

1 Introduction
In recent years, many research groups have explored molecular structures [1,2] using techniques based on experimental spectroscopy [3] or theoretical calculations, such as those derived from ab initio formalism or density functional theory [4]. Of the well-known approaches, the conformation/configuration of some molecular structures has gained importance due to the influence of chemical interactions [5], particularly those involving hydrogen bonds, which can be either intermolecular (formed between Lewis acids/bases) or intramolecular, where resonant effects are present [6]. In fact, the interaction strength is the benchmark that determines the stabilization of intermolecular systems [7–10], some involving weak or median interaction and others being strongly stabilized with high attraction energies. In this regard, systems with hydrogen bonds are of particular interest, as well as other similar interactions [11–15] involving dihydrogen bonds or halogen hydride bonds, among others [16,17].
As is widely known, the interaction strengths of dihydrogen bonds and hydride bonds vary widely, with values of 10 kJ·mol−1 (typical of weakly bound systems) to 50 kJ·mol−1 (for strongly bound systems) being reported [18–20]. This wide range of interaction strengths is verified by the stretching modes observed on the infrared spectra [21,22], where the depletion of the electronic density along the bonds of the proton donors is restored by charge transfer from the Lewis bases [23–25], which influences the stretch frequencies (e.g., with a redshift or blueshift). Redshift and blueshift are modes related to frequencies shifted downward and upward [26–29], respectively, although it should be noted that a direct relationship with the interaction strength has not been established [30–32]. In dihydrogen-bonded complexes [33,34], however, the appearance of red shifted frequencies has been observed in the NaH⋯HCF3 dimer [35], and this interaction is stronger in comparison to that of complexes with BeH2, MgH2 and LiH hydrides [36]. Optimally, hydrogen complexes have the strongest bonds, but some dihydrogen complexes also have high interaction energies, and the threshold of covalency can be reached or even exceeded. In this study, the formation of NaH⋯n(HF) (n = 2 or 4) complexes was investigated. The aim was to determine whether or not the intermolecular system becomes more stable due to the formation of multiple interactions with higher energies distributed non-additively along the electronic structure. As expected, the formation of stronger dihydrogen bonds between sodium hydride and hydrofluoric acid appears to occur, and also the hydrogen bond strength in the HF⋯HF dimer and HF⋯HF⋯HF trimer increases [37].
In this study, the standard electronic structure method was used, which is based on the second-order Møller-Plesset perturbation theory (MP2) in combination with the 6-311++G(d,p) Pople's split-valence double-zeta basis set with valence, diffuse and polarization functions [38]. Hybrids of the Density Functional Theory (DFT) have been used extensively in studies on intermolecular systems [39], but calculations with the Hamiltonian perturbation at the MP2 level have provided accurate results for weakly bound complexes [40]. In addition to the optimized geometries of deep potential energy surfaces without imaginary frequencies in the infrared spectra, the MP2/6-311++G(d,p) level of theory is employed to investigate the stretch frequencies and absorption intensities. The topologies of the charge density are obtained using Quantum Theory of Atoms in Molecules (QTAIM) [41] and, finally, orbital analysis is carried out to calculate the Natural Bond Orbital (NBO) [42] in order to determine the interaction strengths and their influence on the structures of NaH⋯2(HF) and NaH⋯4(HF) systems. A direct comparison with the interaction profiles and vibration modes of other similar systems, such as the NaH⋯HF dimer, the trimolecular complexes of NaH⋯2(HCF3) and NaH⋯2(HCCl3), and linear and bifurcate dihydrogen complexes formed by BeH2 and MgH2, is also carried out.
2 Computational details
The optimized geometries of NaH⋯2(HF) and NaH⋯4(HF) complexes were determined by means of the MP2/6-311++G(d,p) level of theory, with all calculations carried out using the GAUSSIAN 03W quantum program [43]. With the use of this computational package the ChelpG [44] and NBO [45] parameters as well as the values of the Boys and Bernardi's Basis-Sets Superposition Error (BSSE) [46] were also computed. The QTAIM topological calculations were processed using the AIMAll 11.12.19 suite of codes [47].
