

1 Introduction
Les halophytes sont des plantes qui ont la capacité de se développer en milieu salin (environ 200 mM de NaCl). Il existe des halophytes strictes, qui ne se développent naturellement que sur des sols salés, et des halophytes facultatives, qui peuvent tolérer une forte salinité, ce qui est le cas de l’espèce choisie comme modèle pour cette étude. Pour tolérer cette salinité, les halophytes développent différentes stratégies, notamment des adaptations morphologiques (transformation ou réduction de l’appareil aérien pour limiter l’évapotranspiration), physiologiques (prélèvement et compartimentation contrôlés des ions Na+ et Cl–) ainsi que métaboliques [1]. En effet, les halophytes synthétisent de façon accrue des composés appelés osmolytes, car capables d’ajuster la pression osmotique intracellulaire. Ces composés sont stockés dans la vacuole pour contrebalancer le potentiel osmotique des ions Na+ et Cl– accumulés dans cet organite. Leur nature chimique est assez variée : sucres (mannitol), acides aminés (proline), ammoniums quaternaires (bétaïne), notamment. Par ailleurs, le stress salin engendre aussi un stress oxydatif. Ainsi, les halophytes synthétisent de façon accrue des molécules anti-oxydantes [2–4] en plus d’augmenter la synthèse et l’activité d’enzymes détoxifiantes, comme la superoxyde dismutase [5]. Ces métabolites secondaires antioxydants, souvent de nature polyphénolique, sont porteurs de nombreuses activités biologiques [6,7], qui en font des ingrédients naturels d’intérêt, notamment dans le domaine de la cosmétique [8–10].
Dans un souci d’innovation, l’industrie cosmétique est en quête permanente de nouveaux actifs végétaux susceptibles de rentrer dans la composition de différentes formulations topiques, que ce soit sous forme d’extraits totaux, d’extraits enrichis ou encore de molécules purifiées. Une grande diversité de polyphénols est retrouvée dans les halophytes, mais les composés majoritaires sont les acides phénoliques et les flavonoïdes [8]. Un large spectre d’activités a déjà été mis en évidence pour ces composés : anti-âge, anti-inflammatoire [11], anti-collagénase, anti-élastase [12], anti-tyrosinase [13], par exemple. De façon plus précise, les acides caféiques et leurs dérivés, sélectionnés dans notre étude, ont déjà montré des activités in vitro sur des cibles telles que l’inflammation (TNFá, NO, COX-2) [14], ou encore la tyrosinase [15]. Grâce à leurs propriétés anti-oxydantes décrites ci-dessus, les acides dicaféoylquiniques(diCQA) peuvent aussi être utilisés dans des formulations cosmétiques en alternative à des conservateurs d’origine chimique [16].
Les polyphénols sont des molécules largement répandues dans le monde végétal. De nombreuses études ont été réalisées sur ces métabolites secondaires, leurs propriétés physico-chimiques sont très bien décrites, notamment leurs propriétés spectrales (UV et masse). Ainsi, il est possible de les caractériser en ligne sans les isoler au préalable. Cette approche, appelée déréplication, peut être réalisée par chromatographie en phase liquide couplée à un détecteur UV à barrette de diodes et à un spectromètre de masse à haute résolution (HPLC–DAD–HRMS). Cet équipement permet d’obtenir le spectre UV des métabolites détectés dans un extrait, ainsi que le spectre de masse avec l’ion de la molécule protonée ou déprotonée à partir duquel la formule brute de la molécule est déterminée. Les différents ions fragments apportent aussi des informations précieuses sur la structure du métabolite détecté.
Classiquement, les composés phénoliques sont extraits par macération ou extraction à reflux avec un mélange hydro-alcoolique. Néanmoins, le développement de techniques plus respectueuses de l’environnement devient un enjeu majeur. En effet, le recours à des méthodes plus efficaces en termes de rendement, de temps et de coût, mais aussi qui limitent la consommation de solvants, devient une priorité pour de nombreuses entreprises [17]. De plus, les solvants utilisés doivent eux aussi être le plus « vert » possible, notamment pour protéger l’environnement et l’expérimentateur tout comme le consommateur. Dans le cas de produits finis destinés au marché de la cosmétique, la réglementation REACH impose des contraintes justifiant le choix de procédés d’extraction écoresponsables. Enfin, les techniques d’extraction et/ou de purification doivent aussi être pratiques et limiter le nombre d’étapes pré- et post-traitement, qui sont chronophages, génératrices de déchets et de variabilité dans les résultats. C’est dans ce contexte que nous avons choisi d’évaluer les performances de trois techniques d’extraction écoresponsables et de n’utiliser comme solvant d’extraction que de l’eau, de l’éthanol et du CO2, qui sont des solvants « verts ».
