

1 Introduction
There are multidisciplinary approaches nowadays focused on N-substituted amino-s-triazines with hydroxy-phenyl units [1–4]. Actually, the use of p-aminophenol in the classic SN2-Ar amination of cyanuric chloride, yielding m-trivalent branched melamine (2,4,6-triamino-1,3,5-triazine)-polycarbonates, has been known since 1987 [1a]. Later on, the patented applications of these macromolecules as new plasticizers promoted subsequent findings in the domain of organic materials, for example thermoplastic compounds [1b] and ingredients for lubricants or lubricant compositions [1c,1d]. More recently, the synthesis and structural properties of arborescent architectures built around trimesic(1,3,5-tricarbonylbenzene) [2a] or s-triazine [2b] cores by means of p-aminophenol units were also described. The first synthesis of a (G-1) dendritic melamine comprising p-aminophenol as an internal linker and 2-aminopyridine as a peripheral unit, reported by Gamez and co-workers in 2002, targeted a novel polydentate and polynucleating N-donor ligand [3]. On the other hand, it is also worth mentioning the bioimpact of some N-substituted melamines with (O-protected)hydroxyphenyl motifs conjugating 4-aminoquinoline as antimalarial agents [4].
In the above multi-facet context, we consider of interest to extend our expertise in the field of dendritic melamines' preparation and structure [5] (including electrochemical approach [5e]) by using a versatile-redox and bidentate nucleophile, p-aminophenol. The last one was designed to play the role of a peripheral unit, in an iterative-convergent type synthesis, a strategy previously developed by Simanek's group [6]. Next, cyclic voltammetry has been employed to study the electrochemical behaviour of the first (G-2) dendrimer encompassing p-aminophenol as the peripheral unit and of its precursors on the Pt electrode in DMSO.
To our knowledge, no similar investigation has been reported so far.
Indeed, dendrimers containing redox active functionalities are especially important because of their interesting electron-transfer properties [7]. Thus, the peripheral redox active units, undergoing multiple electron transfer and redox properties, can be modulated by the size and nature of the dendritic branches [8]. Aminophenols are well-documented electrochemically active compounds since they have two groups (–NH2 and –OH), which could be oxidized [9]. Moreover, the electropolymerization of p-aminophenol itself on a Pt electrode in organic solvents, providing a soluble electroactive polymer, was also reported [9a].
2 Results and discussion
2.1 Synthesis
The chemistry we followed is resumed in Scheme 1 and the reaction conditions are presented in Table 1.

Reaction conditions and results in the synthesis of compounds 1–7 (see Scheme 1).
| Entry | Reaction | Conditions | Products, yields (%) and isolation: direct crystallisation (d.c.), column chromatography on silica gel (c.c.)a |
| 1 | p-AP → 1a | i) 0.30 equiv C3N3Cl3, butanone/0–5 °C (30 min); ii) 1.00 equiv AcONa, H2O/5–9 °C (30 min); iii) reflux (3 h) | 1a: 91 (d.c.) |
| 2 | p-AP → 1b | i) 0.50 equiv C3N3Cl3, butanone/0–5 °C (30 min); ii) 1.00 equiv AcONa, H2O/5–9 °C (30 min); iii) r.t. (24 h) | 1b: 56 (c.c.) |
| 3 | p-AP → 1b | i) 0.50 equiv C3N3Cl3, acetone/0–5 °C (2 h); ii) 1.00 equiv NaHCO3, H2O/5–9 °C; iii) 45 °C (3 h); r.t. (15 h) | 1b: 73 (d.c.)b |
| 4 | 1b → 2 | 2.23 equiv Ac2O, 1.00 equiv K2CO3, THF/r.t. (4 h); reflux (10 h) | 2: 90 (d.c.) |
| 5 | 1b → 3a | 4.00 equiv piperazine, 1.00 equiv K2CO3, THF/r.t. (15–20 h) | 3a: 83 (c.c.); 85 (d.c.) |
| 6 | 1b → 3a, 4a | 1.00 equiv piperazine, 1.00 equiv K2CO3, 1,4-dioxane/reflux (8 h) | 3a: 17 (c.c.); 4a 83 (c.c.)c |
| 7 | 1b → 3b | 4.00 equiv 4,4′-bipiperidine, 1.00 equiv K2CO3, THF/r.t. (36–42 h) | 3b: 45 (c.c.); 86 (d.c.) |
| 8 | 3b → 4b | 1.00 equiv 1b, 1.00 equiv K2CO3, 1,4-dioxane/reflux (13 h) | 4b: 64 (d.c.) |
| 9 | 3a → 5a | i) 1.00 equiv 3a, 1.00 equiv K2CO3, THF/−15 °C (20–24 h); ii) 1.00 equiv 3a, 1.00 equiv K2CO3, THF/r.t. (20–24 h); iii) 1,4-dioxane, reflux (36 h) | 5a: 90 (d.c.) |
| 10 | 3b → 5b | i) 1.00 equiv 3b, 1.00 equiv K2CO3, THF/−15 °C (22 h); ii) 1.00 equiv 3b, 1.00 equiv K2CO3, THF/r.t. (24 h); iii) 1,4-dioxane, reflux (36 h) | 5b: 47 (c.c.); 86 (d.c.) |
| 11 | 5a → 6 | i) 4.00 equiv piperazine, 1.00 equiv K2CO3, THF/r.t. (20–30 h); ii) reflux (12 h) | 6: 55 (c.c.); 88 (d.c.) |
| 12 | 6 → 7 | i) 0.30 equiv C3N3Cl3, 1.00 equiv K2CO3, 1.4-dioxane/r.t. (24 h); ii) reflux (48 h) | 7: 85 (d.c.) |
a On partially deactivated silica gel (eluent EtOH: aq. NH3 25%) for compounds 3a, 4a, 3b and 6.
b 67% Yield according to Refs. [4b] and [4c].
c Partial conversions of 1b into the depicted compounds, calculated based on effective amounts isolated by column chromatography. Full details of these syntheses are given in the SI (Supporting information).
First, we tested the chemoselectivity in the amination of cyanuric chloride by p-aminophenol (p-AP) in the synthesis of melamine 1a (Table 1, entry 1) versus that of chlorodiamino-s-triazine 1b (Table 1, entries 2 and 3). Although the preparation of 1a was of applied interest for several authors [1a–d,3], in our hands only the procedure of Negoro and Kawata [1c,1d] gave satisfaction as excellent and reproducible yield, 91% [10]. Under similar but milder conditions (Table 1, entry 2), amination of cyanuric chloride performed with two molar equiv. of p-aminophenol yielded compound 1b contaminated with traces of melamine 1a. Hence, 1b required purification by column chromatography when an important loss of the material due to its relative retention on silica gel was observed, affecting the yield. Our option of choice was then the method of Bhat and co-workers [4b,4c] (Table 1, entry 3) which produced 1b with good yield, 73%, in an expeditious manner.1 Compound 1b exhibited moderate solubility in organic solvents usually recommended for the synthesis of amino-s-triazines (THF, 1,4-dioxane, acetone etc.) [6], most likely because of its high polarity. That is, in order to adopt a protective–deprotective strategy, 1b was diacetylated (Table 1, entry 4) in high yield (90%) with complete O,O′-chemoselectivity. Unfortunately, the resulting O,O′-diacetyl derivative 2 was inappropriate for our envisaged iterative syntheses (Scheme 1). Thus, the reaction between 2 and piperazine (not depicted in Scheme 1), besides the expected amination of the s-triazine chlorine, consisted as well of the partial transfer of the acetyl group (O-phenol → N-piperazine) affording a multicomponent reaction mixture. By contrast, compound 2 was of crucial relevance in electrochemical investigations (Section 3.1.) To conclude, for the present report, we had to manage our strategy limited to (G-0) chlorodendron 1b.