3 Results and discussion
3.1 Structures and infrared spectra
The geometries of both NaH⋯2(HF) (I) and NaH⋯4(HF) (II) complexes, optimized at the MP2/6-311++G(d,p) level of theory, are shown in Fig. 1, in which the values of the bond lengths are given. Firstly, by taking into account the length of the dihydrogen bond in the NaH⋯HF dimer, the result of 1.3692 Å obtained at MP2/6-311++G(d,p) is in good agreement with the value of 1.371 Å reported by Grabowski [48] following a study carried out employing the same level of theory. Moreover, 1.3692 Å is shorter than the lengths obtained via higher-level ab initio calculations, e.g., MP4(SDQ)/6-311++G** (1.412 Å) and QCISD/6-311++G** (1.426 Å). As mentioned above, we reproduced the optimized geometry of the NaH⋯HF dimer since its parameters will be used for comparison with the NaH⋯2(HF) trimer and NaH⋯4(HF) pentamer. In the formation of these two multimolecular complexes, the distances of the H⋯H dihydrogen bonds are considerably shortened, with values of 1.2402 (I) and 1.2290 Å (II). Regarding the hydrogen bond distance in the 2HF dimer, surprisingly the value of 1.5167 for I is shorter than 1.5609 Å for II, although both are shorter than the value of 1.8736 Å for the isolated dimer. As a reference, this latter value is in good agreement with the result of 1.8100 Å obtained via CCSD(T)/aug-cc-pVTZ calculations, for which further details have been provided by Ireta et al. [49]. From the structural viewpoint, the formation of multimolecular complexes often results in strengthening of the intermolecular interactions followed by much greater variations in the bond lengths of the proton donors and acceptors [50–52]. In I, the lengths of the Na–H, (H–F)a and (H–F)b bonds vary (0.0555, 0.0892 and 0.037 Å, respectively). Although (H–F)a can behave as a proton donor or acceptor, its bond length undergoes the greatest alteration. In II, however, the profile of the H–F bond differs, because the variation of 0.2264 Å in the Na–H bond is the greatest difference. In both I and II, a halogen-hydride interaction, F⋯Na, is observed, for which the lengths of 2.2980 Å (I) and 2.2851 Å (II) are shorter than the values of 2.4345 and 2.900 Å obtained for the NaH⋯2(HCF3) and NaH⋯2(HCCl3) complexes [35] through the B3LYP/6-311++G(3df,3pd) calculations.

Optimized geometries of the NaH⋯2(HF) (I) and NaH⋯4(HF) (II) complexes obtained at the MP2/6-311++G(d,p) level of theory. Variations are placed in parentheses.
Table 1 shows the values for the vibration modes of complexes I and II. Although it is not common to obtain a good correlation with the variations in the bond lengths, the values for the frequency shifts and absorption intensity ratios are unusual and have not been previously reported for any type of intermolecular system [53–55]. Taking into account the stretch frequency of 4198.6 cm−1 for the hydrofluoric acid in isolated form, even though the Δυ(H–F)b redshifts of −477.6 (I) and −511.7 cm−1 (II) are very large, they are not in agreement with the bond length enhancements of 0.037 and 0.0308 Å, respectively. For complex II, the unexpected redshift of −1270.3 cm−1 reveals the level of deformation of the (H–F)a bond after complexation. By way of comparison, for the NaH⋯HF dimer, the values of the stretch frequency and redshift are 3253.3 and −945 cm−1, respectively. In relation to complex I, considering a variation of 0.0892 Å in the (H–F)a bond, the appearance of a large redshift was expected. However, the absorption intensity ratio of 1670.9 followed by a very large blueshift of +1758.4 cm−1 is anomalous and not in agreement with the bond length variation mentioned above.
Values of the stretch frequencies and absorption intensities of the complexes NaH⋯2(HF) (I) and NaH⋯4(HF) (II) complexes obtained at the MP2/6-311++G(d,p) level of theory.
| Vibration modes | Complexes | |
| I | II | |
| υH⋯H | 1305.5 | 1573.9 |
| IH⋯H | 660.6 | 2844.5 |
| 369.6 | 302.7 | |
| 125.4 | 128.3 | |
| 254.5 | 186.3 | |
| 14.1 | 0.04 | |
| 5957 | 2928.3 | |
| +1758.4 | −1270.3 | |
| 236440 | 895.5 | |
| 1670.9 | 6.4 | |
| 3721 | 3686.9 | |
| −477.6 | −511.7 | |
| 12450 | 3062.8 | |
| 88.1 | 21.6 | |
| 958.9 | 701.9 | |
| −223.5 | -480.5 | |
| 63.8 | 35.6 | |
| 0.20 | 0.1 |
This specific case is not consistent with a direct relationship between the bond length variations and frequency shifts [56], where decreasing and increasing bond lengths correlate with blueshifts and redshifts [57], respectively. On the other hand, the bond lengths of Na–H are increased and their frequencies are shifted to the red region, with values of −223.5 (I) and −480.5 cm−1 (II). Specifically, this is a case where a direct relationship between these redshifts and the respective bond length increases of 0.055 and 0.2264 Ǻ is established. Regarding complexes I and II, although the lengths of the H⋯H dihydrogen bonds are short, their new vibration modes of 1305.5 and 1573.9 cm−1 and absorption intensities of 660.6 and 2844.5 km·mol−1 are anomalous and do not lie within the ranges of 100–200 cm−1 for median strength or 50–100 cm−1 for weakly bound complexes. Nevertheless, the stretch frequencies of the halogen-hydride interactions present values of 254.5 (I) and 186.3 (II) cm−1, in satisfactory agreement with the intermolecular distances of 2.2980 and 2.2851 Å, respectively.