L’extraction assistée par micro-ondes (MAE) correspond à une soumission du mélange solvant/matrice à un champ électromagnétique de 2,45 GHz. Sous l’influence du courant alternatif, les molécules possédant un moment dipolaire vont changer d’orientation 4,9·109 fois par seconde [18]. Le transfert d’énergie au mélange se fait grâce à la rotation des dipôles et à la conduction ionique. Dans le cas d’un mélange hydro-alcoolique alliant deux solvants polaires, le mélange va s’échauffer très rapidement. La MAE permet ainsi d’augmenter la pénétration du solvant dans la matrice. Dans le cas de matériel végétal frais, l’eau intrinsèque au végétal possède un moment dipolaire (1,85 D) et peut jouer aussi un rôle de solvant. Ganzler et al. [19] citent, comme avantages généraux de cette technique, son fort rendement d’extraction, sa rapidité d’exécution ainsi que son coût réduit.
L’extraction par fluide pressurisé (PFE), aussi appelée extraction accélérée par solvant (ASE), permet quant à elle de réaliser des extractions à température élevée (jusqu’à 200 °C) et surtout à pression élevée. L’échantillon à extraire est placé dans une cellule, complétée par de la terre de diatomée. La cellule est ensuite remplie de solvant, puis maintenue sous pression (100 bar) pendant une certaine durée grâce au solvant. Puis le solvant, chargé en métabolites, est collecté et l’échantillon rincé avec une quantité souhaitée de solvant frais, qui sera également collecté. Successivement, plusieurs cycles d’extraction peuvent être réalisés sur un même échantillon. La pression permet de maintenir le solvant d’extraction sous forme liquide à haute température, et l’augmentation de la température engendre une diminution de la viscosité et de la tension de surface du solvant, l’ensemble favorisant la pénétration du solvant dans la matrice et améliorant ainsi les temps et rendements d’extraction.
L’extraction par fluide supercritique (SFE) consiste à faire passer du CO2 à l’état supercritique au travers d’une cartouche contenant du matériel végétal (extraction dynamique). À l’état supercritique, le CO2 a une densité qui peut varier de celle des gaz jusqu’à celle des liquides, suivant les conditions de pression et de température, une viscosité plus proche des gaz et une diffusivité intermédiaire. Ainsi, il possède une importante capacité de transfert. Cette technique, très respectueuse de l’environnement, présente l’inconvénient de l’apolarité du CO2, qui limite l’extraction des composés polaires. Cependant, l’addition au CO2 de co-solvants permet de modifier la polarité du mélange et rend possible l’extraction de métabolites secondaires polaires, comme c’est le cas pour la majorité des polyphénols [20,21]. Généralement, ce sont des alcools qui sont utilisés comme co-solvants, l’éthanol présentant de plus l’avantage d’une très faible toxicité [22]. D’autres paramètres, comme la température, la pression ou le débit du fluide, peuvent également être modifiés pour accroître les rendements d’extraction ou la sélectivité.
2 Matériel et méthodes
2.1 Matériel végétal
La plante choisie pour cette étude est une Asteracée halophile achetée à la société Elma par BiotechMarine-Seppic. Son identité n’est pas dévoilée ici pour des raisons de confidentialité. Un spécimen est déposé à l’herbier de l’université de Strasbourg. Les parties aériennes en fleurs sont lyophilisées, puis finement broyées à l’aide d’un broyeur ultra-centrifuge ZM 200 Retsch.
2.2 Solvants
Les solvants utilisés en LC-HRMS sont de qualité HPLC-MS, achetés chez Sigma-Aldrich (Saint-Quentin-Fallavier, France). Les solvants utilisés en HPLC–DAD–DEDL et pour l’extraction sont de qualité HPLC et proviennent de VWR (Fontenay-sous-Bois, France). L’eau est purifiée (résistance < 18 MΩ) par un système Elgastat UHQ II (Elga, Antony, France). Pour les extractions MAE et PFE, le mélange standard de solvants est constitué d’éthanol/eau (80:20 ; v/v).
