

1 Introduction
Cyclodextrins (CDs) are a series of cyclic oligosaccharides formed through α (1–4) ether linkages of glucopyranose units [1,2]. α-, β- and γ-cyclodextrins, which are 6-, 7-, and 8-membered sugar ring molecules, respectively, are the most common naturally occurring cyclodextrins [2]. The hydroxyl groups are oriented to the outside of the ring with the primary hydroxyls of the sugar residues at the narrow edge of the cone and the secondary hydroxyl groups at the wider edge (see Fig. 1a). This combination gives cyclodextrins a hydrophobic inner cavity and a hydrophilic outer surface. The hydrophobic internal cavity of cyclodextrins enables the formation of inclusion complexes with a variety of guest hydrophobic molecules such as aromatics, alcohols, halides, fatty acids, esters, etc. [3–5]. The use of CDs in both food and pharmaceutical industries is increasing due to their ability to improve the bioavailability, stability, and hydrosolubility of a wide range of biologically active compounds [6,7].
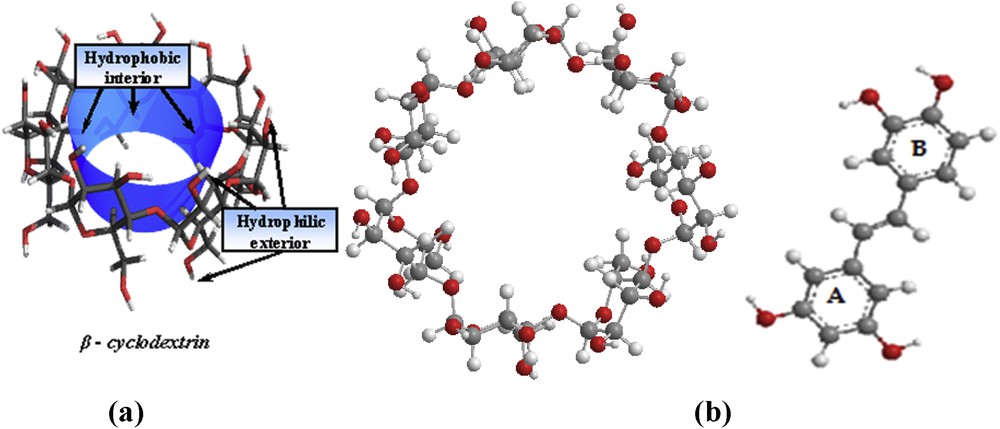
(a) Schematic representation of β-CD. (b) Molecular structures of β-CD (left) and PIC (right).
The property of CDs to form inclusion complexes with a variety of molecules has been the subject of significant research. Theoretical studies have used both quantum semi-empirical PM3 calculations and spectroscopic methods because the results of both methods are in good agreement [8]. The use of molecular modeling techniques can help to reinforce experimental results, including the stoichiometry, geometry and thermodynamic parameters of the complexation process. These techniques also provide information regarding the driving forces responsible for such processes [9].
Piceatannol (Fig. 1 b, right), or trans 3,5,3′,4′-tetrahydroxystilbene (PIC), is a naturally-occurring polyphenol belonging to the stilbenes subclass, found in the skin of grapes, different parts of the grapevine, rhubarb wine, berries and sugar cane [10]. Potter et al. [11] suggested that piceatannol is produced by resveratrol metabolism via cytochrome P450 1B1 [11]. Like most resveratrol-derived compounds, piceatannol has been reported to have several biological functions, including chemoprevention, antioxidant properties, anti-inflammatory properties, anti-leukemic properties, anti-cardiovascular properties and anti-cancer activities. It has also been reported to be an inhibitor of protein-tyrosine kinase with immunosuppressive action, which could contribute to the prevention of graft rejection [12–16]. Nevertheless, other researchers have shown that piceatannol possesses low bioavailability and shows poor absorption through the intestinal tract. This limitation is the handicap for the use of piceatannol in pharmaceutical and food industries, and it is the case for resveratrol and many other naturally derived compounds [17]. The inclusion of host molecules with cyclodextrins could potentially solve this problem.
Previous studies [18–20] have discussed the inclusion mode between CDs and resveratrol based on force field computation, molecular dynamic simulation and experimental observations. The results showed that resveratrol and β-cyclodextrin form an axial inclusion complex and that part of the A-ring and the B-ring of resveratrol are placed in the cavity of β-cyclodextrin while the hydroxyl groups are projected outside.
Lu et al. [20] have studied the complexation of resveratrol with CDs by density functional theory calculations at the B3LYP/6-311G (d, p) level of theory. The equilibrium geometries, electronic structures, and antioxidative capabilities were investigated to determine the influence of inclusion by CDs on the properties of resveratrol.