3.2 QTAIM topography
Tables 2 and 3 show the values of the QTAIM properties, namely electronic density (ρ), Laplacian (∇2ρ), kinetic (G) and potential (U) electronic density energies and their sum, referred to as the total electronic energy density (H), ellipticity (ε) and atomic radius (r) [95,96]. In complex I, a significant result is the topological characterization of the H⋯H dihydrogen bond as shared contact, since the Laplacian is negative, with a value of −0.0120 e.ao−5, revealing an interaction of high energy and strength [58,59], for which, optimally, the electronic density is 0.0710 e.ao−3. In the case of the NaH⋯HF dimer, however, the modeling of its dihydrogen bond reveals a closed-shell profile, with a Laplacian value of 0.0556 e.ao−5 and an electronic density of 0.0432 e.ao−3. With regard to the trimer, the value for its intermolecular electronic density (0.0710 e.ao−3) is lower than those of 0.2569 and 0.3112 e.ao−3 for (H–F)a and (H–F)b, respectively. It should be noted that all of these QTAIM values were obtained through the previous localization of the Bond Critical Points (BCPs) (Fig. 2) [60], and the charge densities are greater than those determined via the Ring Critical Points (RCPs) (Fig. 2). This can be observed through a comparison between the electronic densities, with the values of 0.0059 (I) and 0.0050 e.ao−3 (II) (Table 4) being lower than those computed for the bond path of the BCPs. In fact, the contributions of G and U are useful, but the –G/U ratio deserves greater attention since this can reveal the intermolecular covalent character, at least qualitatively. Although for H⋯H, the –G/U ratio of 0.4743 indicates a profile of total covalence, the covalent character of the Fa⋯H hydrogen bond appears to be partial, since the –G/U result is 0.9051, even though its Laplacian value is positive (0.2151 e.ao−5). Regarding the halogen-hydride, Fb⋯Na, this interaction also presents a positive Laplacian value (0.1245 e.ao−5), although the character of the bond is non-covalent, with a –G/U ratio of 1.2460. In terms of the intermolecular distance of the dihydrogen bonds (A) and hydrogen bonds (B) and the halogen-hydride interactions (C), the relationship with the –G/U ratios (Eq. 1) exposes an exponential behavior, with the dihydrogen bonds showing the strongest interactions, as can be seen in Fig. 3:
| (1) |
Values of the QTAIM parameters computed in each BCP of the NaH⋯2(HF) complex.
| Parameters | BCPs of the complex NaH⋯2(HF) (I) | |||||
| Na–H | H⋯H | H–Fa | Fa⋯H | H–Fb | Fb⋯Na | |
| ρ | 0.0281 (−0.0036) | 0.0710 | 0.2569 (−0.1164) | 0.0548 | 0.3112 (−0.0621) | 0.0178 |
| ∇2ρ | 0.1265 (−0.0043) | −0.0120 | −1.6351 (1.4352) | 0.2151 | −2.5818 (0.4885) | 0.1245 |
| G | 0.0293 (−0.0021) | 0.0286 | 0.0889 (0.0120) | 0.0601 | 0.0697 (−0.0072) | 0.0260 |
| U | −0.0270 (0.2730) | −0.0603 | −0.5867 (0.3347) | −0.0664 | −0.7850 (0.1364) | −0.0209 |
| H | 0.0023 (0.0009) | −0.0317 | −0.4978 (0.3467) | −0.0063 | −0.7153 (0.1292) | 0.0051 |
| −G/U | 0.4743 (0.0386) | 0.4743 | 0.1515 (0.0681) | 0.9051 | 0.0887 (0.0053) | 1.2460 |
| ε | 0.0334 (0.0334) | 0.0174 | 0.0000 (0.0000) | 0.0128 | 0.0006 (0.0006) | 0.0262 |
Values of the QTAIM parameters computed in each BCP of the NaH⋯4(HF) complex.