Based on our earlier published methodology [5a–d], the chemoselective mono-attachment of piperazine, as the first linker, to 1b (Table 1, entry 5) gave the (G-0) melamine 3a. Thus, during the portionwise addition of 1b to a 300% molar excess of piperazine, the TLC monitoring of the amination revealed a very clean evolution towards 3a only, i.e. the complete absence of the dimeric melamine 4a as a side product (Scheme 1). As shown in Table 1 (entry 5), it was the single situation, that of 3a, in which both techniques for its purification (direct crystallisation or column chromatography on partially deactivated silica gel) provided good and comparable quantitative results. Our attempt to prepare compound 3a by applying the method of Bath and co-workers [4c] (equimolar ratio 1b: piperazine, 82% claimed yield of 3a) failed (Table 1, entry 6). The resulting mixture 3a (minor) and 4a (major) could be successfully separated by column chromatography.
The protocol for the non-symmetric anchorage of the 4,4′-bipiperidine linker to 1b (Table 1, entry 7) producing melamine 3b was the same as in the case of piperazine against 1b but the amination reached completion in a much longer time. The yield of 3b was critically influenced by the work-up used for its isolation as a pure analytical sample, 86% (direct crystallisation from boiling ethanol) versus 45% (column chromatography on a partially deactivated silica gel). Our tentative explanation relates, once again, to the high relative retention on column chromatography of 3b (see also the next examples).
The symmetric analogue 4b of 3b was also prepared from the latter in reaction with 1b (Table 1, entry 8).
As expected, the one-pot synthesis of (G-1) chlorodendrons 5a and 5b (Table 1, entries 9 and 10 respectively) required mild conditions for the first step amination. However, the second step was mandatory to taxing reaction parameters (36 h in refluxing 1,4-dioxane), suggesting the low reactivity of the intermediates due to their strong solvation in the depicted solvents. Except for DMF and DMSO, both compounds 5a and 5b manifested low solubility in common organic volatile solvents. Therefore, in order to avoid the loss of the material on column chromatography (i.e. compound 5b, 47% yield), purifications were successfully realised by crystallisations from boiling ethanol (86% optimised yield of 5b). Even so, the (G-1) chlorodendron 5b showed an unexpected weak reactivity in subsequent aminations attempted with piperazine or 4,4′-bipiperidine. Therefore, for the present work, we had to limit the study by using henceforth (G-1) chlorodendron 5a only.
In so doing, we effected, in accordance with the same methodology as that in the case of (G-0) melamine 3a, the selective anchorage of the next piperazine linker on 5a (Table 1, entry 11) and obtained (G-1) melamine 6. We detected no dimeric melamine as a side product and isolated 6 by column chromatography on a partially deactivated silica gel or, as an optimal decision, by crystallisation (88% yield).
The final installation of the trivalent s-triazine core on 6 (Table 1, entry 12) was performed in a one-pot procedure with good global yield (85%). Compound 7, the first (G-2) dendrimer comprising p-aminophenol as the peripheral unit, could not be eluted on TLC due to its low solubility in common organic solvents. Its identity was fully confirmed by spectral methods.
2.2 Preliminary structural assignments
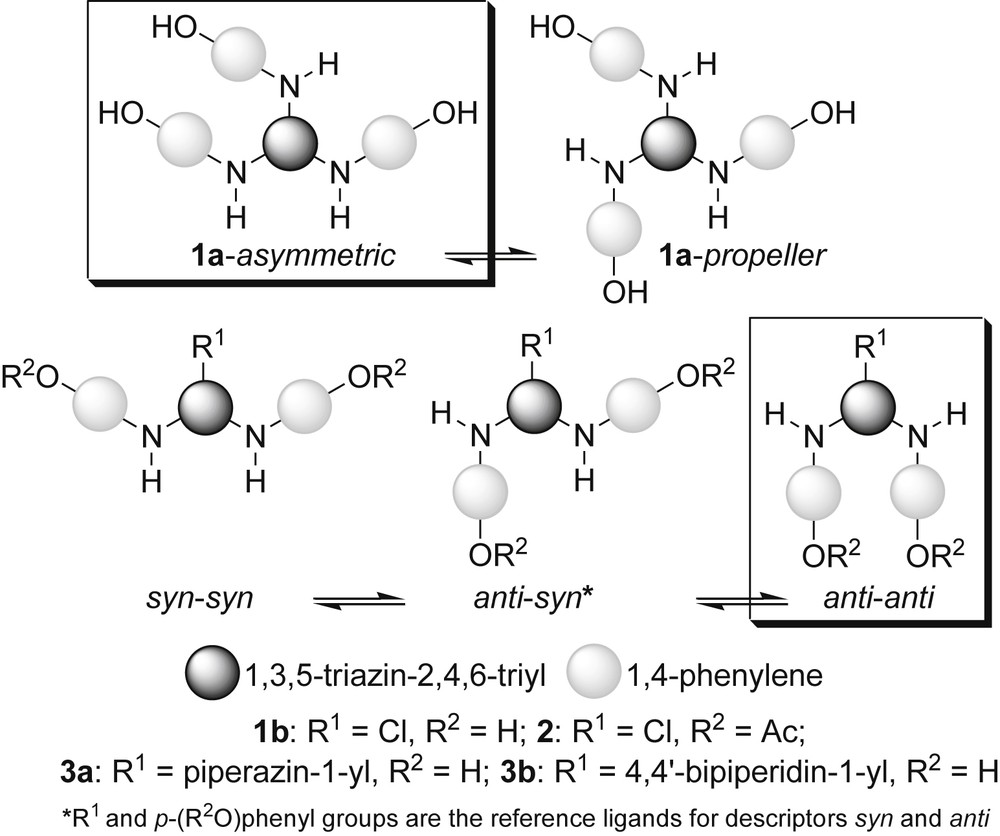
As early as 1971 [11a], it was established that the C(s-triazine)-N(exocyclic) linkages in amino-s-triazines have a partial double bond character due to the p(N)→π(CN) conjugation between the lone pair of the exocyclic N-atom in conjunction with the high π-deficient s-triazine ring [11]. There is a restricted rotation thus induced. Variable temperature NMR spectroscopy is an appropriate technique for monitoring this stereodynamism [12], including examples of arylamino-s-triazines [12d–f,12h]. As for other amino-s-triazines, in the case of monomeric (G-0) dendrons 1a, 1b, 2, 3a and 3b, at room temperature, a topological idealised model [5a–d,12g] predicted, for 1a, a two term rotational diastereomerism (asymmetric ⇆ propeller) and three terms (syn–syn ⇆ anti–syn ⇆ anti–anti) for 1b, 2, 3a and 3b (Scheme 2).
Therefore, before any electrochemical investigation, a study by means of DFT [13–20] calculations in solution (DMSO) was implemented for compounds 1a, 1b, 2, 3a, 3b and 7, selected as the representative. The results are shown in Tables 2 and 3 and Fig. 1 together with those referring to the starting p-aminophenol (p-AP) and its N-acetyl derivative, the well-known anti-inflammatory drug paracetamol (PA).