Recently, and as part of our ongoing interest on properties of polyphenolic compounds, we investigated the complexation of PIC with β-CD [21] using an experimental approach based on High Performance Liquid Chromatography coupled to a UV–VIS detector. The results indicated that PIC could be complexed with β-CD following a 1:1 stoichiometry. It was also demonstrated in that study that the use of the drug carrier CD increased the aqueous solubility and stability of PIC.
In order to provide complementary information, a theoretical investigation of the inclusion process of PIC in β-CD using PM models was undertaken. Our aim was to provide additional insight into the formation mechanism of the complex, particularly on the structural geometry of the inclusion complex. We focused our attention on the position of hydroxyl groups to evaluate the thermodynamic and electronic properties of the stable complex.
2 Computational methods
The theoretical calculations in this work were performed using Mopac software (Version 2009) and the Gaussian 09 software package [22]. Cambridge Chem Bio 3D Ultra (version 11.0) software was used for building the initial piceatannol structure. Initial β-CD coordinates were extracted from the database of the same software (Fig. 1). Optimization of the structure and the inclusion process were performed at the B3LYP/6-31G* level of theory. Training models of inclusion complexes were performed with manual and systematic searches [23]. The coordinate system used to define the process of complexation is shown in Fig. 2.
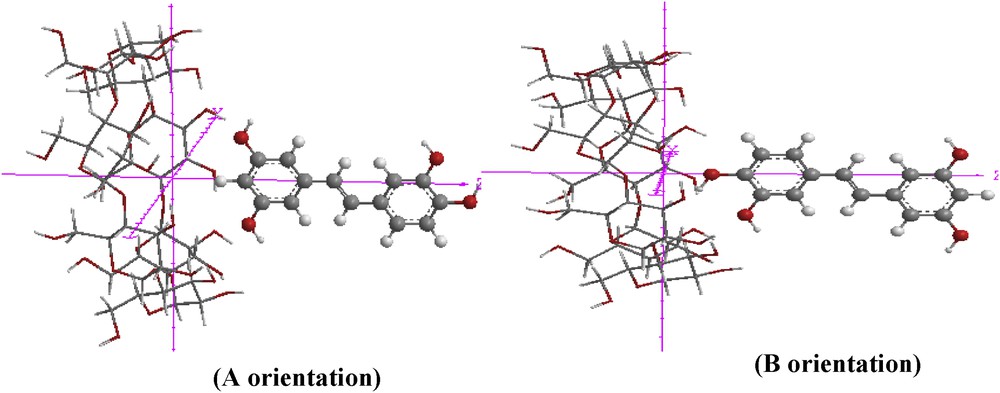
Coordinate systems used to describe inclusion processes of PIC with β-CD for ‘A’ orientation and ‘B’ orientation.
The origin of the coordinate system was defined by the center of the glycosidic oxygen atoms of β-CD located on the XY-plane. The secondary hydroxyl groups of β-CD were placed pointing towards the positive Z-axis. Then, the guest molecule was placed on the Z-axis and allowed to approach and pass through the β-CD cavity from −6 Å to +6 Å with a step of 1 Å.
In order to find an even more stable structure of the complex, the guest molecule was rotated around the Z-axis at 20° intervals from 0° to 360°.
The structures generated at each step were optimized with the PM3 (Parametric Model 3) method without imposing any symmetrical restrictions.
The corresponding frequencies were calculated to ensure that the calculated stationary points were true minima. These semi-empirical methods are convenient for modeling large molecular systems such as cyclodextrin inclusion complexes [24,25], and they are much less time-consuming than ab initio methods. The most stable complexes were optimized by PM3 semi-empirical methods under no constraints [26].
All optimizations during the docking process were carried out in a vacuum.
Two possible orientations were investigated (Fig. 3). For simplicity, the orientation in which the A aromatic ring was oriented toward the center of β-CD was named the ‘A’ orientation, while the other one, in which the B aromatic ring was oriented toward the β-CD cavity, was called the ‘B’ orientation.