| Parameters | BCPs of the complex NaH⋯4(HF) (II) | |||||
| Na–H | H⋯H | H–Fa | Fa⋯H | H–Fb | Fb⋯Na | |
| ρ | 0.0185 (−0.0132) | 0.0654 | 0.2680 (−0.1053) | 0.0482 | 0.3192 (−0.0541) | 0.0184 |
| ∇2ρ | 0.0839 (−0.0469) | 0.0253 | −1.8012 (1.2691) | 0.1985 | −2.6993 (0.3710) | 0.1299 |
| G | 0.0186 (−0.0128) | 0.0318 | 0.0814 (0.0045) | 0.0532 | 0.0681 (−0.0088) | 0.0271 |
| U | −0.0162 (0.0138) | −0.0573 | −0.6132 (0.3082) | −0.0567 | −0.8111 (0.1103) | −0.0217 |
| H | 0.0024 (0.0010) | −0.0255 | −0.5318 (0.3127) | −0.0053 | −0.7430 (0.1015) | 0.0054 |
| −G/U | 1.1481 (−0.8985) | 0.5550 | 0.1327 (0.0493) | 0.9383 | 0.0840 (0.0006) | 1.2488 |
| ε | 0.0182 (0.0182) | 0.0105 | 0.0002 (0.0002) | 0.0174 | 0.0004 (0.0004) | 0.0407 |
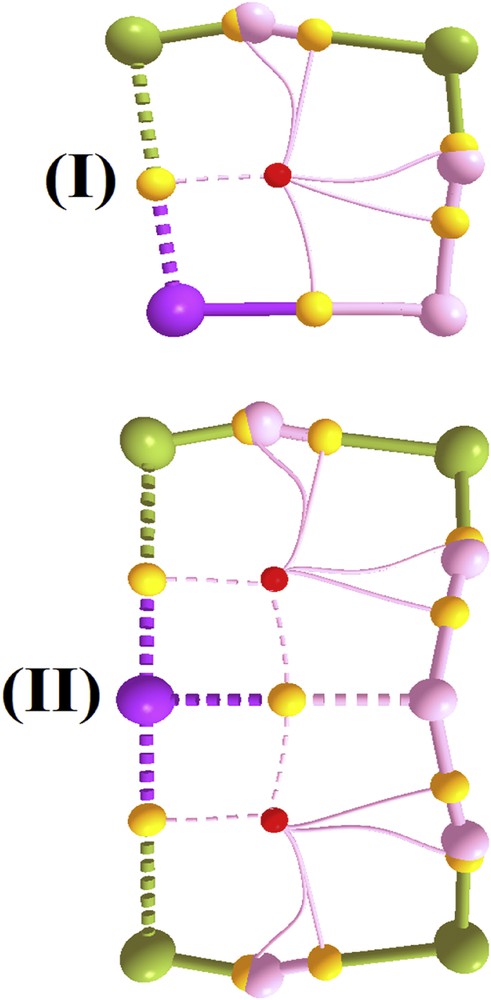
QTAIM topography with bond paths, BCPs (small yellow balls) and RCPs (small red balls) of the NaH⋯2(HF) (I) and NaH⋯4(HF) (II) complexes.
Values of the QTAIM parameters computed in each RCP of the NaH⋯2(HF) (I) and NaH⋯4(HF) (II) complexes
| Parameters | Complexes | |
| I | II | |
| ρ | 0.0059 | 0.0050 |
| ∇2ρ | 0.0312 | 0.0234 |
| G | 0.0063 | 0.0047 |
| U | −0.0049 | −0.0035 |
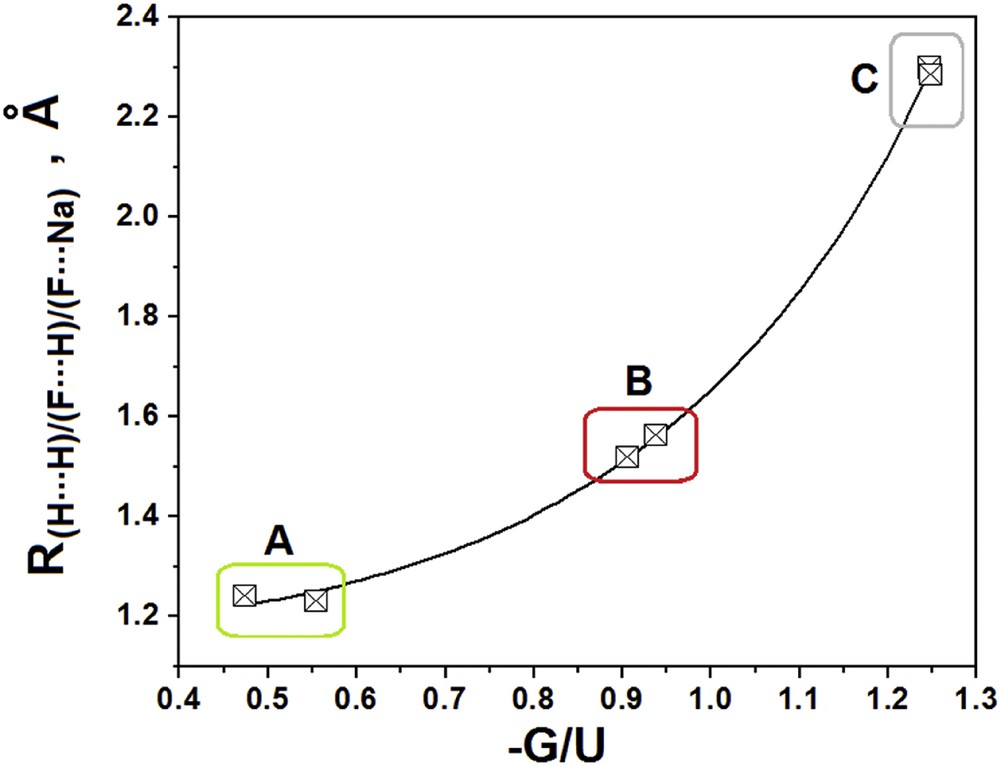
Relationship between the intermolecular distances and QTAIM energy ratios.
Continuing the reasoning detailed above, both the (H–F)a and (H–F)b are recognized as shared interactions because the Laplacian fields are negative, although they present intense charge density reductions, with values greater than those of the Na–H hydride. However, the greatest reduction in the electronic density is observed in the case of the (H–F)a bond, with a value of −0.1164 e.ao−3. Even though these profiles were predicted by means of structural analysis and spectral description, a good relationship with the decrease in the bond length was observed, although this was not consistent with the blueshift in the frequencies.
Continuing with the same reasoning applied to complex I, for II the greatest variation in the charge density is the reduction of −0.1053 e.ao−3 in the (H–F)a bond. Although lower than −0.1164 e.ao−3 (I), this result rationalizes one of the two HF moieties. However, this reduction of −0.1053 is fully in line with the larger redshift of −1270.3 cm−1. For the remaining bonds, that is, Na–H and (H–F)b, although the results of −0.0132 and −0.0541 e.ao−3 show slight charge density variations, they are in agreement with the redshift values of −480.5 and −511.9 cm−1, respectively. In contrast to complex