

1 Introduction
Deprotonation next to carbonyl groups gives rise to enolates that are one of the most extensively used carbon-centred nucleophiles in synthetic chemistry [1]. Enolates are planar species, so if the proton to be removed is at a stereogenic centre then this stereochemistry present in the original carbonyl compound will be lost. The same scenario is possible with nitriles 1 if, on deprotonation, they form metallated ketene-imine-type structures such as compound 2 (Fig. 1). This is believed to occur in many cases, particularly with lithium as the counterion, where the lithium typically coordinates to the nitrogen atom of the nitrile, although the CN bond is thought to maintain considerable triple bond character [2]. Indeed there is X-ray crystallographic evidence for this structure on lithiation of phenylacetonitrile [3]. However, examples of metallated nitriles in which the metal resides on the carbon atom (structure 3) are known, even for lithium but especially for softer metals such as palladium or ruthenium [4,5]. Therefore, there exists the possibility that nitriles could maintain their configuration on metallation, should structures of type 3 be formed directly from the chiral nitrile 1 and if this metallated species does not racemise rapidly.
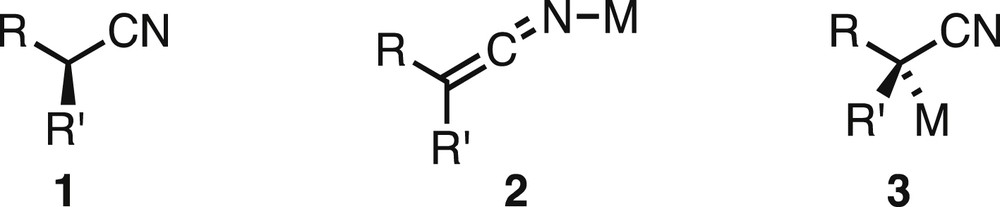
A generic chiral nitrile and possible metallated structures (M = metal).
The first example of such chemistry was reported by Carlier and co-workers [6,7]. They showed that the chiral cyclopropylnitrile 4 (Fig. 2) could be formed by bromine–magnesium exchange with i-PrMgCl and reacts with D2O to give high enantiomer ratio (er) values of the deuterated product. Fleming and co-workers [8] have found very different selectivities between lithiated and magnesiated nitriles and that magnesium counterions favour attachment to the carbon atom of the nitrile. Therefore, we were interested in whether it might be possible to take an acyclic chiral nitrile and carry out enantiospecific metallation and then substitution, particularly using a base centred on the metal magnesium. We reported our preliminary findings in this area recently in which the chiral nitriles 5 and 6 successfully undergo such chemistry [9]. This work originated from the observation by Takeda et al. [10] that low er values were possible by treatment of the nitrile 6 with lithium diisopropylamide (LDA) and in situ benzyl bromide. Since then Takeda and co-workers [11] reported much improved selectivities using more reactive electrophiles (in situ quench with acid chlorides or ethyl cyanoformate) with the nitriles 5 and 6, with LDA as the base. Herein, we describe further results with the nitrile 6 and with related compounds that demonstrate the importance of the carbamate group. With the base TMPMgCl, we show that high er values of new substituted products are possible after quenching with different electrophiles [12].