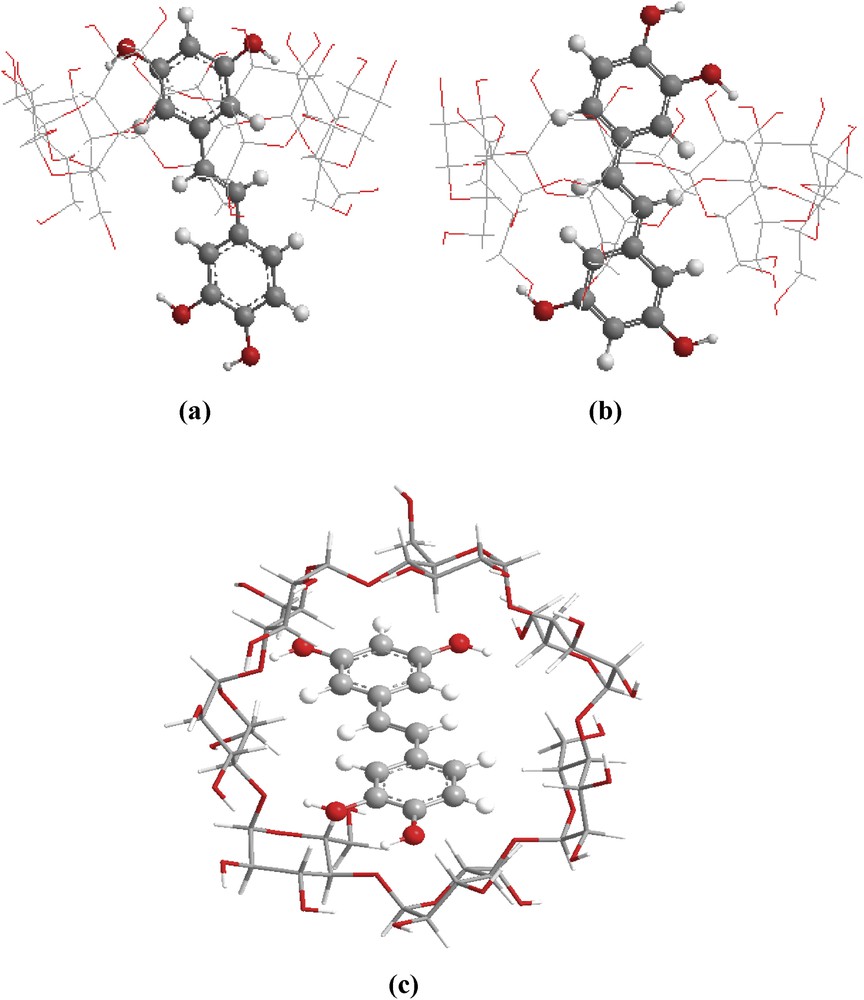
Structure of minimum energy obtained by the PM3 calculations, in vacuum for: (a) ‘A’ orientation and (b) ‘B’ orientation (side view) (c) ‘A’ orientation (top view).
In this work, different formulas were used to characterize the inclusion complexes.
The binding energy (ΔEbinding) is defined as the difference between the energy of the complex and the energy of the individual components in their optimized geometry [27]:
| (1) |
The energy of stabilization (complexation) between the guest molecule and β-CD calculated for the minimum energy structure can be obtained by Eq. 2 [28]:
| (2) |
| (3) |
3 Results and discussion
3.1 Docking process
The complexation energy calculated by the semi-empirical PM3 method is shown in Fig. 3. Graphical representations of the binding energies involved during the inclusion process of PIC into β-CD, at different distances and for both orientations, are displayed in Fig. 4. This figure shows that all of the energy values are negative, which indicates that the PIC/β-CD complex is thermodynamically favorable.
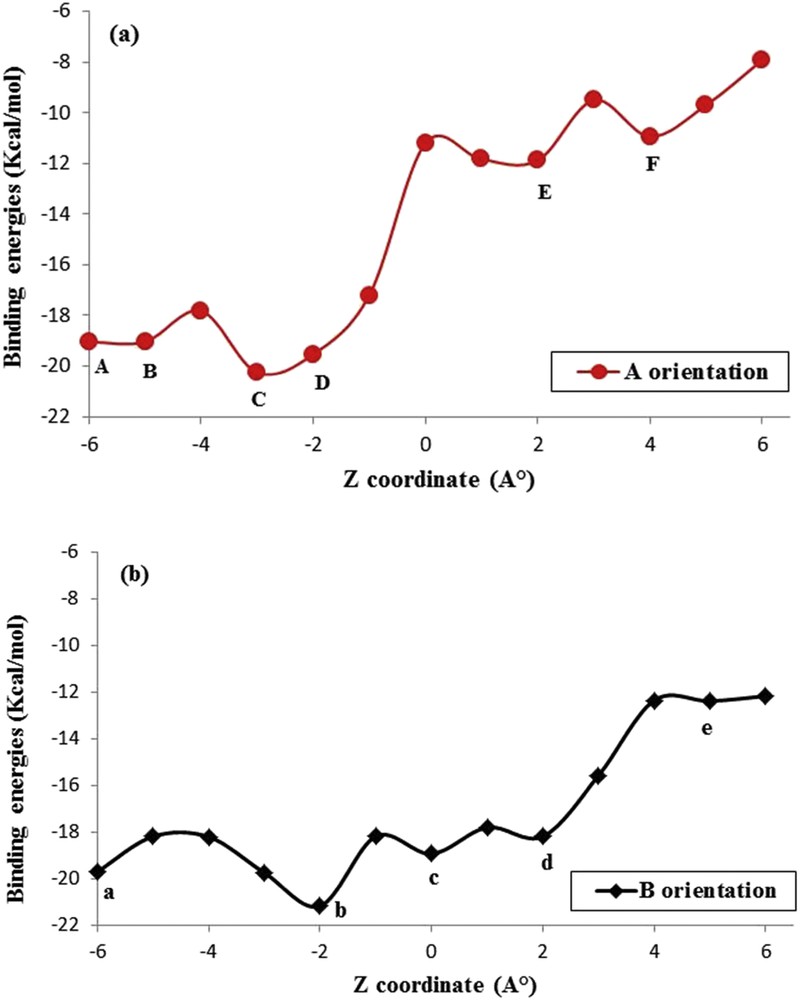
Binding energies of PIC/β-CD inclusion complexes at different positions Z (Å) and orientations: (a) ‘A’ orientation; (b) ‘B’ orientation. Points A, B, C, D, E, and F represent ZA = −6 Å, ZB = −5 Å, ZC = −3 Å, ZD = −2 Å, ZE = 2 Å, and ZF = 4 Å, respectively. Points a, b, c, d, and e represent Za = −6 Å, Zb = −2 Å, Zc = 0 Å, Zd = 2 Å, and Ze = 5 Å, respectively, using the PM3 method.