Le CO2 utilisé en SFE est de qualité industrielle 99,5% (Messer, Puteaux, France).
2.3 Extraction standard (sonication)
Un extrait qualifié de standard est réalisé dans un mélange éthanol/eau (80:20 ; v/v), avec un ratio poudre de plante/solvant de 1 g pour 25 mL. Le mélange est placé 15 min dans un bain à ultra-sons, puis le marc est ré-extrait deux fois de la même manière. Les trois extraits successifs sont rassemblés, puis séchés à l’évaporateur rotatif, sous vide, avec une température de bain-marie de 40 °C.
2.4 Appareillage
2.4.1 HPLC
La colonne utilisée est une Kinetex 2,6 μm C18 100 Å 100 × 3,00 mm (Phenomenex, Le Pecq, France).
Les solvants utilisés sont (A) l’eau, (B) l’acétonitrile, tous deux additionnés de 0,05 % d’acide formique. Le gradient est le suivant : à 0 min, 5% de B, à 5 min, 10% de B, à 10 min, 20% de B, à 15 min, 40% de B, puis 100% de B de 25 à 38 min.
La déréplication est réalisée par LC–DAD–ESI–QTOF (Liquid Chromatography – Diode Array Detector – Electrospray Ionization – Quadrupole Time-Of-Flight) Agilent 1200 équipée d’une pompe quaternaire. La détection par spectrométrie de masse est effectuée en ESI––QTOF piloté par le logiciel Mass Hunter. La pression du gaz de nébulisation (azote) est de 40 psi. L’azote est également utilisé comme gaz de collision. L’électronébuliseur (electrospray) est réglé à –3500 V, en mode négatif. L’énergie de collision évolue en fonction du rapport m/z. La fonction selon laquelle elle évolue est déterminée par l’équation suivante : (slope)×(m/z)/100 + offset (avec slope = 3 et offset = 5 lorsque z = 1). Les spectres sont enregistrés entre 115 et 2000 m/z. Le BPC (Base Peak Chromatogram) est utilisé pour le traitement des données.
Les composés phénoliques sont caractérisés en fonction de leur spectre UV et de leur spectre de masse, en comparaison avec la littérature.
Le suivi analytique des extractions est réalisé sur un chromatographe HPLC–DAD–DEDL. La chaine HPLC est une LaChrom Elite (VWR) équipée d’une pompe quaternaire. Le détecteur évaporatif à diffusion de la lumière (DEDL) est un SEDEX 85 (SEDERE, Orléans, France), réglé à une température de 50 °C, à une pression de 3 bar et à un gain de 7. Le pilotage se fait grâce au logiciel EZChrom Elite. Pour le DAD, les hauteurs de pics sont exprimées en mAU et pour le DEDL en mV.
2.4.2 MAE
Le four micro-ondes utilisé est un modèle MycroSYNTH (Milestone, Shelton, USA) piloté par une console « easyCONTROL ». Deux cents milligrammes de poudre de plante sont placés dans un réacteur en verre de 12 mL contenant 5 mL du mélange éthanol/eau (80:20 ; v/v). Un barreau aimanté est placé dans le réacteur pour assurer une bonne homogénéisation poudre–solvant. La température maximale de consigne autorisée pour cet appareil est de 180 °C.
Plusieurs paramètres sont étudiés : la durée d’extraction (15, 30, 45, 60 et 120 s), la puissance (200, 500 et 1000 W) et le nombre de cycles (1, 2 ou 3). Dans le cas où plusieurs cycles d’extraction sont réalisés, le réacteur est placé dans un bac de glace entre chaque cycle pour un retour à température ambiante, ce qui permet d’éviter de dépasser les 180 °C (température de consigne du fabriquant). Les extraits sont ensuite centrifugés et le surnageant récupéré, puis évaporé à sec sous flux d’azote.
Les conditions initiales utilisées lors de l’optimisation des différents paramètres sont les suivantes : trois cycles de 30 s à 1000 W sans renouvellement de solvant à chaque cycle, tous les paramètres étant par la suite optimisés indépendamment.