Full geometry optimisation and calculated ɛHOMO, ɛLUMO in p-aminophenol (p-AP), paracetamol (PA) and of (G-0) dendrons 1a, 1b, 2, 3a and 3b in DMSO.a
| No. | HOMO | LUMO | ɛHOMO (eV) | ɛLUMO (eV) | Gap (eV) |
| p-AP | −6.24 | +0.68 | 6.92 | ||
| PA | −7.00 | +0.50 | 7.50 | ||
| 1a | −6.81 | +0.31 | 7.12 | ||
| 1b | −6.99 | −0.03 | 6.96 | ||
| 2 | −7.47 | −0.23 | 7.24 | ||
| 3a | −6.80 | +0.35 | 7.15 | ||
| 3b | −6.81 | +0.35 | 7.16 |
a The full geometry optimisation has been carried out at the DFT level of theory considering the M06-2X [13] exchange-correlation functional together with the def2-TZVP [14] basis set in the presence of a solvent environment implemented in the Gaussian 09 [15] program package. The solvent effects have been taken into account via the Polarizable Continuum Model (PCM) using the integral equation formalism variant (IEFPCM) [16] considering the DMSO (ɛ = 46.826) as the solvent environment.
Indicative Bond Order (BO) indexes, Bond Lengths (BLs) and Lone Pair (LP) orbital populations in p-aminophenol (p-AP), paracetamol (PA) and (G-0) dendrons 1a, 1b, 2, 3a and 3b in DMSO.
| No. | C(s-triazine)-N(H) | C(Ph)-N(H) | C(s-triazine)-N< | Lone Pair (LP) orbital populations (e)a | ||||||
| N(H) | O(H, Ac) | -N< | s-Triazine | |||||||
| BOb | BL (Å)c | BO | BL (Å) | BO | BL (Å) | N(1, 3, 5) | ||||
| p-AP | — | 1.15 | 1.375 | — | 1.82 | 3.88 | — | — | ||
| PA | — | 1.05 | 1.410 | — | 1.64 | 3.86 | — | — | ||
| 1a | 1.21 | 1.353 | 1.04 | 1.411 | — | 1.68 | 3.86 | — | 1.90 | |
| 1b | 1.25 | 1.351 | 1.03 | 1.410 | — | 1.64 | 3.86 | — | 1.90 | |
| 2 | 1.24 | 1.343 | 1.04 | 1.406 | — | 1.64 | 3.76 | — | 1.90 | |
| 3a | 1.20 | 1.360 | 1.04 | 1.407 | 1.23 | 1.353 | 1.68 | 3.86 | 1.67 | 1.90–1.91 |
| 3b | 1.20 | 1.361 | 1.04 | 1.407 | 1.23 | 1.352 | 1.68 | 3.86 | 1.67 | 1.90–1.91 |
a Obtained by considering the NBO electron population analysis at the M06-2X/def2-TZVP level of theory where e is the elementary electric charge carried by a single electron.
b Calculated at the M06-2X/def2-TZVP level of theory considering the Wiberg definitions of bond order (see Ref. [19]) implemented in the NBO analysis module.
c Calculated at the M06-2X/def2-TZVP level of theory.
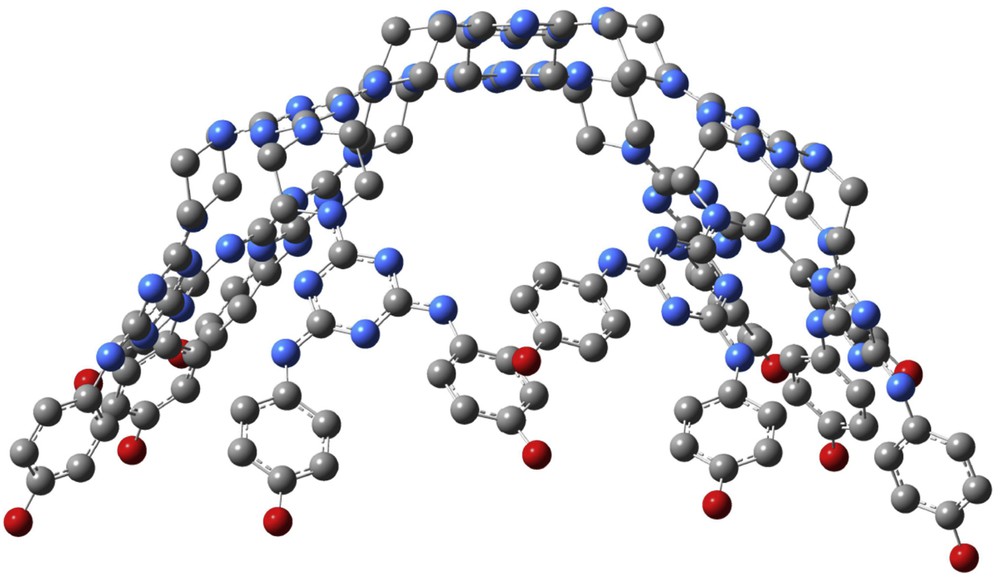
The optimised geometry structure of compound 7 obtained at M06-2X/def2-TZV level of theory [14] (the H atoms were omitted for reasons of simplicity).
They deserved the below comments:
- (i) The optimised geometry of melamine 1a was found to be of type asymmetric (Table SI-1, Supporting information); meanwhile anti–anti was the preferred rotameric arrangement adopted by the p-(R2O)phenyl units in compounds 1b, 2, 3a and 3b in DMSO (Table SI-2, Table 2). The same anti–anti stereochemistry was local at the periphery of dendrimer 7 which disclosed a vaulted global shape [21] (Fig. 1).
- (ii) The increasing order of ɛHOMO energies (Table 2) correlated with the decreasing strength of the EWG N(O)-substituting the p-(R2O)phenylamino units (Scheme 1), namely acetyl and variable π-deficient s-triazine ring, i.e. 2 < paracetamol ∼ 1b. However, an equalisation of ɛHOMO energies in the melamine series 1a ∼ 3a ∼ 3b resulted from calculation. Obviously, the highest ɛHOMO level was that of p-aminophenol.
- (iii) The partial double bond nature of connexions C(s-triazine)-N(exocyclic) was disclosed by Bond Order (BO) values ranging between 1.20 and 1.25, anyhow much greater than those of connexions C(Ph)–NH. The calculated Bond Lengths (BLs) fully supported their inverted dependence with respect to the corresponding BO indexes (Table 3).
- (iv) The lowest nitrogen Lone Pair (LP) orbital populations (1.64–1.68) were found in LPN(H) → π(CO) (paracetamol) and LPN(exocyclic) → π(s-triazine) conjugated compounds 1a, 1b, 2, 3a and 3b. The LP orbital population of the phenolic oxygen was less diminished (3.76–3.88) as a consequence of the LPO(H, Ac) → π(Ph) or LPO → π(CO) delocalisation.
Furthermore, data issued from (VT) 1H NMR investigations (Table 4) completed those provided by theoretical anticipations.