The curves show several local minima. The lowest minimum energies, corresponding to the most stable structures for ‘A’ and ‘B’ orientations, are precisely located at Z=−3 Å and Z=−2 Å, respectively. The corresponding energy values are −21.16 and −20.26 kcal/mol, respectively (Table 1). Both orientations are favorable for complexation because the energy difference between them is only −0.90 kcal/mol. This minimal difference is due to the nearly symmetrical structure of piceatannol.
Optimized (Eopt) energies of free B-CD and PIC and complexes, deformation energies (EDEF,β-CD) and (EDEF,PIC), binding (Ebinding) and complexation (Ecomplex) energies of PIC/B-CD complexes for the two orientations (A and B) by the PM3 method.
| Energies (Kcal/mol) | PIC | β-CD | ‘A’ orientation | ‘B’ orientation | ΔE |
| Eopt | −117.636 | −1453.78 | −1592.589 | −1591.683 | |
| Ebinding | −21.164 | −20.257 | −0.90 | ||
| Ecomplex | −21.173 | −20.267 | −0.90 | ||
| EDEF,PIC | 87.65 | 0.399 | |||
| EDEF,β-CD | 99.69 | 4.32 |
However, the results indicated that the deformation energies for both β-CD and PIC are significantly more important in ‘A’ orientation than in ‘B’ one. Additionally, the deformation energies of β-CD in both models are higher than those of the guest molecule, indicating that during the inclusion process β-CD is deformed, allowing the guest molecule to be easily accommodated into its cavity.
3.2 Thermodynamic parameters for the complexation process
Statistical thermodynamic calculations of the binding process were carried out at 1 atm. and 298.15 K, using harmonic frequency analysis in the PM3 method for the most stable structures and characterizing them as true minima on the potential energy surface. Thermodynamic parameters, such as the standard enthalpy change (ΔH°), the standard entropy change (ΔS°), and the standard Gibbs free energy (ΔG°) for most stable PIC complexes with β-CD are summarized in Table 2.Table 2 shows that the enthalpy values are negative, indicating that the complexation reactions of PIC with β-CD are exothermic. The enthalpy change for the A-orientation is less than that of the B-orientation; this difference could be due to stronger van der Waals interactions, and it means that the inclusion process is enthalpically favorable. The negative entropy change means that the complexation of PIC with β-CD is spontaneous [30]. These theoretical results are in good agreement with those obtained by the experimental investigation of PIC/β-CD complexation in solution, by reversed phase high performance liquid chromatography [21].
Thermodynamic parameters of PIC and its inclusion complexes by the PM3 method.
| Properties (kcal mol−1) | PIC | β-CD | ‘A’ orientation | ‘B’ orientation |
| H | 40.651 | −666.319 | −466.543 | −634.430 |
| ΔH°a | −159,125 | −8762 | ||
| G | 1.392 | −784.919 | −784.916 | −778.335 |
| ΔG°a | −1.389 | −5192 | ||
| S | 131.678 | 397.788 | 268.429 | 392.689 |
| ΔS°b | −261,037 | −136,777 |
a ΔA°=Acomplex − (Aβ-CD + APIC), A = H, G.
b ΔS°= (ΔH − ΔG)/T, at P = 1 atm and T = 298.15 K.
3.3 Frontier molecular orbitals analysis
The Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO) each play an important role in the optical and electric properties, kinetic stability and hardness of molecules [31]. The HOMO represents the ability of a molecule to donate an electron, while the LUMO, as an electron acceptor, represents the ability to accept an electron. In simple molecular orbital theory approaches, the HOMO energy (EHOMO) is related to the Ionization Potential (IP) by Koopman's theorem, and the LUMO energy (ELUMO) has been used to estimate the electron affinity (EA). Both EHOMO and ELUMO are used for calculating the electronic chemical potential (μ), which is half of the energy of the HOMO and LUMO. The average value of the HOMO and LUMO energy is related to the electronegativity (χ) defined by Mulliken [32]:
| (4) |
| (5) |
Calculations showed that the HOMO and LUMO energy levels for the complexes are lower than those for free PIC (Fig. 5). HOMO–LUMO values are almost the same for both orientations.