2.4.3 PFE
L’appareil utilisé est un ASE 150 Dionex (Thermo-Fisher, Les Ulis, France) équipé d’une cellule d’extraction de 5 mL. Celle-ci est remplie avec 200 mg de poudre de plante préalablement mélangée avec de la terre de diatomée. Le solvant d’extraction utilisé est un mélange éthanol/eau (80:20 ; v/v). La pression, qui est fixe, est de 100 bar.
Plusieurs paramètres sont étudiés: la durée de contact (5, 10, 20, 30 min), la température (40, 65 et 100°C), le volume de rinçage (30, 60 et 100%, exprimé par rapport au volume total de la cellule) et le nombre de cycles (1 ou 3). Les extraits sont évaporés à sec sous flux d’azote.
Les conditions initiales utilisées lors de l’optimisation des différents paramètres sont les suivantes : 3 cycles de 10 min à 40 °C avec un volume de rinçage de 60%, tous les paramètres étant par la suite optimisés indépendamment.
2.4.4 SFE
L’extracteur supercritique est un MV-10 ASFE (Waters, Guyancourt, France). Cet extracteur est équipé de 10 cellules d’extraction de 5 mL et de 12 flacons de collecte. La cellule est remplie de la façon suivante : coton, sable de Fontainebleau, 500 mg de poudre de plante, sable de Fontainebleau et coton. Le CO2 est à débit constant (5 mL/min) et la pression de sortie est maintenue à 150 bar. La température peut être modifiée (30 ou 60°C dans cette étude), ainsi que la durée d’extraction. Un co-solvant dont la nature peut varier (éthanol, éthanol/eau 80:20, v/v) peut être ajouté au CO2,et ce dans différentes proportions (5, 10 et 15 %).
3 Résultats et discussions
3.1 Déréplication
La déréplication permet de caractériser des molécules déjà connues sans procéder à un fractionnement pour les isoler au préalable. Cette démarche réduit considérablement le temps nécessaire ainsi que le coût, puisque l’on évite la consommation des solvants due au fractionnement [23]. Néanmoins, cette approche a aussi ses limites, car elle ne permet pas d’identifier précisément certaines molécules, notamment les isomères, qui présentent souvent des spectres UV analogues et qui sont isobares (même masse pour l’ion moléculaire).
Un extrait hydro-alcoolique standard est réalisé pour extraire au maximum les acides phénoliques [24]. Ensuite, cet extrait est analysé en LC–DAD–ESI–QTOF. Les composés d’intérêt sont caractérisés sur la base de leur spectre UV et de leur spectre de masse (ion de molécule déprotonée, fragments et formule brute générée) en comparaison avec les données de la bibliographie [25,26] et avec celles répertoriées dans le dictionnaire des produits naturels [27]. La stratégie de déréplication mise en place a permis de caractériser les acides phénoliques majoritaires de la plante choisie comme matrice pour cette étude. Il s’agit des acides dicaféoylquiniques avec [M–H–] = 515,120 m/z et des fragments de 191 et 179 m/z, qui correspondent respectivement aux acides quinique et caféique. L’acide caféique et ses dérivés présentent un spectre UV caractéristique avec des maxima d’absorption à 218 et 325 nm, avec un épaulement à 241 nm (±2 nm) [26]. Les diCQA sont les molécules phénoliques d’intérêt dans cette étude et serviront de traceurs lors du suivi analytique des extractions. La présence de leurs précurseurs, les acides caféique et caféoylquinique, a également été démontrée (Tableau 1). Leurs structures sont présentées sur la Fig. 1.
Caractérisation par HPLC-DAD-ESI-QTOF des acides phénoliques d'un extrait standard de l'Asteraceae halophile étudiée
| Temps de rétention (min) | [M-H]- (m/z) | Fragments (m/z) | Λmax (nm) | Composé caractérisé |
| 4,4 | 353,087 | 191 [acide quinique – H]- 179 [acide caféique – H]- | 217 – 240 – 325 | Acide caféoylquinique (CQA) |
| 4,6 | 179,034 | 135 | 218 – 241 – 325 | Acide caféique |
| 10,6 10,9 10,9 | 515,120 | 353 [M-H-acide caféique]- 191 [acide quinique – H]- 179 [acide caféique – H]- | 220 – 239 – 326 | Acides dicaféoylquiniques(DiCQA) |

Structures des acides quinique, caféique, caféoylquinique et dicaféoylquinique.