Relevant (VT) 1H and 2D-1H-DOSY-NMR data (500 MHz, DMSO-d6) of compounds 1a, 1b, 2, 3–5a, 3–5b, 6 and 7.
| No. | Relevant δH values (ppm) (298 K) | Relevant δH values (ppm) (363 K) | Temperature gradients (TGs) as (ΔδNH/ΔΤ)×103 (ppb/K)a | D (μm2/s) | dHb (nm) | ||
| NH | OH | NH | OH | ||||
| 1a | 8.74 | 9.06 | 8.37 | 8.74 | −5.69 | 152 | 1.44 |
| 1b | 9.70 | 9.27 | 9.46 | 8.91 | −3.69 | 218 | 1.00 |
| 9.83 | −5.69 | ||||||
| 9.91c | −6.92 | ||||||
| 2 | 10.18 | — | 9.97d | — | −3.82 | 230 | 0.95 |
| 10.34c | −6.73 | ||||||
| 3a | 8.71 | 9.01 | 8.34 | 8.68 | −5.69 | 152 | 1.44 |
| 4a | 8.81 | 9.04 | 8.41 | 8.69 | −6.15 | 137 | 1.59 |
| 3b | 8.72 | 9.02 | 8.33 | 8.66 | −6.00 | 131 | 1.67 |
| 4b | 8.72 | 9.01 | 8.36d | 8.70 | −6.55 | 125 | 1.75 |
| 5a | 8.83 | 9.04 | 8.54 | 8.79 | −4.46 | 90 | 2.43 |
| 5b | 8.72 | 9.01 | 8.32d | 8.66 | −7.27 | 72 | 3.03 |
| 6 | 8.78 | 9.04 | 8.47 | 8.78 | −4.77 | 82 | 2.66 |
| 7 | 8.79 | 9.05 | 8.39 | 8.70 | −6.15 | 70 | 3.12 |
a Calculated as [(δH298K–δHTK)/(298 K–T K)] × 103 < 0.
b dH (Hydrodynamic diameter) issued from D [diffusion coefficient observed in 2D-1H-DOSY NMR charts in 5 mM DMSO-d6 (η, dynamic viscosity 2.00 × 10−3 kg m−1 s−1) at 298 K] by applying the Stokes–Einstein equation.
c Multiple δH values due to more than one (anti–anti) species found in a frozen rotational equilibrium in agreement with the highest π-deficiency of the s-triazine ring in the analysed series (Scheme 2, Figs. SI-4 and SI-8).
d At 353 K.
Unsurprisingly, the most π-deficient diamino-chloro-s-triazines 1b and 2 displayed, at room temperature, the most convincing 1H NMR spectral appearance, consistent with the nearly frozen nature of their rotational equilibria (Scheme 2) on the NMR time scale (Figs. SI-4 and SI-8). In contrast, under the same conditions, the rotamerism of all melamines (G-0, -1, -2) was identified by NMR as a classically entitled “slow exchange status between (un)equally populated sites” [22]. Upon heating to 80–90 °C (Table 4), all compounds became single mediated structures, in a fast freely rotating status about all C(s-triazine)-N(exocyclic) bonds.
Temperature Gradients (TGs) of the NH protons were also indicative because they revealed the influence of different types of N-ligands on solvation. Although this parameter is appropriate for amide –N(H)–C(O)– ↔ –N+(H)C(–O−)– protons in peptides and proteins [23], it can also be applied to amino-s-triazines, –N(H)–C(N–)– ↔ –N+(H)C(–N−–)– as established by Simanek and Moreno [21b]. Following this extrapolation, if TG values of “amide-like” protons in amino-s-triazines are more negative than −4 ppb/K in strong hydrogen bond acceptor solvents, such as DMSO-d6 [12g], the NH groups are exposed to the solvent rather than forming intramolecular hydrogen bonds. On the other hand, a TG value less negative than −4 ppb/K indicates that the NH group preferentially forms intramolecular hydrogen bonds at room temperature. If so, besides the normal aptitude for binding of p-HO phenolic functionalities, the contribution to solvation of NH groups, i.e. Me2SO⋯H⋯N<, was also significant. Indeed, as one can see, almost all TGs in Table 4 were more negative than −4 ppb/K in conjunction with an anti–anti arrangement of phenolic (peripheral) units (Scheme 2, Table 2) in all types of compounds. Consistent with some of our previous data [5b], when the rotamerism was 1H NMR detected as a mixture of stereoisomers (compounds 1b and 2, Table 4), this situation disclosed the major rotamer anti–anti (Table 2) being the most solvated as well (TGs as −6.92 ppb/K and −6.73 ppb/K, respectively, Figs. SI-4 and SI-8).
Surprisingly, the different conformational nature of the internal linker, anancomeric2 [22b] (rigid, 4,4′-bipiperidin-1,1′-diyl) against chair ⇆ chair flipping (piperazine-1,4-diyl) determined lower TGs in 4b (−6.55 ppb/K) and 5b (−7.27 ppb/K) with respect to 4a (−6.15 ppb/K) and 5a (−4.46 ppb/K) (Scheme 3).
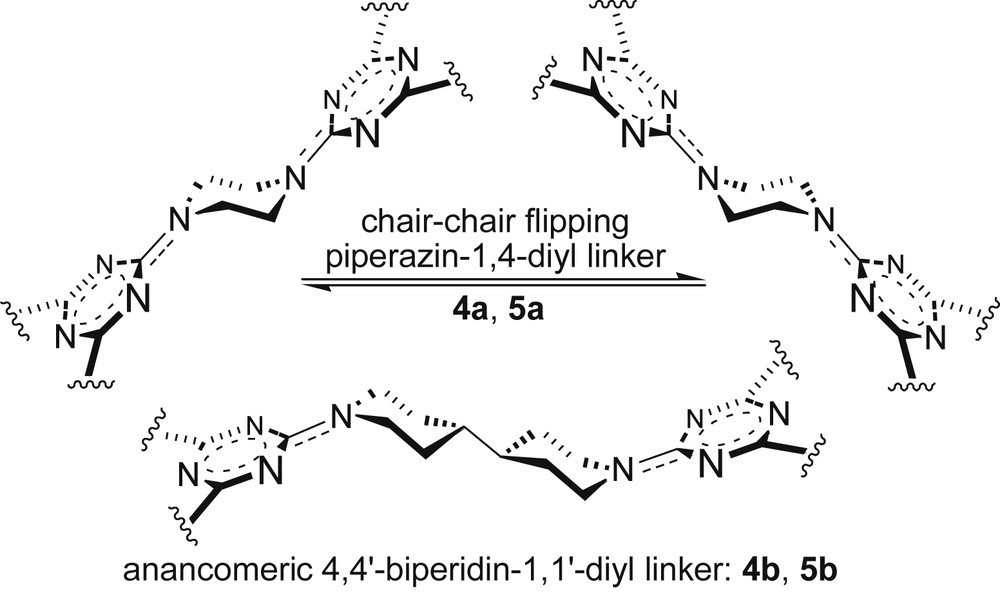
Dimeric (G-0) melamines 4a and 4b were more Me2SO⋯H⋯N< solvated in comparison with their monomeric precursors 3a (TG as −5.69 ppb/K) and 3b (TG as −6.00 ppb/K), respectively.
To what extent the above structural findings have a corresponding electrochemical impact, we will discuss hereafter.
2.3 Electrochemical characterisation
2.3.1 Cyclic voltammetry
Next, the electrochemical behaviour of our p-aminophenol-based amino-s-triazines was investigated by means of cyclic voltammetry. The cyclic voltammograms recorded on the Pt electrode in DMSO, 0.1 M KCl solution are presented in Fig. 2.