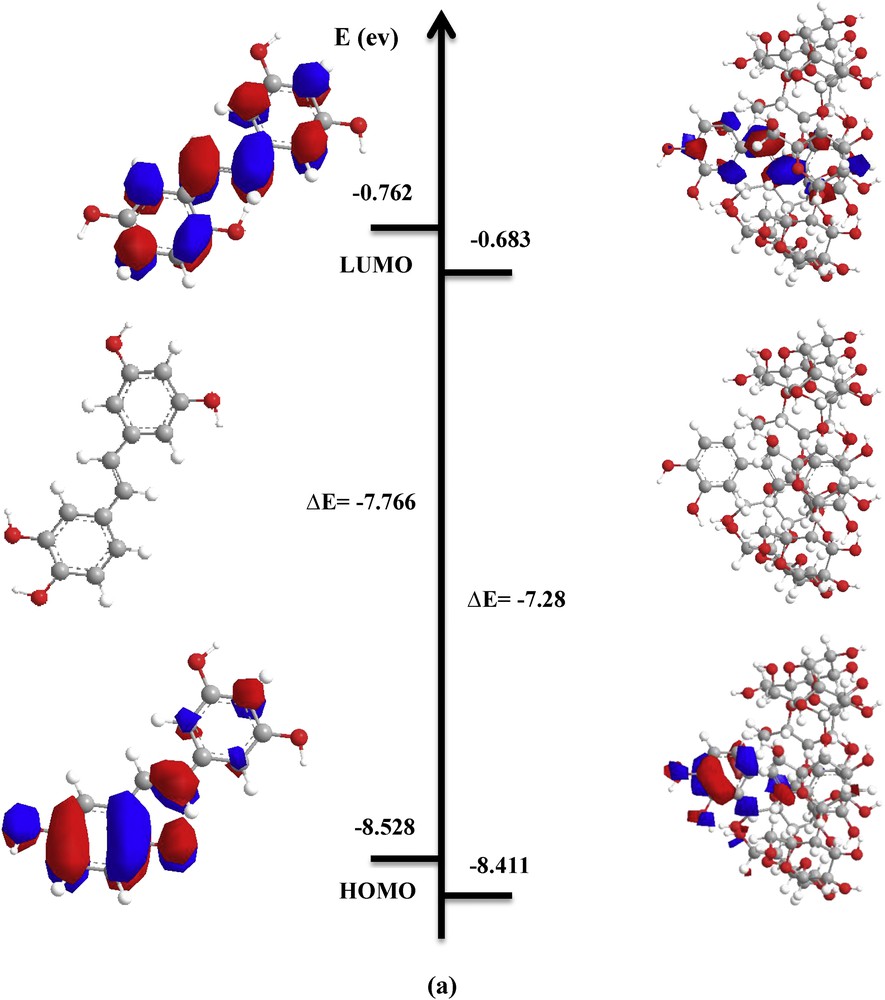
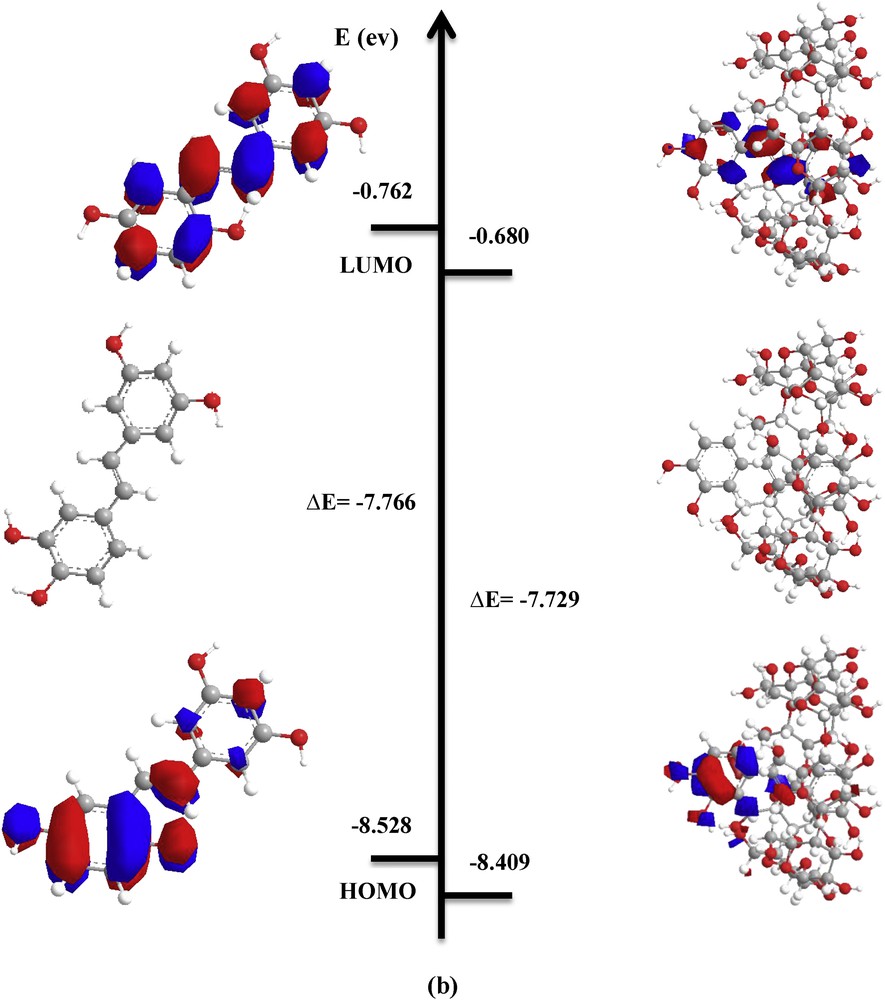
PM3 optimized structures and HOMO and LUMO orbitals' density distribution for free and complexed PIC in vacuum: a) ‘A’ orientation; b) ‘B’ orientation.
The energy gap is the difference between the HOMO and LUMO energies, and the hardness (η) is the half of the energy gap between HOMO and LUMO energies. The gap and η were calculated using the following expressions [33]:
| (6) |
| (7) |
The electrophilicity (ω), which is the measure of the electrophilic power of the drug molecule and its complexes, was calculated using Eq. 8 below:
| (8) |
Global softness (S) is a property of molecules that measures the extent of chemical reactivity. It is the reciprocal of hardness [34] and can be defined as:
| (9) |
As shown in the results in Table 3, the energy gap (EHOMO − ELUMO), which is an important scale of stability and chemical activity, is almost the same for both orientations. This result is in good agreement with the complexation energy.
Frontier molecular orbitals and electronic parameters of PIC and its inclusion complexes for the two orientations (A and B) by the PM3 method.
| Energetic terms (eV) | PIC | β-CD | ‘A’ orientation | ‘B’ orientation |
| EHOMOa | −8.528 | −10.70 | −8.411 | −8.409 |
| ELUMOb | −0.762 | 1.56 | −0.683 | −0.680 |
| EHOMO − ELUMO | −7.766 | −12.26 | −7.728 | −7.029 |
| EHOMO + ELUMO | −9.290 | −9.14 | −9.094 | −9.089 |
| μc | −4.645 | −4.57 | −4.547 | −4.544 |
| χd | 4.645 | 4.57 | 4.547 | 4.544 |
| ηe | 3.833 | 6.13 | 3.864 | 3.864 |
| ωf | 2.8145 | 1.7034 | 2.6753 | 2.6781 |
| Si | 0.2608 | 0.1631 | 0.2587 | 0.2587 |
| Dipoleh (D) | 1.85 | 4.57 | 4.49 | 4.32 |
| Δ Dipoleg (D) | – | – | −1.93 | −2.1 |
| Mullikan charges | 0.00 | 0.00 | 0.00 | 0.00 |