I, in which the covalent character of the Na–H bond is totally due to the –G/U ratio, in II the Na–H bond is purely non-covalent, since the –G/U value is 1.1481. This is not observed in the case of the (H–F)a and (H–F)b bonds, since the values for the –G/U ratios are 0.1327 and 0.0840, respectively. In fact, the dihydrogen bond distances are shorter in II, although the –G/U values of 0.555 (for H⋯H) and 0.9383 (for the longer Fa⋯H hydrogen bond) are higher than those computed for I, but, even so, they also suggest a partial covalent character. The –G/U ratio of 1.2488 indicates that the Fb⋯Na halogen-hydride is non-covalent, and this value is similar to that of 1.260 for I complex. In comparison with the case of halogen-hydride bonds in the NaH⋯2(HCF3) complex [53], although the value of the F⋯Na distance of 2.4345 Ǻ is longer, the –G/U ratio of 1.220 also reveals a non-covalent character. Even though the intermolecular distance of 2.2851 is shorter in comparison with I (2.2980), the electronic density values of 0.0178 e.ao−3 (I) and 0.0184 e.ao−3 (II) corroborate this finding, although a direct relationship with the stretch frequency values could be not established. An exception to the direct relationship between intermolecular density and stretch frequency [8] is the new υH⋯H vibration mode of 1573.9 (II) being higher than 1305.5 cm−1 (I), while the charge densities are 0.0654 and 0.0710 e.ao−3, respectively. Clearly, an important finding in this study is the strengthening of the H⋯H dihydrogen bond in complex I. The conception of ε involves a relationship between the eigenvalues of the Hessian matrix for the Laplacian [61], where any chemical bond with the delocalization of electrons, either σ or π, can be modeled with regard to the charge flux along the bond path [62,63]. For the monomers, the value of ε for the Na–H and H–F bonds is zero. For the complexes, however, the values of ε are 0.0334 (I) and 0.0182 (II) for the Na–H bond. Regarding the (H–F)a and (H–F)b bonds, their ε values of 0.0000 (I) and 0.0006 (I) and of 0.0002 (II) and 0.0004 (II), respectively, display an unchanged behavior of electronic movement [102]. In relation to the intermolecular interactions, the hydride bonds have the highest ε values (0.0262 (I) and 0.0407 (II)). With regard to the partial and total covalent interactions, for the H⋯H dihydrogen bonds and Fb⋯Na halogen-hydride bonds the ε values vary considerably, with no systematic tendency related to the interaction strength being observed.