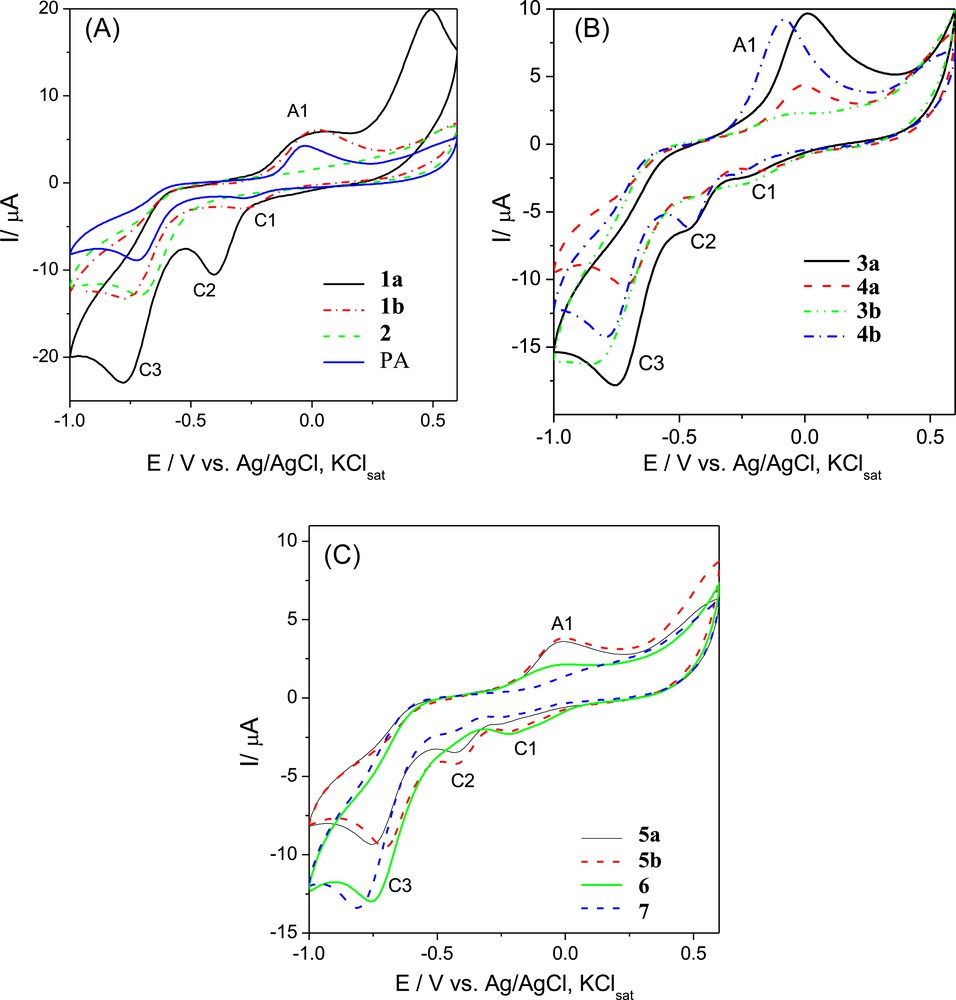
Cyclic voltammograms of 10−3 M paracetamol (PA), 1a, 1b, 2 (2A), 3a, 3b, 4a, 4b (2B) and 5a, 5b, 6, 7 (2C). Experimental conditions: electrolyte, anhydrous and ultrasonicated DMSO/0.1 M KClsat solution deaerated with Ar; starting potential −1 V versus Ag/AgCl, KClsat; scan rate, 0.050 V−1; the 25th cycle is shown.
Except for compounds 2 and 7, all voltammograms revealed the presence of a single well-defined anodic oxidation peak A1 closely located around 0 V versus Ag/AgCl, KClsat. By contrast, 1–3 cathodic reduction peaks, C1-3, were disclosed as C1 and C3 (paracetamol and compounds 1b, 3b, and 6), C2 and C3 (compound 1a) or C1, C2 and C3 (compounds 3a, 4a, 4b, 5a and 5b). The variable incidence of the reduction peaks C1 and/or C2 was attributed to the successive reduction of the species produced during electrochemical oxidation. We note that the reduction peak C3, observed even in anhydrous DMSO 0.1 M KCl ultrasonicated solution in the absence of any organic compound, was related to the activity of the Pt electrode [24] and will be not discussed here. The potential values Ep corresponding to peaks A1, C1 and C2, issued from the voltammetric response, are listed in Table 5.
Values of A1, C1, C2 peaks potential of compounds paracetamol (PA), 1a, 1b, 2, 3a–5a, 3b–5b, 6 and 7 (for Experimental conditions, see Fig. 2).
| Compound Ep (V) vs. Ag/AgCl, KClsat | ||||||||||||
| 1b | 1a | 3a | 4a | 5a | 5b | PA | 6 | 3b | 4b | 2 | 7 | |
| A1 | 0.020 | 0.015 | 0.013 | −0.015 | −0.021 | −0.024 | −0.027 | −0.048 | −0.052 | −0.090 | — | —a |
| C1 | −0.270 | — | −0.230 | −0.180 | −0.245 | −0.210 | −0.275 | −0.205 | −0.220 | −0.275 | — | −0.240 |
| C2 | — | −0.400 | −0.440 | −0.415 | −0.430 | −0.420 | — | — | — | −0.460 | — | −0.440 |
a Too weak to be measured.
Since the A1 anodic potentials values were indicative of the electron donating strength of the compounds, we considered that these potentials also reflect their oxidation easiness, i.e. increasing with the decrease of the oxidation potential value.
Our attention was primarily focused on the pair of peaks A1/C1. Thus, by comparing voltammograms of the bis(p-aminophenol)-based diamino-chloro-s-triazine 1b with its O,O′-diaceyl analogue 2 (Fig. 2A), the disappearance of peak A1, around 0.020 V versus Ag/AgCl, KClsat, proved that this was due to absence of the free p-HO-phenolic group in 1b, involved in the first step of oxidation. That is, the replacement of p-HO (from 1b) by the p-AcO group (in 2) leads to peak A1 vanishing, i.e. the single NH group in 2 was unable to trigger any oxidation process. We note that, in the case of paracetamol, containing just one p-aminophenol unit, a recent well-documented N-acetyl-p-benzoquinonimine oxidation product is mentioned in the literature as a result of a reversible A1/C1 two-electron transfer process (Scheme 4) [25a–c].
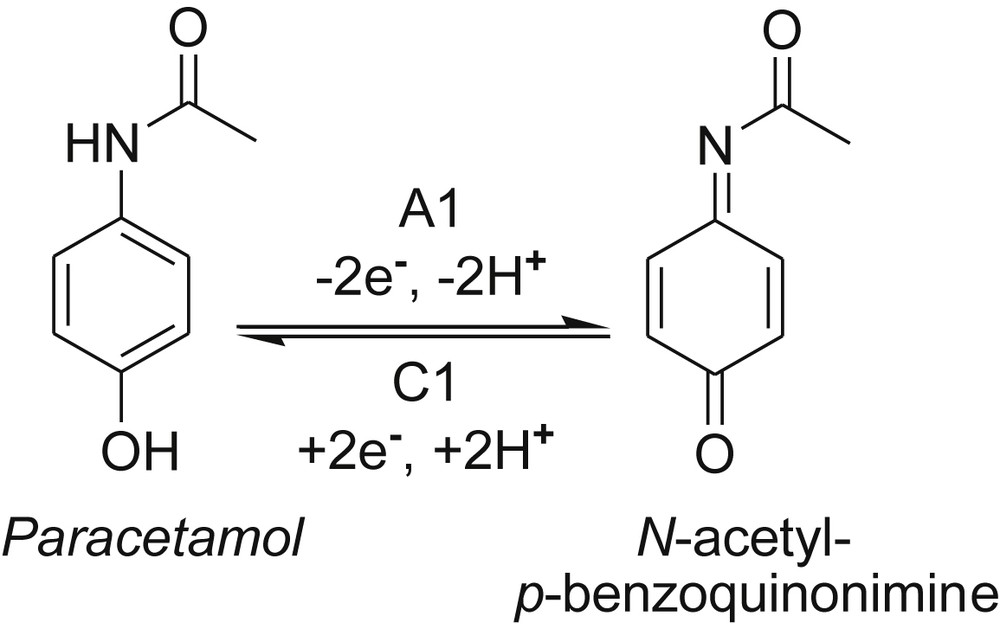
The above redox pathway occurs in spite of the low ɛHOMO level (Table 2) and low LPN(H) orbital population of paracetamol (Table 3). As shown in Table 5, under our conditions, the peak A1 of paracetamol was located at a negative value, −0.027 V versus Ag/AgCl, KClsat.
