those obtained by the Mulliken charge calculations. It was also found that a similar trend exists for different types of aminoalkyl groups.
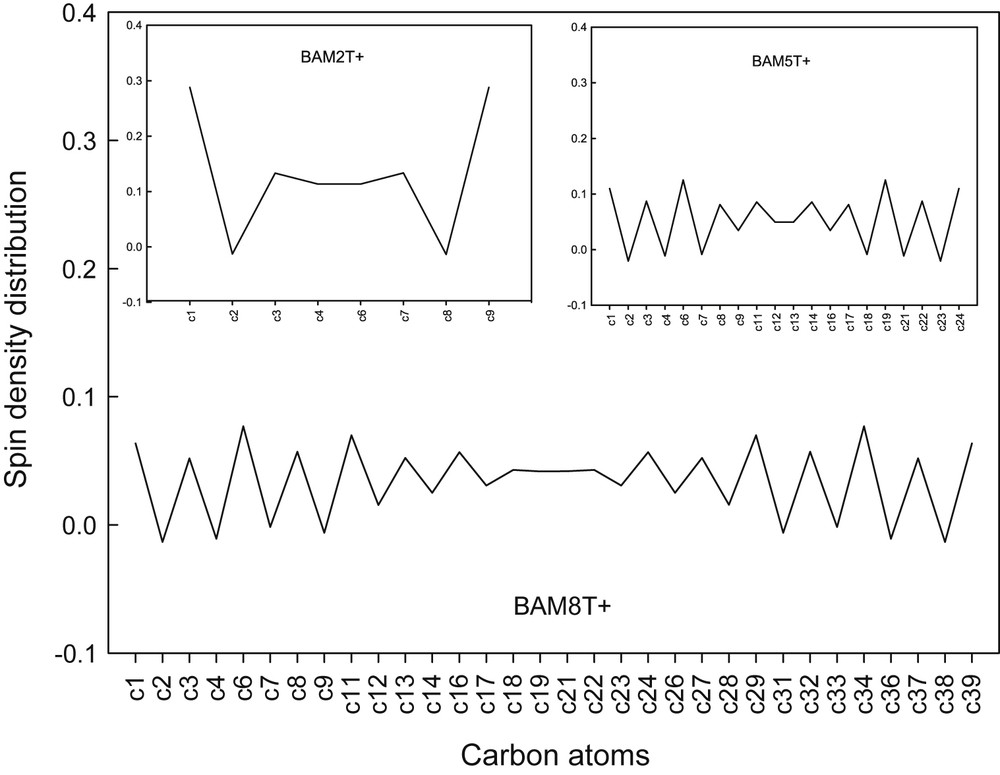
Spin density distribution on carbon atoms (Scheme 1) of BAM2T+, BAM5T+, and BAM8T+ calculated at the B3LYP/6-31G(d,p) level of theory.
Understanding the relationship between the low-lying excited states of the π-conjugated organic semi-conductors and the charge transport properties of the oligomers is a key point in providing good candidates for the design of conducting polymers. In this way, studying the excited states by the TD-DFT technique is an important tool to qualitatively judge the elucidation of the mechanism of the electron and hole conductivities in the BRnT oligomers. The results obtained for the TD-DFT excitation energies (Eg) and longest wavelengths (λmax) for the maximum absorption peaks together with the corresponding oscillator strengths (f) of each oligomer are given in Table S6. For the case of the octamer chain in the neutral and ionic states, the values of simulated absorption spectra properties for the unsubstituted and aminoalkyl-capped octathiophenes were compared, and the results obtained are listed in Table 8. For all the oligothiophene derivatives, attachment of the aminoalkyl groups to the end-sides of oligothiophenes leads to an evident bathochromic shift and a decrease in the Eg values. It is noticeable that by increasing the oligomer chain length from dimers up to octamers, an increase in the λmax value and so a decreasing trend in the Eg values were observed (Table S6). In comparison with the ground state of BAM8T, the ionic states have larger λmax values owning to their higher conjugation lengths and flat structures. This trend was similar to those observed for the other oligomers with different aminoalkyl groups. These results suggest that the titled end-capped oligomers are good candidates for the ambipolar charge transfer semi-conductors under the suitable operating conditions.
Values of λmax, Eg, and oscillator strength (f) in simulated absorption spectra of the unsubstituted (8T) and aminomethyl end-capped octathiophenes (BAM8T) at neutral and ionic states.
| Properties | 8T | BAM8T | ||||
| Neut. | Cat. | Ani. | Neut. | Cat. | Ani. | |
| λmax/nm | 559 | 1693 | 1764 | 569 | 1811 | 1776 |
| Eg/eV | 2.27 | 0.73 | 0.70 | 2.18 | 0.68 | 0.70 |
| f | 2.73 | 0.82 | 0.77 | 2.80 | 0.92 | 0.82 |
Furthermore, the simulated excitation spectra obtained using a Gaussian broadening for the six lowest excitation states of the bis(aminoethyl) end-capped hexathiophenes (BAE6T) in the neutral, cationic, and anionic states are illustrated in Fig. 3. This figure shows that in the neutral and ionic species, attachment of the aminoethyl groups to the end-sides of hexathiophene reduces the Eg values, compared with the unsubstituted oligomer (6T). In comparison with the n-doped state (BAE6T−), the p-doped state (BAE6T+) reveals a larger shift in the decrease of excitation energy which corresponds to its higher conjugation length. As expected, this issue indicates an improved conductivity of the polymer chains of the molecules under study, which are qualitatively in agreement with the results obtained in studying the HOMOs and LUMOs.