3.3 Interaction energy, balance of charge transfer and NBO analysis
Table 5 shows the total interaction energies with corrected (BSSE and ZPE) and uncorrected values [64,65]. Optimally, the values of −94.20 (I) and −218.05 kJ·mol−1 (II) do not reflect the stabilization profile. Thus, each one of the interactions could contribute with −23.73 (I) and −36.34 (II) kJ·mol−1, values which do not accurately represent the energetic cooperativity [66,67]. Also, cooperativity, also known as ‘non-additivity’, is a parameter that indicates uniformity of the electronic distribution within the molecular structure, wherein the energy quantification can be carried out by means of certain algebraic equations, e.g., the Site–Site Function Counterpoise (SSFC) [68]:
| (2) |
Values of the interaction energies of the NaH⋯2(HF) (I) and NaH⋯4(HF) (II) complexes obtained at the MP2/6-311++G(d,p) level of theory.
| Energies | Complexes | |
| I | II | |
| ΔE | −162.16 | −299.50 |
| ΔZPE | 47.28 | 44.14 |
| BSSE | 18.96 | 37.31 |
| ΔEC | −94.92 | −218.05 |
| −363.0229751 | −563.6353435 | |
| ΔEint | −166.91 | −310.20 |
The refinement of the interaction energy using the counterpoise correction of Eq. (2) is determined by Eq. (3). This equation is useful for trimers (complex I) and can be adapted to pentamers (complex II) or larger oligomers. The values of and also are listed in Table 5. The values of −166.91 (I) and −310.20 kJ·mol−1 (II) indicate greater stability than the corresponding results of −94.20 (I) and −218.05 kJ·mol−1 (II), determined by means of the supermolecule approach. It should be noted that the values of −166.91 and −310.20 kJ·mol−1 do not represent the interaction strength but rather cooperative energies distributed along the H⋯H, Fa⋯H and Fb⋯Na interactions.
| (3) |
The charge balance or charge transfer should be discussed based on the variations in the atomic charge [96], for which the values obtained from ChelpG and NBO calculations are shown in Fig. 4. It can be observed that the fluorine and hydride atoms assume negative characters whereas the hydrogen and sodium atoms are positive. On taking into account the hydrofluoric dimer, the ChelpG values of −0.042 (I) and −0.019 e.u. (II) for the hydrogen in the (H–F)b bond confirm the charge transfer from the fluorine of the (H–F)a bond. Unfortunately, the NBO results fail in this respect when applied to II. In the hydride, the values for the balance of charge of 0.171 e.u. (I) and 0.072 e.u. (II) obtained from ChelpG calculations, together with the NBO results of 0.129 e.u. (I) and 0.173 e.u. (II), are associated with a charge transfer from NaH toward the (H–F)a bond of the hydrofluoric acid dimer. Regarding the Fb⋯Na hydride bond, surprisingly the negative balances of charge observed for the fluorine of the (H–F)a bond reveal electron reception by the sodium. On the other hand, sodium presents a depletion of charges, with ChelpG values of 0.107 e.u. (I) and 0.129 e.u. (II) and the values obtained from the NBO-based analysis of 0.108 e.u. (I) and 0.136 e.u. (II) diverge from the charge gain for fluorine.

Values of the ChelpG and NBO atomic charges in the NaH⋯2(HF) (I) and NaH⋯4(HF) (II) complexes. The values of the charge transfers are indicated in parentheses.