Therefore, concerning the oxidation stage, we postulated a similarity between the EW influence of the variable π-deficient s-triazine ring linked to the NH group in our compounds and that of the EW Ac group in paracetamol. If so, as for paracetamol, the pair of peaks A1/C1 also disclosed an overall and reversible two-electron transfer process/p-aminophenol unit even for compounds 1b and 3a exhibiting positive Ep A1 values (Table 5). Thus, in a two-step oxidation process, a novel extended π–π-conjugated system of type (s-triazinyl)-p-benzoquinonimine was formed (Scheme 5).
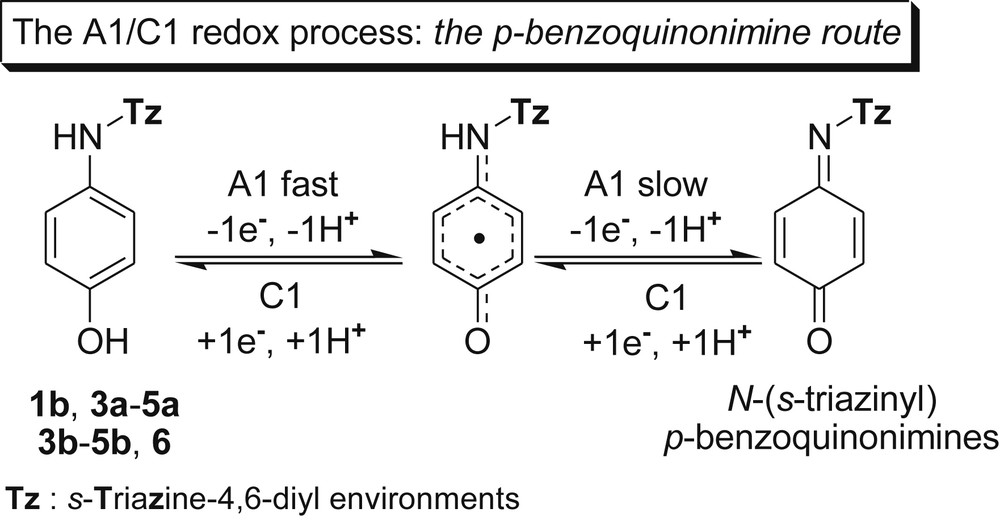
One should observe that the decreasing order of the A1 peak potentials (Table 5), for example in the (G-0) dendron series as 1b > 1a > 3a > 3b, was parallel to the decreasing strength of the π-deficiency of the EWG s-triazine expressed as ɛHOMO values (Table 2) or LPN(H) orbital populations (Table 3). They crucially determined the adjacent NH group ability for oxidation. Therefore, we assumed the p-benzoquinonimine route (Scheme 5) to be valid for all derivatives exhibiting the pair of peaks A1/C1 (Fig. 2) and the unique electronic transfer for compounds 1b, 3b and 6 (Table 5). Nevertheless, to what amount this route involved, simultaneously, all p-aminophenol peripheral units could not be established because of the iterative nature of the oxidation process.
Furthermore, the influence of several other factors concerning the redox process A1/C1 (Scheme 5) was also observed. Thus, (G-0) dimeric melamines 4a and 4b disclosed significantly lower anodic oxidation potentials against (G-0) monomeric melamines 3a and 3b, respectively, most probably because of a better NH-solvation in DMSO expressed by TG values (Table 4).
A comparison between the CVs of (G-0) melamines 3a and 3b (Fig. 2B) showed that the replacement of the piperazine-1-yl ligand with the more electron-donor 4,4-bipiperidin-1-yl dramatically decreased the anodic oxidation potential, i.e. from +0.013 V to −0.052 V versus Ag/AgCl, KClsat. We associated this behaviour with the higher basicity of 3b versus 3a together with the different conformational nature of their diaza-linkers, flipping in 3a but anancomeric in 3b (Scheme 3). Consequently, the adsorption on the electrode surface of 3b was more intimate in comparison with that of 3a. Presumably for the same conformational reason, a similar fluctuation was observed in the case of dimeric (G-0) melamines 4a (−0.015 V) and 4b (−0.090 V). Mutatis-mutandis, the CVs of the (G-0) melamine 3a against its dimer 4a from one hand or of 3b against 4b on the other hand (Fig. 2B) exhibited a comparable enhancement of the oxidation current in the case of dimers due, very likely, to the doubling of the number of peripheral phenolic groups and to the symmetry of the molecule.
Similar concepts applied in the case of angularly built (G-1) dendrons 5a, 5b and 6.
The oxidation potential values corresponding to peak A1 in these compounds revealed that 5a, 5b and 6 (Fig. 2C) behave similarly in what the easiness of the oxidation was concerned. However, the oxidation current intensity was much smaller in the case of (G-1) melamine 6 with respect to its (G-1) chlorodendron precursor 5a, presumably because of the blocking of the electrode surface by adsorption of the compound 6 oxidation products.
It is important to notice that, for compound 7 (Fig. 2C), the oxidation peak A1 almost disappeared, suggesting that the dendrimer was much more difficult to oxidize than all its precursors. This behaviour could be motivated by the vaulted spatial arrangement (Fig. 1) of 7, which impeded the contact between the redox centres of the dendrimer and the Pt electrode surface.
2.3.2 Influence of the starting potential
In order to investigate the influence of the starting potential on the electrochemical response of compounds, dimeric G-0 dendron 4a was selected as a typical example (Fig. 3).
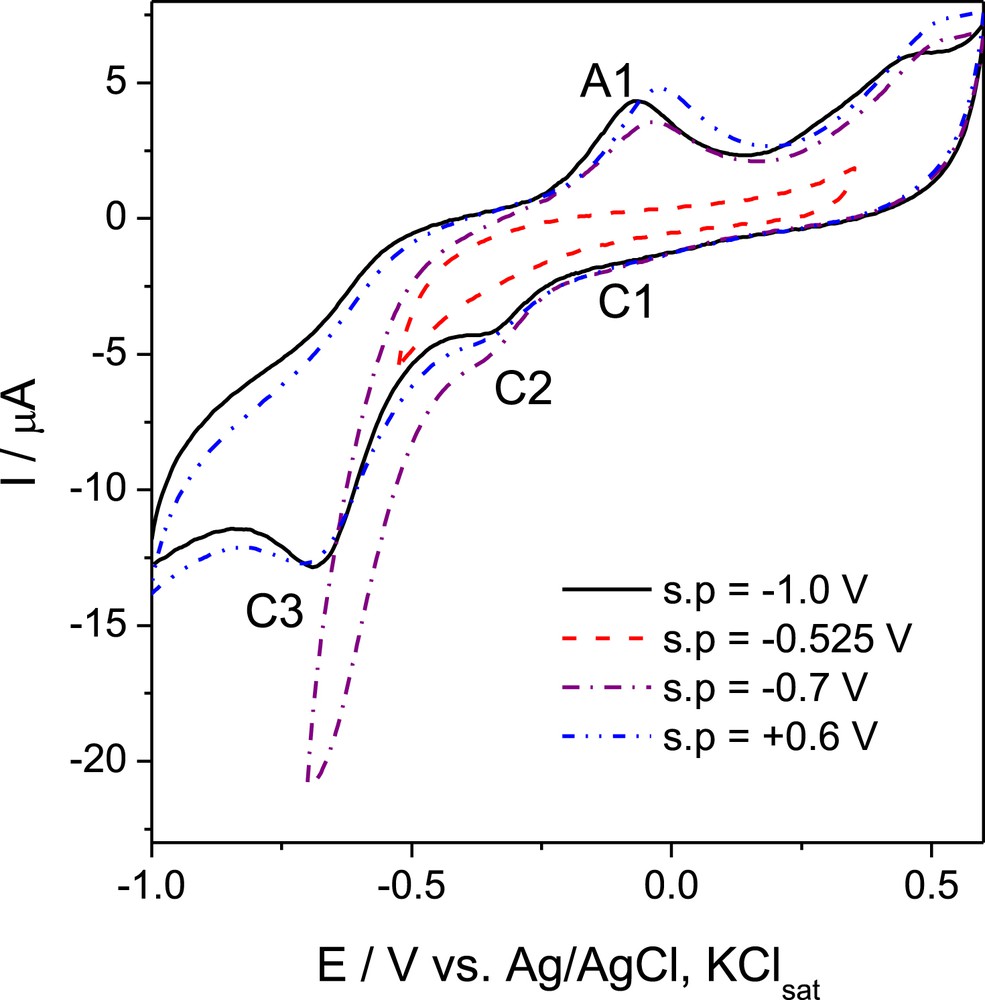
Cyclic voltammogram of 10−3 M 4a at different values of starting potential. Experimental conditions: electrolyte, anhydrous and ultrasonicated DMSO/0.1 M KClsat solution deaerated with Ar; scan rate, 0.050 V s−1; the 7th cycle is shown.