3.2 MAE
Avec le solvant éthanol/eau 80/20 (v/v), plusieurs temps d’extraction ont été testés entre 15 et 120 s. La durée n’influence pas le rendement global d’extraction, calculé en faisant le rapport de la masse de l’extrait sec sur la masse de broyat de plante, il est toujours d’environ 18 %. Par ailleurs, il n’influence que très peu l’extraction du diCQA, qui est le composé majoritaire. Toutefois, lorsque la durée d’extraction augmente, le pic majoritaire se dédouble (diminution de son aire et augmentation de l’aire du pic voisin). De plus, les températures atteintes augmentent avec la durée (à 1000 W : 87 °C pour 15 s, 122 °C pour 30 s 156 °C pour 45 s) (à 500W : 115 °C pour 45 s, 130 °C pour 60 s, 156 °C pour 120 s), ce qui pourrait expliquer le dédoublement du pic majoritaire en HPLC, probablement dû à une isomérisation. En effet, les esters d’acide cinnamique sont connus pour s’isomériser facilement [24]. Il semble donc préférable de retenir une durée intermédiaire pour limiter la potentielle dégradation/modification des composés, soit 30 s à 1000 W ou 60 s à 500 W.
L’étude de l’influence de la puissance montre que pour 45 s, à 1000 W (versus 200 W), un dédoublement de pic est observé, comme dans le cas de l’augmentation de la durée, ce qui laisse penser à une dégradation/isomérisation. Ce phénomène n’est pas observé à 500 W ; cette puissance sera donc privilégiée lorsque la durée dépasse les 30 s.
L’augmentation du nombre de cycles entraîne un accroissement du rendement d'extraction global et de celui des diCQA. Ainsi, trois cycles favorisent de meilleurs rendements d’extraction. En revanche, le renouvellement du solvant entre chaque cycle s’est avéré inutile, car il ne permet pas d’augmenter l’extraction des diCQA. Ce qui signifie que, dans les conditions testées (200 mg dans 5 mL), le solvant n’est pas saturé par les composés extraits.
Ainsi, les meilleures conditions pour les micro-ondes sont : 3 cycles de 30 s à 1000 W sans renouveler le solvant (Fig. 2 et Tableau 2).

Profils HPLC-DAD (à 210 nm) des extraits d’halophyte obtenus par MAE, ASE et SFE dans les conditions optimales pour chacune des trois techniques. a- MAE, b- SFE, c- ASE.
Comparaison des méthodes d’extraction MAE, PFE et SFE optimisées.
| MAE | PFE | SFE | |
| Etape de traitement échantillon | Refroidissement Filtration Evaporation | Préparation cartouche Évaporation | Préparation cartouche Évaporation |
| Durée extraction | 1,5 min | 60 min | 60 min |
| Température d’extraction | 120 °C | 65 °C | 60 °C |
| Consommation solvant par mg de prise d'essai | 5 mL/200 mg | 8 mL/200 mg | 30 mL (co-solvant) / 500 mg |
| Solvant | EtOH 80% | EtOH 80% | 90% CO2 10% EtOH/H2O 80:20 v/v |
| Rendement d’extraction global par masse de plante sèche | 18% | 20% | 15% |
| Extraction des composés phénoliques (analyses HPLC) | Profils phytochimiques similaires |
Un test supplémentaire a été réalisé avec de l’acétate d’éthyle, mais dans nos conditions expérimentales initiales (3 cycles, 30 s, 1000 W), ce solvant n’a pas extrait les diCQA.
Cette étude a permis de démontrer que le choix d’un solvant adapté est primordial, puisque sa polarité gouverne, non seulement la solubilité des composés, mais aussi la température d’extraction. En revanche, une fois le solvant adéquat sélectionné, les paramètres temps d’extraction et puissance se sont avérés peu influents sur l’extraction des diCQA.
Cependant, l’extraction aurait encore pu être améliorée avec du matériel végétal frais. En effet, l’eau endogène du végétal peut rapidement capter l’énergie des micro-ondes et provoquer l’explosion des parois cellulaires [28]. Dans ce cas, les micro-ondes interagissent directement avec le matériel végétal. Si les molécules d’intérêt sont polaires, comme dans cette étude, l’eau intrinsèque peut alors jouer un rôle de solvant et favoriser leur extraction.