a Energy of the Highest Occupied Molecular Orbital.
b Energy of the Lowest Unoccupied Molecular Orbital.
c Electronic chemical potential.
d Electronegativity.
e Chemical hardness.
f Global electrophilicity index.
g Δ Dipole moment = Dipole momentcomplex − (Dipole momentPIC + Dipole momentβ-CD).
h Dipole moment in Debye.
i Softness.
The HOMO energy of the guest molecule is related to the strength of the charge transfer interactions during complexation. Fig. 5, which shows the density charge distribution of HOMO and LUMO orbitals for free PIC and the most stable PIC:β-CD complex in A-orientation demonstrates that both HOMO and LUMO orbitals characterizing the complex are concentrated only on the guest molecule.
As shown in Table 3, the values of electronic chemical potential (μ), hardness (η), electronegativity (χ), electrophilicity (ω) and softness (S) for complexes differ from the individual host and guest molecules.
- (i) The values of the electronic chemical potential (μ) of both inclusion complexes are negative and of approximately the same magnitude; this result indicates that the inclusion process is spontaneous [35,36].
- (ii) The hardness (η) is nearly identical for both orientations. Zhou et al. [37] reported that the stability of molecules is related to their chemical hardness. This one value denotes the resistance of the system to charge transfer, which is less likely to occur for the higher values of η.
- (iii) The values of ω are an important index of electrophilicity; the greater the values of ω, the more electrophilic the compound is. The results reported in Table 3 show that free PIC is the most electrophilic compound in this study and that the complexes PIC:β-CD have almost the same value of ω for both orientations.