As expected, the width of the scanned potential domain was proven to be of great importance. Starting from an enough negative value of potential (e.g., at least −0.7 V versus Ag/AgCl, KClsat), the appearance of the oxidation peak A1 and of the subsequent reduction processes responsible for reduction peaks C1 and C2 was noticed. When we limited the potential range to a narrow domain (e.g., −0.525 V to +0.300 V versus Ag/AgCl, KClsat), no characteristic oxido-reduction peaks appeared. Generally, the starting potential for the electrochemical measurements was −1 V versus Ag/AgCl, KClsat. If the experiment was done within a large potential window (−1.0 V to +0.6 V), reversing the scan direction (e.g., starting from +0.6 V versus Ag/AgCl, KClsat), no influence on the shape of the voltammogram was observed (Fig. 3).
2.3.3 Reproducibility
The reproducibility of the voltammetric measurements was investigated by recording 3 successive voltammograms for the same electrode with cleaning of its surface between measurements. The reproducibility of cyclic voltammetric measurements for compound 4a was good. Thus, for a mean value of the peak current intensity of 9.48 × 10−6 A, for 3 successive measurements the relative standard deviation (RSD) was 11.9% (results not shown).
2.3.4 Influence of repetitive potential cycling
Dimeric (G-0) melamine dendrons 4a and 4b, having a low s-triazine π-deficiency, were selected in order to study the influence of repetitive potential cycling on the electrochemical response under potentiodynamic conditions by continuous cycling of the electrode potential (25 cycles) in the potential range of −1 V ÷ +0.6 V versus Ag/AgCl, KClsat, in DMSO solution, on the Pt electrode. The results are shown in Fig. 4.
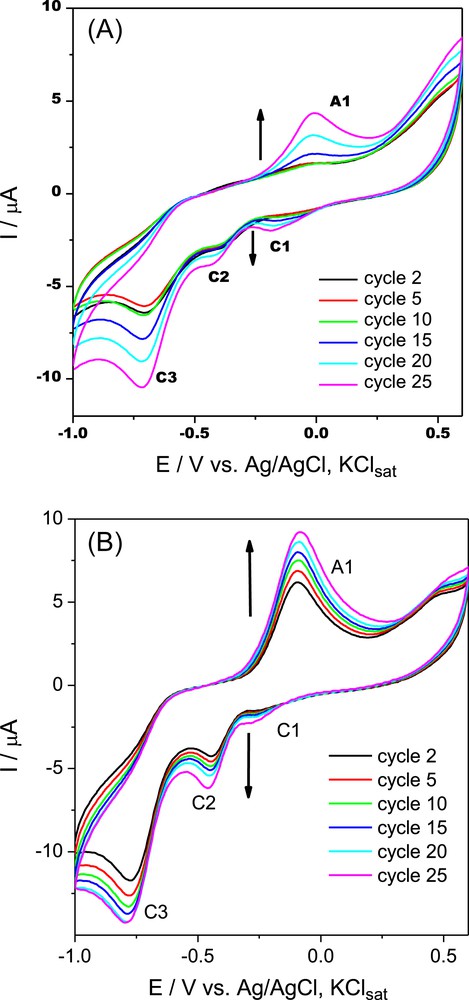
Cyclic voltammograms of 10−3 M 4a (4A) and 4b (4B) on the Pt electrode. Experimental conditions: electrolyte, anhydrous and ultrasonicated DMSO/0.1 M KClsat solution deaerated with Ar; starting potential, −1 V versus Ag/AgCl, KClsat; scan rate, 0.050 V s−1.
A progressive increase of the peak current was observed suggesting a possible polymerization taking place on the Pt electrode surface. We note that the electropolymerization of p-aminophenol on the Pt electrode in organic media leading to a soluble and electroactive polymer was reported by Taj et al. [9a] as early as 1992. As proposed in Scheme 6, in our case, the electrochemical pathway should be (i) similar to that of p-aminophenol [9h], irrespective of the presence of the s-triazinyl fragment and (ii) related to that of paracetamol [25d].
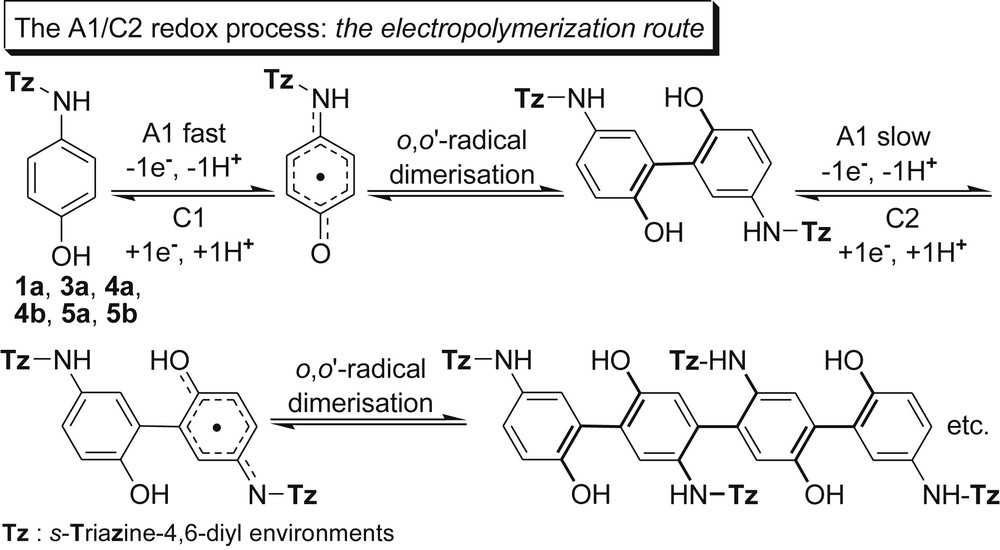
Thus, the electrochemical significance of peak A1 was almost the same as that in Scheme 5, fast p-HO phenolic against slow NH oxidation, the last one being the most sensitive to the EWG s-triazine vicinity. As a result, a radical o,o′-coupling, similar to that previously reported for paracetamol [25d], occurred to produce dimeric and then linear tetrameric species, respectively, with a s-trans arrangement of the s-triazine bulky motifs. One must observe that the cathodic peak potential C2 (Table 5) was located at much more negative values in comparison with C1, in agreement with the decreasing order of stability against reduction of the involved π–π-conjugated system, aromatic (C2) > p-benzoquinonimine (C1).