3.3 PFE
Différentes températures ont été testées: 40, 65 et 100 °C. Une augmentation du rendement global d’extraction est observée avec l’augmentation de la température (14, 20 et 24% respectivement). Les profils HPLC–DEDL montrent, quant à eux, que le diCQA est mieux extrait à 65 °C, qu’un doublement de la hauteur du pic du diCQA majoritaire est observé entre 40 et 65 °C. Entre 65 °C et 100 °C, cette hauteur de pic diminue, la différence de masse est alors due à une meilleure extraction des composés qui sortent dans le volume mort en HPLC, donc des composés très polaires au vu de la méthode en phase inverse qui est utilisée. Il pourrait s’agir de métabolites primaires, comme des sucres ou des acides aminés, sans intérêt pour cette étude.
L’impact de la durée d’extraction a également été regardé (5, 10 et 20 min). Il a été démontré par comparaison des profils chromatographiques qu’une extraction pendant 20 min permet de mieux extraire le diCQA majoritaire (12 mV pour 10 min, 16 mV pour 20 min). Néanmoins, dans les conditions initiales, une augmentation de la température (65 °C) est beaucoup plus efficace qu’une augmentation de la durée (20 min), avec des hauteurs de pic de 41 mV et 16 mV, respectivement. La température permet en effet d’augmenter la diffusivité du solvant dans la matrice.
Le volume de rinçage de la cellule a également été testé (30, 60 et 100 %). Peu de différences sont observées entre 60 et 100 % pour l’extraction du diCQA (respectivement 22,5 et 25 mV en hauteur de pic) ; en revanche, la valeur 60 % donne de meilleurs résultats que 30 % (17,5 mV). Ainsi, un volume de rinçage de 60 % est un bon compromis entre efficacité et quantité de solvant utilisée.
Une comparaison est faite entre un seul cycle de 30 min et trois cycles de 10 min. Trois cycles de 10 min permettent une meilleure extraction du diCQA, alors que, qualitativement, aucune différence n’est observée. Ceci est dû au fait que, pour chacun des trois cycles, du solvant frais est injecté dans la cellule au moment du rinçage, ce qui favorise la diffusion des solutés vers le solvant d’extraction.
Ainsi, les meilleures conditions pour l’extraction du diCQA ont été fixées à 3 × 20 min à 65 °C, avec 60 % de rinçage (Fig. 2 et Tableau 2).
Un test a été réalisé avec de l’acétate d’éthyle dans les conditions expérimentales initiales, mais ce solvant n’a pas permis d’extraire les diCQA de la matrice végétale.
Il est important de souligner que, contrairement aux extraits obtenus par la MAE, aucune modification qualitative n’a été observée avec cette technique, notamment pas de dédoublement de pic.
Cette technique se rapproche davantage de la SFE que de la MAE, puisque l’extraction est réalisée sous pression (100 bar), mais dans tous les cas, lors de la PFE les conditions utilisées sont en dessous des conditions permettant au fluide d’atteindre son point supercritique (240°C et 6,14 MPa pour l’éthanol pur ; 373°C et 22,1 MPa pour l’eau pure).