According to the ChelpG and NBO protocols, electronic transfer was not observed for the Fb⋯Na halogen-hydride bond. However, although the NBO results did not demonstrate intermolecular charge transfer, this method did provide important information regarding the electronic structure. The contribution of each s and p orbital to the formation of chemical bonding is one of the significant benchmarks in this regard. In other words, the most useful results are obtained from an analysis of the s and p hybridizations for the Na–H, (H–F)a and (H–F)b bonds, although values are only available for the latter two cases (Table 6). In the case of complex II, the greatest variations in the s character are the considerable increase of 10.51% and the smaller increase of 5.81% for the fluorine in the (H–F) and (H–F)b bonds, respectively. In line with this, the p contributions reduced in a similar manner and the slight variations in the s and p orbitals of the hydrogen atoms are not accounted for. Since these greater s orbital variations would result in a reduction in the polarity of the chemical bond, this also leads to weakening of the chemical bond, in agreement with the redshift values of −1270.3 and −511.7 cm−1. The same behavior was observed for the (H–F)b bond of complex I, in which the Δ%s and Δ%p contributions of the fluorine atom present values of 6.54 and −6.19%, respectively. As noted above, the formation of the H–H bond could be caused by the strong H⋯H dihydrogen bond and the QTAIM modeled this as a totally covalent shared interaction. In the infrared analysis, however, the harmonic oscillators of the H–H bond were not detected. On the other hand, the stretch mode and absorption intensity of the H⋯H dihydrogen bond were clearly observed. Nevertheless, instead of an H⋯H dihydrogen bond the NBO results indicate an H–H bond, whose values (Table 6) were comparable with those of the H2 of the monomer. In fact, the values show slight variations, ranging between −0.02 and 0.15% and, as shown herein, they are not comparable with the results obtained for the (H–F)a and (H–F)b bonds. These values suggest the formation of an H2 molecule as a new entity in complex I.
Values of the percentages of the s and p orbitals for the covalent bonds of the NaH⋯2(HF) (I) and NaH⋯4(HF) (II) complexes.
| Hybridizations | Bonds of the complex NaH⋯2(HF) (I) | ||
| H–Fa | H–Fb | H–H | |
| %sA | — | 99.54 (−0.21) | 99.70 (−0.15) |
| %pA | — | 0.46 (0.21) | 0.30 (0.15) |
| %sB | — | 27.55 (6.54) | 99.87 (0.02) |
| %pB | — | 72.35 (−6.19) | 0.13 (−0.02) |
| Hybridizations | Bonds of the complex NaH⋯4(HF) (II) | ||
| H–Fa | H–Fb | H–H | |
| %sA | 99.48 (−0.27) | 99.57 (−0.18) | — |
| %pA | 0.52 (0.27) | 0.43 (0.18) | — |
| %sB | 31.85 (10.51) | 27.12 (5.81) | — |
| %pB | 68.07 (−10.47) | 72.78 (−5.76) | — |
The QTAIM atomic radius has been used as an important tool in studies on chemical bonds [12,13,28,31], in particular, those typical of proton donors. The locations of the BCPs can be used to determine the distance between the atomic coordinates, the measured value for which is known as the QTAIM atomic radius. Table 7 shows the variations in the values for the atomic radius for the sodium, hydride, fluorine and hydrogen atoms. In the case of complex I, the results of 0.0443 (Na–H), 0.0731 (H–F)a and 0.0428 Å (H–F)b indicate that the atomic radii of hydride and fluorine atoms show the greatest increases. Although a larger variation in the atomic radius could be related to the highly weighted orbital, in this case, regardless of whether the fluorine has an s or p character, the reduction of p within the hybrid orbital of the H–F bonds leads to an enlargement of the fluorine atom and to chemical bonding with a higher polarity. In contrast, the smallest variations in the hydrogen atoms are in direct correlation with the slight changes in the %s contributions (A), whereas the median (B) and the largest (C) modifications are related to the fluorine in the (H–F)b and (H–F)a bonds, respectively. This yields a linear relationship, described by Eq. (4) and graphically illustrated in Fig. 5.
| (4) |
Values of the QTAIM atomic radii.