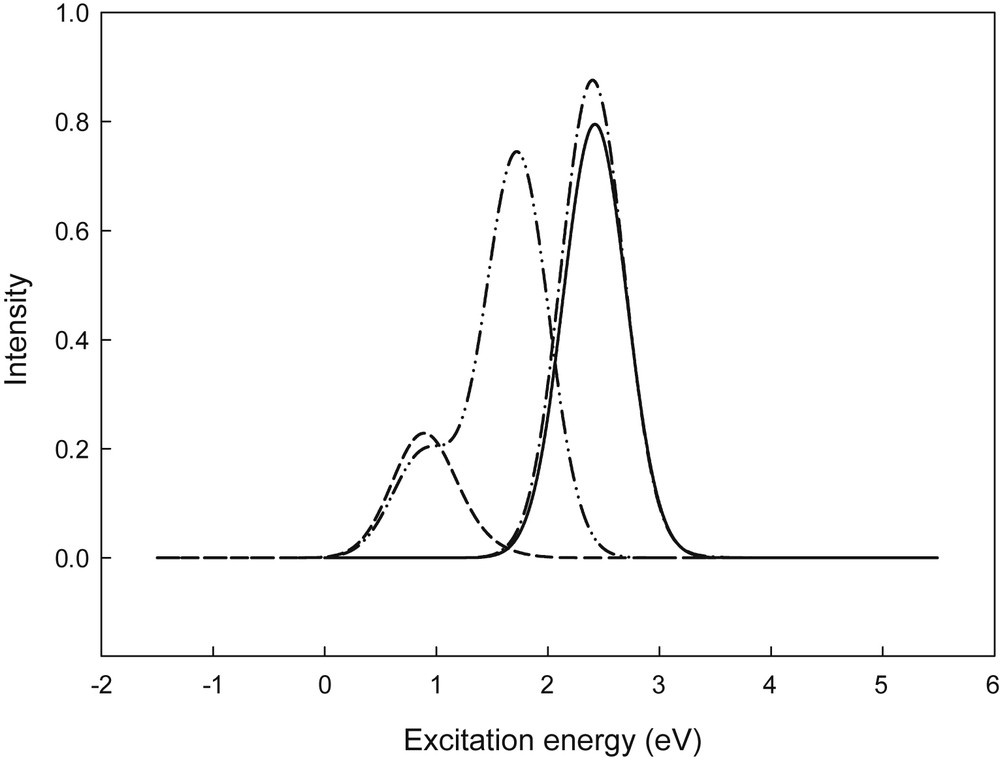
Simulated absorption spectra of 6T (Solid line), BAE6T (Dash-dot line), BAE6T+ (Short dash line), and BAE6T− (Dash-dot-dot line) species, calculated at the TD-B3LYP/6-31G(d,p) level of theory (relative oscillator strengths are given in arbitrary units).
As mentioned earlier, the NBO computations can be used qualitatively to evaluate the electron conjugation effect by the stabilization of interaction energies, E(2), between a donor bonding orbital and an acceptor anti-bonding orbital. Strong conjugation occurs between the two bonds when the E(2) value increases [67]. The calculated average stabilization energies,
Average interaction energies of interaction between electron-donating and electron-accepting CC bonds, E(2)π(out) → π*(in), over carbon backbone structures of BRnTs (kcal mol−1).
| Species | H | CH2–NH2 | C2H4–NH2 | C3H6–NH2 |
| 2T | 13.75 | 14.45 | 14.58 | 14.26 |
| 3T | 14.85 | 15.30 | 15.22 | 15.18 |
| 4T | 14.85 | 15.25 | 15.20 | 15.02 |
| 5T | 15.30 | 15.60 | 15.71 | 15.25 |
| 6T | 15.38 | 15.52 | 15.48 | 15.24 |
| 7T | 15.39 | 15.80 | 15.98 | 15.33 |
| 8T | 15.41 | 15.61 | 15.75 | 15.33 |
4 Conclusion
In the present work, the effect of end-substitution of aminoalkyl groups (including –CH2–NH2, –C2H4–NH2, and –C3H6–NH2) on the geometry and electron conjugation properties of a series of oligothiophenes (from dimer up to octamer) was investigated using the B3LYP/6-31G(d,p) level of theory. The results obtained revealed that the presence of an electron-donating aminoalkyl substituent at the end-sides of the oligothiophene chains played fine-tune effects on their planar structures, π-electron conjugated system, and excitation states. The calculations carried out on both the neutral and doped aminoalkyl end-capped oligothiophenes showed that injection of electrons or holes into these oligomers induced a better conjugation length and that the whole molecules tend to be more planar than their corresponding ground states. In comparison, the hole injection to form p-type semi-conductors is more favorable than the electron injection to form the n-type ones. The calculated Mulliken charge distribution and spin densities, HOMO-LUMO energies, electron affinities, and ionization potentials show that with increase in the oligomer chain length the doped oligomers have a more satisfactory structural and electronic characteristics for the conducting polymers. The results obtained from the absorption spectra evaluated at the TD-DFT level are in good agreement with the results obtained for the HLG investigations. In addition, the results obtained from the NBO analysis show that the average stabilization energy values for all the studied species are near 15.0 kcal mol−1, indicating a good electron delocalization over the backbone structure of BRnTs. Generally, the influence of the aminoalkyl end-substitution does not significantly depend on the size of the alkyl group. It could be anticipated that the aminoalkyl end-capped substitution is an effective technique for designing novel materials with desirable conduction properties.
Acknowledgment
The authors wish to thank Shahrood University of Technology for the financial support of this work.