The dipole moment of a molecule is primarily used to study the intermolecular interactions involving non-bonded type dipole–dipole interactions because higher dipole moment values indicate stronger intermolecular interactions.
The dipole moments of free host and guest molecules and inclusion complexes can be arranged in the following order: β-CD > PIC:β-CD (A) > PIC:β-CD (B) > PIC. The inclusion complexes showed higher dipole moment values than those for the corresponding isolated guest molecule, but lower than those for β-CD. This result indicates that the polarity of the β-CD cavity changed after the entrance of the drug, which indicates that the dipole moment of the complex is closely related to the polarity of the guest molecules (Table 3).
3.4 Structural parameters
The structural parameters of the guest molecule before and after complexation in β-CD obtained from the quantum mechanical PM3 calculations in a vacuum are reported in Table 4. The results indicate that the PIC in the complexes has completely altered its initial geometry.
Geometrical parameters of PIC before and after inclusion in β-CD: bond distances (Å), bond angles (°) and dihedral angles (°), calculated by the PM3 method.
| Parameters | Free PIC | PIC/β-CD ‘A’ orientation | PIC/β-CD ‘B’ orientation |
| Bond length (Å) | |||
| H(177)O(165) | 0.97 | 0.97 | 0.96 |
| H(176)O(164) | 0.97 | 0.97 | 0.97 |
| H(175)C(163) | 1.09 | 1.08 | 1.09 |
| H(174)C(160) | 0.97 | 1.10 | 1.10 |
| H(172)C(157) | 1.35 | 1.09 | 1.09 |
| H(170)O(155) | 1.33 | 0.97 | 0.97 |
| H(169)O(154) | 1.33 | 0.97 | 0.97 |
| O(164)C(162) | 1.10 | 1.35 | 1.35 |
| O(155)C(150) | 1.35 | 1.36 | 1.35 |
| C(163)C(162) | 1.10 | 1.34 | 1.33 |
| C(162)C(161) | 1.10 | 1.33 | 1.33 |
| C(157)C(156) | 1.33 | 1.34 | 1.33 |
| Bond angle (°) | |||
| H(177)O(165)C(161) | 120.01 | 120.00 | 119.99 |
| H(176)O(164)C(162) | 120.00 | 119.99 | 119.98 |
| H(174)C(160)C(161) | 119.99 | 119.98 | 119.95 |
| H(172)C(157)C(156) | 119.99 | 120.00 | 120.01 |
| H(169)O(154)C(148) | 120.50 | 120.36 | 120.52 |
| O(164)C(162)C(161) | 120.84 | 120.34 | 120.02 |
| O(165)C(161)C(162) | 119.98 | 119.99 | 120.00 |
| C(162)C(163)C(158) | 120.00 | 119.97 | 119.98 |
| C(162)C(161)C(160) | 120.00 | 120.00 | 119.99 |
| C(157)C(156)C(152) | 120.96 | 120.78 | 120.45 |
| C(152)C(153)C(148) | 119.52 | 119.20 | 119.56 |
| Dihedral angle (°) | |||
| H(176)O(164)C(162)C(161) | 179.98 | 179.99 | 179.99 |
| H(176)O(164)C(162)C(163) | −0.0067 | 0.0034 | 0.0027 |
| O(164)C(162)C(161)C(160) | −179.51 | −179.39 | −179.15 |
| C(158)C(163)C(162)C(161) | 179.81 | 0.0031 | −0.0050 |
| C(158)C(163)C(162)O(164) | 0.0080 | 179.56 | 179.18 |
| C(163)C(162)C(161)C(160) | −0.0049 | −0.0013 | 0.0053 |
This alteration is demonstrated by the difference between the values of the bond lengths, the torsion and dihedral angles. A significant distortion in dihedral angles compared to other parameters was found. This result indicates that PIC adopts a specific conformation inside the host cavity to form a more stable inclusion complex in a vacuum [38].
3.5 FT-IR spectra
The possible interaction between PIC and β-CD in solid state was investigated by Fourier-transform infrared (FT-IR) spectroscopy.
In Fig. 6, we summarize the computed infrared spectra of PIC/β-CD, complex A and complex B obtained by PM3 calculation.