| QTAIM atomic radii | Bonds of the complex NaH⋯2(HF) (I) | ||
| Na–H | H–Fa | H–Fb | |
| rA | 1.0133 (0.0119) | 0.1570 (0.0161) | 0.1350 (−0.0059) |
| rB | 0.9513 (0.0443) | 0.8488 (0.0731) | 0.8185 (0.0428) |
| QTAIM atomic radii | Bonds of the complex NaH⋯4(HF) (II) | ||
| Na–H | H–Fa | H–Fb | |
| rA | 1.0718 (0.0704) | 0.1540 (0.0131) | 0.1342 (−0.0067) |
| rB | 1.0631 (0.1561) | 0.8445 (0.0688) | 0.8132 (0.0375) |
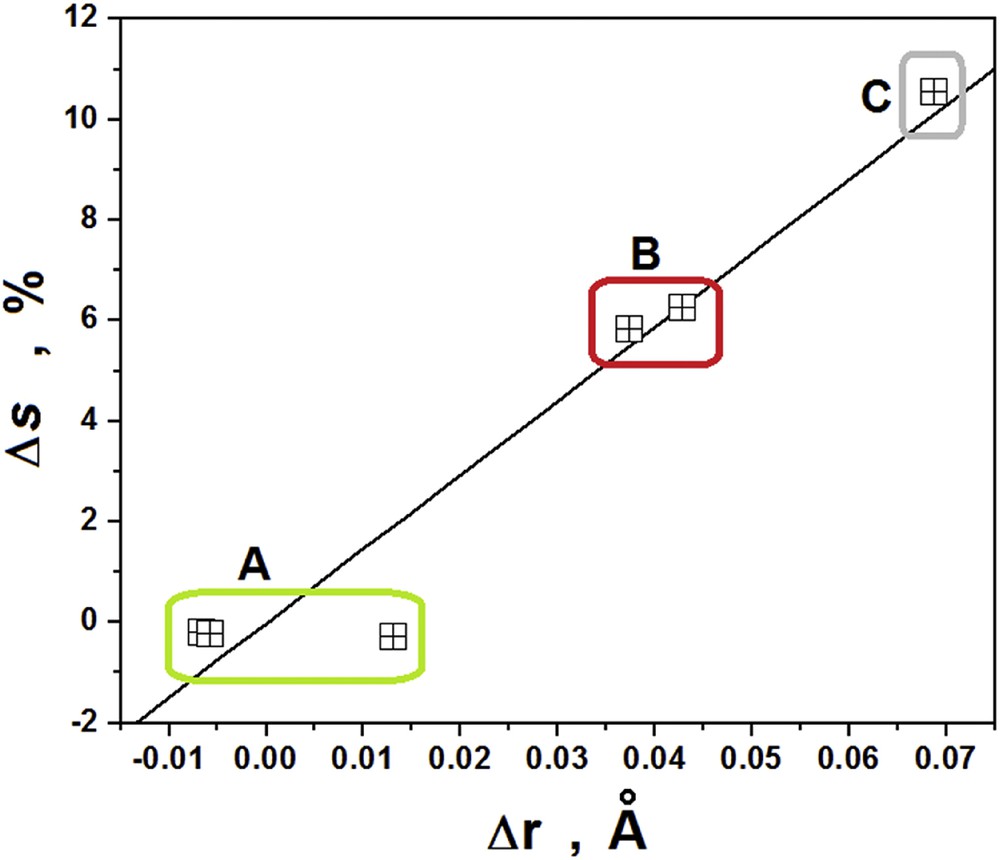
Relationship between the variations in the s orbital (hydrogen and fluorine) and QTAIM atomic radii.
In some studies, the variations in the s and p orbitals considered in combination with the QTAIM radii aid an understanding of the main changes in the electronic structure and also help to explain the frequency shifts of the proton donors. The large redshift in the (H–F)b bond and the anomalous blueshift in the (H–F)a oscillator, together with the formation of the H2 subunit, in the case of complex I, inhibit the interpretation of vibration modes based on the QTAIM atomic radii.
4 Conclusions
The theoretical study of the molecular and intermolecular parameters of the NaH⋯2(HF) and NaH⋯4(HF) complexes at the MP2/6-311++G(d,p) level of theory revealed systematic tendencies and benchmarks with profiles not previously observed. The structural analysis and the examination of the infrared modes show that the dihydrogen bond distances are shorter than those of the hydrogen bonds and halogen-hydride bonds. In the case of hydrofluoric acid bound to sodium and fluorine, the computed redshifts are unusual because the stretch frequencies relocate beyond −500 cm−1 in the infrared spectrum. Besides these redshifts, the hydrofluoric acid bond with hydride shows a surprising blueshift of 1758.4 (I) and redshift of −1270.3 cm−1 (II). This is not consistent with the shortening of the bond length. The interaction energies determined by the SSFC approach are cooperative, particularly for the pentamer NaH⋯4(HF). The QTAIM descriptors calculated for the dihydrogen bonds, hydrogen bonds and halogen-hydride bonds revealed a strongly bound system profile with high electronic density, negative Laplacians as well as the appearance of a covalent character, since the potential electronic density energies surpass the kinetic energy. Although not identified in the vibration analysis or characterized by the QTAIM descriptors, the NBO computations of the s and p orbitals indicate the formation of a new entity, the H2 molecule. This suggests the Na⋯H–H⋯F⋯H–F configuration for complex I, since the F⋯Na halogen-hydride interaction was taken into account in this regard. Finally, the variations in the percentage in the s hybrid orbital are in agreement with the increase in the atomic radii of fluorine and hydrogen.
Acknowledgments
CAPES, CNPq and FAPESB Brazilian Funding Agencies.