3.4 SFE
La contre-pression, qui permet de définir la densité du fluide à l’intérieur de la cellule d’extraction, donc son pouvoir solvant, a été fixée pour toutes les expériences à 15 MPa. L’influence d’un co-solvant sur l’extraction a été testée à 30 °C. Pour ce faire, l’extraction a été réalisée avec 100% de CO2, 90% de CO2 additionné de 10% d’éthanol et 90% de CO2 additionné de 10% d’un mélange éthanol/eau (80:20 ; v/v). Le choix de l’éthanol et de l’eau comme co-solvants polaires permet de conserver des solvants identiques à ceux utilisés en MAE et PFE. De plus, ce choix réside aussi dans une volonté de faire de la SFE une technique verte et non dangereuse pour le manipulateur, comme pour les deux techniques précédentes. Les résultats obtenus sont présentés sur la Fig. 3. Le CO2 seul n’extrait pas les acides phénoliques. En effet, dans ces conditions, les molécules extraites majoritairement sont beaucoup plus apolaires (leur présence apparaît sur les chromatogrammes après 19 min d’analyse). L’ajout d’éthanol dans le CO2 permet d’augmenter la polarité du fluide d’extraction, mais n’extrait pas les dérivés d’acide caféique. L’ajout de 20% d’eau dans l’éthanol permet d’obtenir un extrait plus enrichi en molécules d’intérêt (vers 13,5 min). Le profil HPLC de ce dernier extrait est très similaire à celui d’un extrait obtenu par sonication ou d’un extrait obtenu par MAE ou PFE ; les diCQA sont donc très bien extraits (Fig. 2). Ainsi, l’ajout d’eau dans le co-solvant change de façon significative la polarité du fluide d’extraction, permettant ainsi d’extraire des métabolites de polarités différentes par rapport au CO2 seul. Le méthanol, qui est souvent décrit comme co-solvant dans la littérature [22,29], ne s’est pas avéré aussi efficace que le mélange éthanol/eau. De plus, les caractéristiques CMR (cancérigène, mutagène et reprotoxique) de ce solvant limite aujourd’hui son utilisation, au sein des laboratoires comme à l’échelle industrielle.

Profils HPLC-DAD (à 280 nm) des extraits d'halophyte obtenus par SFE dans différentes conditions, (a) 100% CO2, (b) 90% CO2 + 10% éthanol, (c) 90% CO2 + 10% éthanol/eau (80:20; v/v).
En parallèle, des essais ont aussi été réalisés à 60 °C. Cette augmentation de la température n’influe pas sur l’empreinte phytochimique de l’extrait obtenu, mais l’extraction du diCQA est légèrement supérieure (510 mV au lieu de 460 mV), sans générer de dédoublement de pic. L’extraction peut donc avoir lieu à 60 °C pour être plus efficace.
Après avoir démontré le pouvoir extractant du co-solvant éthanol/eau (80:20 v/v), l’impact de son ratio par rapport au CO2 a été étudié à raison de 5, 10 ou 20%. Avec 5%, la réponse pour le diCQA atteint 74 mV/min, avec 10% 171 mV/min et 211 mV/min avec 20 % (Fig. 4). L’extraction du diCQA augmente donc avec le pourcentage de co-solvant. Néanmoins, la proportion utilisée peut être discutée pour que la SFE ne soit finalement pas utilisée comme une PFE ; c’est pourquoi des ajouts de co-solvant supérieurs à 20% n’ont pas été testés. Ainsi, pour le reste des tests, le pourcentage a été fixé à 10%.

Influence du pourcentage de co-solvant dans l'extraction SFE des diCQA.
Comme le système MV-10ASFE Waters permet la récupération du solvant d’extraction dans différents flacons de collecte, il a été possible de réaliser une cinétique d’extraction pendant 3 h, avec des durées de collecte par flacon de 30 min. Les résultats obtenus sont présentés sur la Fig. 5. La première heure, on obtient 80% de la masse finale de l’extrait, contre seulement 16% la deuxième heure et 3% la troisième heure.
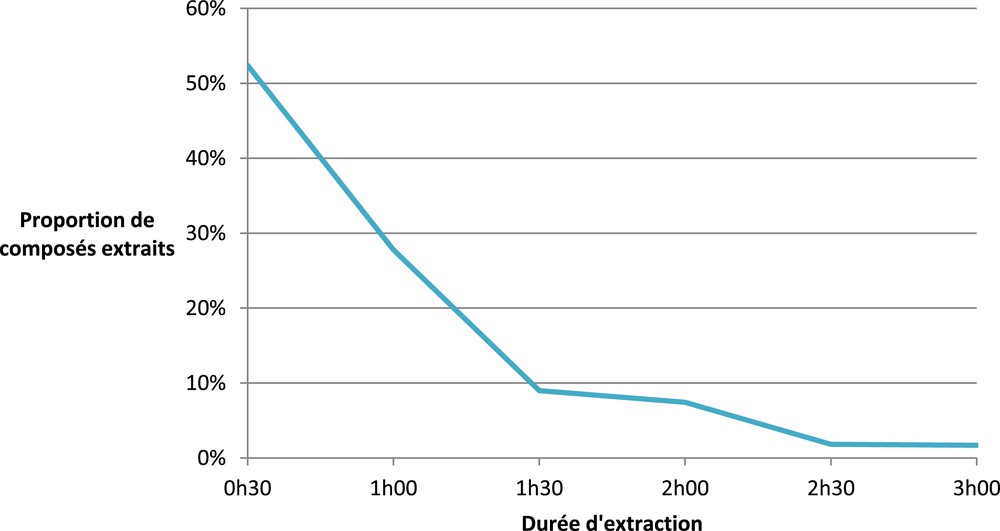
Cinétique d’extraction supercritique avec un co-solvant (CO2 + 10% EtOH/eau, (80:20 ; v/v)).