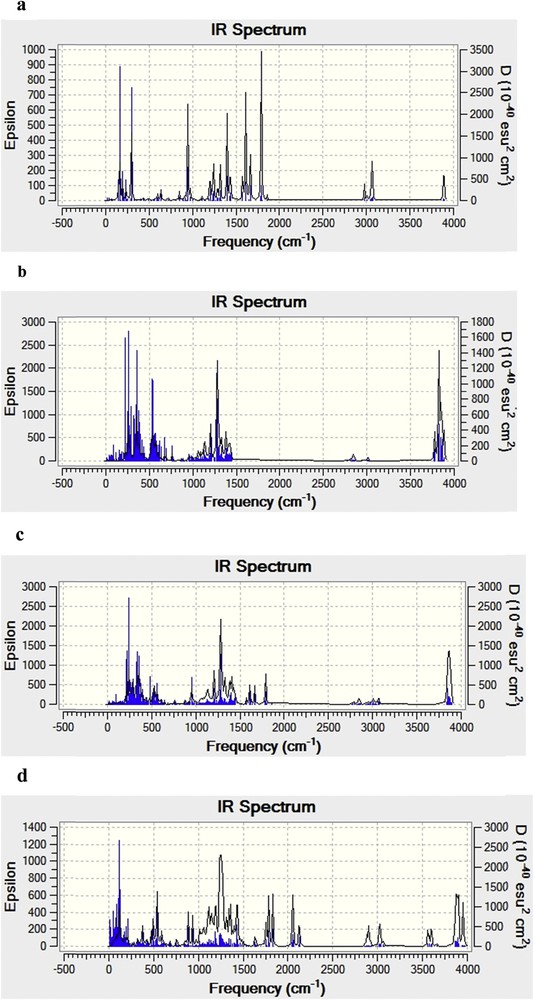
Calculated IR spectra of free PIC (a) and β-CD (b), ‘A’ orientation complex (c) and ‘B’ orientation complex (d) by the PM3 calculations.
The experimental [39,40] and theoretical intensities of selected bands of PIC and PIC/β-CD at PM3 calculations are given in Table 5. The calculated intensity of the bands is higher than their corresponding experimental values; this difference could result from the fact that the experimental values are anharmonic while the calculated values are harmonic ones [38]. Additionally, the spectrum of the inclusion complex was equivalent to the spectrum of the simple combination of piceatannol and β-CD, and some characteristic absorption peaks of piceatannol at 3300, 1625, 1600, 1515, 1295, 1145 and 960 cm−1 were easily observed (Fig. 6), suggesting that the natural structure of piceatannol still existed without any interactions with β-CD. These results indicate that a piceatannol/β-CD inclusion complex was obtained [41], but the intrinsic properties of both the host and guest did not undergo any obvious changes [42].
Experimental [39,40] and calculated (using the PM3 method) IR frequencies of free PIC and the PIC/β-CD complex.
| Bands | Experimental frequencies | Calculated frequencies | ||
| Free PIC | PIC/β-CD | Free PIC | PIC/β-CD | |
| OH | 3300 | 3390.47 | 3885.08 | 3892.35 |
| CC bond stretching | 1625 | 1650.09 | 1794.09 | 1796.04 |
| CC olefinic stretching Aromatic CC | 1600 1515 | 1640 1530 | 1614.18 1575.27 | 1664.73 |
| CO stretching | 1295 1145 | 1311.37 1160 | 1290.45 1200.84 | 1331.73 1200 |
| trans Olefinic | 960 | 965 | 945.71 | 950.20 |
4 Conclusions
In this study, the inclusive complexation of piceatannol with β-CD was investigated using the PM3 method. Two possible orientations for the inclusion of PIC in β-CD were considered, and the results suggest that the ‘A’ orientation is slightly more favorable than the ‘B’ orientation, by an energy difference of −0.90 kcal/mol. Thermodynamic parameters (ΔG°, ΔH°, ΔS°) and HOMO and LUMO orbital calculations confirm the stability of the inclusion complex. Additionally, during the inclusion process, β-CD is deformed, facilitating the accommodation of the guest molecule in its cavity. The energy gap (EHOMO − ELUMO) is approximately the same for both orientations. This result is in good agreement with the complexation energy. The values of the electronic chemical potential (μ), hardness (η), electronegativity (χ), electrophilicity (ω) and softness (S) for complexes differ from those for the individual host and guest molecules, but are approximately the same for both orientations.
The complexation reactions of PIC with β-CD are exothermic and spontaneous. Moreover, the results of the calculations for the PIC/β-CD complex obtained in this study are in accordance with the experimental study of the piceatannol/β-CD complex using RP-HPLC, which required a mobile phase composed of a mixture of methanol and water in order to achieve a reasonable retention time. Additionally, in the experimental study we have shown that the apparent formation constants (K) for the piceatannol/β-CD complex were strongly dependent on both the concentration of methanol and the temperature. The stoichiometry of the complex piceatannol/β-CD was found to be 1:1 for all conditions studied. Piceatannol/β-CD complex applications include its use as an ingredient in pharmaceutical and food industries thanks to its improved water-solubility. Additionally, the piceatannol/β-CD complex could have an impact on the pharmacokinetics and pharmacodynamics of the drug by slowing down its rapid metabolism and elimination.
Acknowledgements
This study was supported by the Algerian Ministry of Higher Education and Scientific Research through project CNEPRU (N°E01520120010).