Besides 4a and 4b, the electropolymerization route could be extrapolated to all melamines exhibiting the reduction peak C2 namely (G-0) 1a and 3a, (G-1) 5a and 5b. The electropolymerization route appeared to be the single one in the case of (G-0) melamine 1a (Fig. 2A); however it was not observed for melamines (G-0) 3b (Fig. 2B) and (G-1) 6 (Fig. 2C).
In order to perform a quantitative evaluation of the polymer matrix permeability, cyclic voltammograms of compounds 4a and 4b were recorded for different potential scan rates on the Pt/4a electrode, immersed in a 5 mM K3[Fe(CN)6] solution, containing 0.1 M KCl (results not shown).
The voltammetric wave corresponding to the anodic and cathodic peak currents of the [Fe(CN)6]3−/[Fe(CN)6]4− couple obeys Equation (1):
| (1) |
Using Equation (1), the K3[Fe(CN)6] diffusion coefficient at the Pt electrode was found to be equal to 3.3 × 10−6 cm2/s which is in concordance with those reported in the literature [26]. Assuming this value for the K3[Fe(CN)6] diffusion coefficient, the area of the modified electrode Pt/4a was estimated to be 0.2226 cm2, which is greater with a factor of 1.13 than the geometrical surface of the electrode (0.19625 cm2), confirming that during the 25 cycling process an immobilization of compound 4a on the Pt surface occurred.
In addition, the relative increase of the charge transfer on the electrode surface can be estimated by the Equation (2):
| (2) |
3 Conclusions
3.1 Synthesis
Avoiding any protective–deprotective step, the iterative synthesis of a new 10 member family of (G-0, -1, -2) dendritic melamines comprising p-aminophenol (peripheral unit) and piperazine or 4,4′-bipiperidine (linkers) was reported. The chemoselective amination of cyanuric chloride performed with the above amino-nucleophiles was more accessible if piperazine was the internal linker of choice. Overall, the low solubility of these amino-s-triazines in common volatile organic solvents and their high relative retention on column chromatography were the two main obstacles to be overcome, successfully, with good yields.
3.2 Structure
DFT calculations in DMSO (with global energy minima, indicative bond order indexes, bond lengths and lone pair orbital populations) helpfully established and discriminated the rotational diastereomerism of p-aminophenol peripheral units about the partial double bonds C(s-triazine)-N(exocyclic) as being of type asymmetric or anti–anti. The last one was also localised at the periphery of the first p-aminophenol-based (G-2) dendritic melamine which adopted a vaulted global shape. Calculation revealed a lower N-lone pair orbital population in Ph–NH units in comparison with that of the phenolic oxygen atom. (VT) 1H NMR spectroscopy disclosed, by means of temperature gradients, our p-aminophenol-based amino-s-triazines being not only OH but also strongly >N⋯H⋯OSMe2 solvated molecules. Solvation was higher if the internal linker was anancomeric (4,4′-bipiperidin-1,1′-diyl) against flipping (piperazine-1,4-diyl); however it was less dependent on the molecular dimension (dH).
3.3 Electrochemical characterisation
Almost all the above structural assignments had an impact on electrochemical characterisation of our amino-s-triazines by cyclic voltammetry (Pt electrode/DMSO). A two-electron transfer process was observed. Depending on the variable strength of π-deficiency of the s-triazine ring acting as the EWG on the adjacent NH group and the modulation ability of the latter to undergo redox processes in tandem with the phenolic p-HO group, two electrochemical pathways, similar to those already reported for paracetamol and p-aminophenol were proposed, (i) the p-benzoquinonimine route and (ii) the electropolymerization route.
4 Experimental
4.1 General
Melting points were carried out on an ELECTROTHERMAL instrument. NMR spectra were recorded on a Bruker AM 400, 500 or 600 instrument operating at 400, 500 or 600 and 100, 125 or 150 MHz for 1H and 13C nuclei, respectively. All chemical shifts (δ values) are given in parts per million (ppm); all homocoupling patterns (nJH,H values) are given in Hertz. In the NMR descriptions, some specific abbreviations were used: “br s” (broad singlet), “br d” (broad doublet), “dd app. t” (doublet of doublets appearance as a triplet), “ddd app. td” (doublet of doublets of doublets appearance as a triplet of doublets), Pip (Piperazine linker) and Bipip (4,4′-Bipiperidine linker). TLC monitoring was performed by using aluminium sheets with silica gel 60 F254 (Merck) (visualisation in UV at λ = 254 nm). Column chromatography was conducted on Silica gel Si 60 (40–63 mm, Merck). IR spectra were recorded on a JASCO FT-IR 6100 Spectrometer. Only relevant absorption maxima are listed, throughout, in cm−1: s (strong), m (medium) and w (weak). Microanalyses were performed on a Carlo Erba CHNOS 1160 apparatus. Mass spectra were carried out on a LTQ ORBITRAP XL (Thermo Scientific) instrument which was externally calibrated using the manufacturer's ESI(+) calibration mix. The samples were introduced into the spectrometer by direct infusion.
Synthesis of compounds 1a, 1b, 2, 3a, 3b, 4a, 4b, 5a, 5b, 6 and 7 together with their full analytical and spectral data are given in the SI (Supporting information).
4.2 Computational details
The full geometry optimisation of compounds 1a, 1b, 2, 3a, 3b and 7 has been carried out at the DFT level of theory considering the M06-2X [13] exchange-correlation functional together with the def2-TZVP basis set (def2-TZV basis set for compound 7) [14] in the presence of a solvent environment implemented in the Gaussian 09 [15] program package. The solvent effects have been taken into account via the Polarizable Continuum Model (PCM) using the integral equation formalism variant (IEFPCM) [16] considering the DMSO (ɛ = 46.826) as the solvent environment. No negative wave numbers were obtained for all optimization cases, proving that true minima of the potential energy surfaces were found in the optimisations. Next, the charge distribution and the Wiberg's bond-order index analyses (Table 3) were performed considering the natural population analysis (NPA) [17–20] method through the NBO module built in the Gaussian09 package.
4.3 Electrochemical investigation. Reagents and methods
The supporting electrolyte for electrochemical measurements was ultrasonicated DMSO (Merck) containing 0.1 M KCl (Reactivul Bucuresti). The reagents were of analytical grade and used as received.
4.4 Electrochemical measurements
Cyclic voltammetry was performed on a PC-controlled electrochemical analyser Autolab-PGSTAT 10, EcoChemie, The Netherlands. Electrochemical experiments were carried out using an undivided cell equipped with three-electrodes: a platinum electrode as the working electrode, a platinum wire as the counter electrode and Ag/AgCl, KClsat as the reference electrode, respectively. The electrolyte solution was deoxygenated by bubbling argon for 15 min before measurements.
Acknowledgement
The financial support from a Grant provided by the Research Council Romania (Project PN-II-ID-PCE-2011-3-0128) is gratefully acknowledged. A.B. acknowledges the financial support from the Romanian National Authority for Scientific Research and Innovation (ANCSI) through the Core Program 2015.
1 Our analytical sample 1b was different with respect to that of Bath and co-workers [4b,4c]. Our diagnosis concerned the aspect (white powder in our case against reported black crystals), melting point (343.7–344.1 °C in our case, against reported 251–252 °C) and the appropriate solubility for NMR analysis (only DMSO-d6 in our case, against reported CDCl3); see Figs. SI-4-7 in SI (Supporting information). Similar observations apply for compound 3a with respect to the sample of Bath and co-workers [4c]; see Figs. SI-12-14.
2 According to the definition from Ref. [22b]: “Fixed in a single conformation either by geometric constraints or because of an overwhelmingly on-sided conformational equilibrium.”