Les conditions optimales sont donc : 1 h à 60°C avec 10% de co-solvant éthanol/eau (80:20 ; v/v) (Tableau 2).
3.5 Praticité des méthodes d’extraction (Tableau 2)
La MAE est une technique très facile à mettre en œuvre, très rapide et peu consommatrice de solvant. Cependant, pour une évaluation comparative des méthodes d’extraction testées (durée totale, consommation de solvants, étapes intermédiaires), il est important de prendre en compte les temps de refroidissement du mélange entre chaque cycle d’extraction ainsi que le traitement de l’échantillon après extraction, qui comprend une étape de filtration ou de centrifugation, puis une étape de concentration de l’échantillon. La difficulté à récupérer l’extrait dans le réacteur de 12 mL est aussi à souligner, car elle peut engendrer des pertes.
La PFE et la SFE sont des méthodes entièrement automatisées, qui présentent l’avantage de permettre d’obtenir un extrait déjà filtré, qu’il ne reste plus qu’à concentrer. Cependant, pour la PFE et la SFE, une étape préliminaire de remplissage des cellules d’extraction est nécessaire. Dans le cas de la PFE, les cellules sont complétées avec de la célite (terre de diatomée), qui est un CMR ; ainsi, l’étape de préparation est dangereuse pour le manipulateur en plus d’être chronophage. Pour la SFE, c’est du sable de Fontainebleau qui est utilisé, selon les recommandations du fournisseur ; il pourrait aussi être utilisé pour la PFE.
La SFE présente un avantage certain en termes de temps de manipulation. En effet, le retour à l’état gazeux du CO2 supercritique permet de concentrer une grande partie de l’extrait (la fraction CO2). Par exemple, pour une extraction de 1 h 30 à 5 mL/min de solvant d’extraction contenant 10% de co-solvant, le volume final de l’extrait est de seulement 45 mL, d’où une concentration d’un facteur 10 par rapport au volume de fluide utilisé. De plus, c’est la seule méthode d’extraction réellement dynamique, ce qui favorise le transfert de masse des composés en maintenant au maximum le gradient de concentration entre la matrice et le fluide d’extraction.
4 Conclusion
Les différents paramètres testés pour la MAE et la PFE n’influent pas de façon significative sur l’extraction des diCQA. Dans les conditions utilisées, ces deux techniques permettent une extraction facile et rapide de ces molécules d’intérêt, qui sont fortement solubles dans le solvant éthanol/eau (80:20 ; v/v) choisi et présentes en grande quantité dans l’halophyte sélectionnée pour cette étude.
L’ensemble des paramètres testés en SFE se sont révélés plus influents, notamment l’ajout de quelques pourcents de co-solvant hydro-alcoolique, qui permet d’extraire des polyphénols très polaires, comme les acides phénoliques. Comme pour une extraction solide/liquide par MAE ou PFE, la composition du fluide influe sur la polarité et permet d’extraire sélectivement certains composés. De plus, l’utilisation d’eau dans le co-solvant n’est que très peu citée dans la littérature [30] ; cette étude a permis de confirmer son intérêt. Ainsi, la SFE, surtout décrite dans la littérature pour l’extraction de métabolites végétaux apolaires, se révèle aussi être une méthode de choix pour l’extraction des diCQA contenus dans l’halophyte sélectionnée. Enfin, cette technique de chimie verte, qui ne nécessite que peu de solvant, présente l’avantage d’être transposable à l’échelle industrielle ; elle pourra donc être utilisée dans l’industrie pour l’extraction de molécules à haute valeur ajoutée et ce, en respectant l’environnement.