Chiral nitriles 4–6.
2 Results and discussion
The nitrile 6 was prepared by a method reported by Takeda et al. [10]. This involved conversion of the commercially available aldehyde 7 to the cyanohydrin 8, followed by acylation to give the ester 9 (Scheme 1). Treatment of the ester 9 with Amano lipase from pseudomonas fluorescens (Amano lipase PS) effected a kinetic resolution to give the desired alcohol 8 with a high er [13]. The data matched those reported for the (S) enantiomer [10]. Conversion of the alcohol 8 to the carbamate 6 was carried out with triphosgene and diisopropylamine. The er of the product 6 was verified by chiral stationary phase high performance liquid chromatography (CSP-HPLC) (er 99:1). In addition, alcohol 8 was converted, in unoptimised yields, to the novel compounds 10 and 11 using dimethoxymethane and pivaloyl chloride, respectively. These compounds were used to probe the importance of the carbamate-protecting group on the oxygen atom.
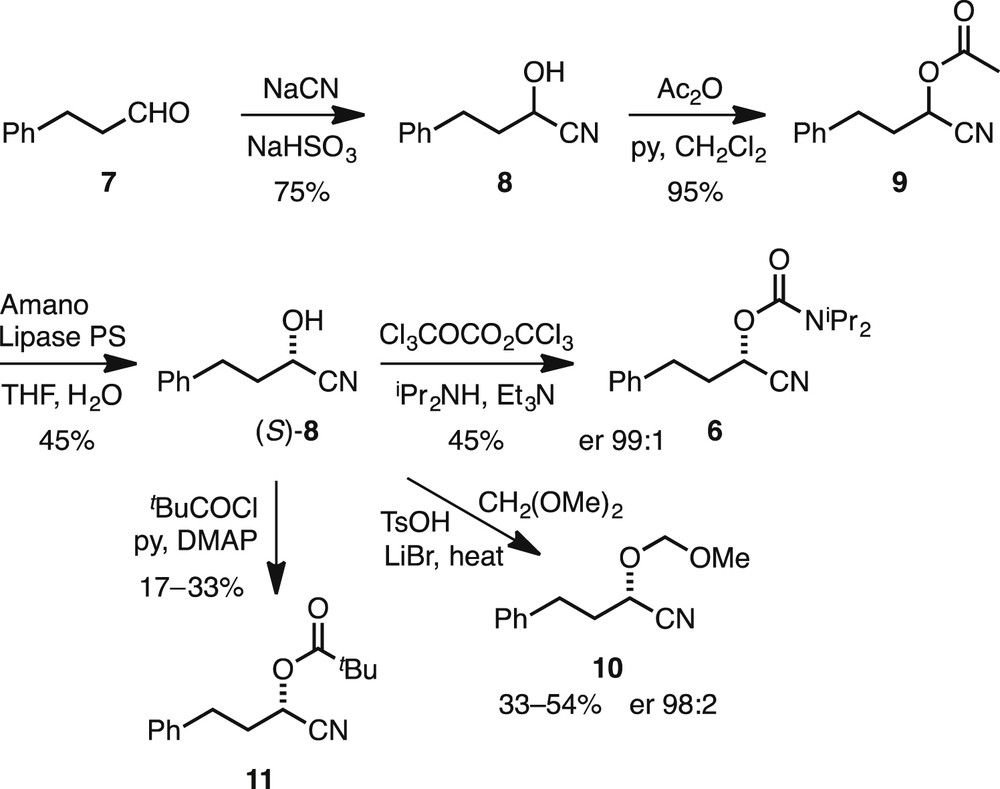
Preparation of chiral nitriles 9–11.
The key chemistry of interest is whether the compounds 6, 10 and 11 will undergo metallation and then substitution and to what extent, if at all, the enantiopurity will be transferred to the product. We anticipated that the magnesium base TMPMgCl would be better than group 1 bases such as LDA. However, initially we confirmed whether metallation was successful.
Treatment of the racemic ether 10 with LDA in tetrahydrofuran (THF) at −78 °C followed by addition of acetone or methyl cyanoformate as the electrophile gave the expected products 12 and 13 in reasonable yields (Scheme 2). The enantiomers of these products could be resolved by CSP-HPLC. The best conditions reported previously for the enantiospecific metallation–quench of 5 or 6 made use of the magnesium base TMPMgCl at −107 °C [9]. However, attempts to conduct the metallation of the ether 10 with TMPMgCl were unsuccessful, and after addition of the electrophile (or by using in situ MeOCOCN) only starting material 10 was recovered. It was possible to carry out the lithiation–quench of ether (S)-10 with in situ MeOCOCN using LDA at −107 °C to give the product 13 (34% yield). Unfortunately, this product was essentially racemic (er 52:48 by CSP-HPLC).

Metallation of nitrile 10.
We then turned our attention to the ester 11. This compound has a carbonyl group that could help to stabilise the metallated intermediate, although it would not be as good a coordinating group as the carbamate carbonyl in 5 or 6. Attempted deprotonation adjacent to the nitrile in racemic 11 with LDA followed by addition of MeOCOCl, MeOCOCN or acetone failed to give any of the desired substituted products. However, addition of TMPMgCl in THF–Et2O at −107 °C and then MeOCOCN was successful and gave the nitrile product 14 in 90% yield. The enantiomers of this product could be resolved by CSP-HPLC. Therefore, we screened enantioenriched nitrile 11 with TMPMgCl at −107 °C, followed after 10 min by addition of MeOCOCN (Scheme 3). This gave the desired product 14 in high yield but as a racemic mixture.

Metallation of nitrile 11.
We then turned our attention to the carbamate 6, which is able to undergo the desired transformation with high er by using TMPMgCl and electrophilic quench with acetone, MeOCOCN or BnOCOCN [9]. To extend this study, we screened a variety of bases and some new electrophiles to explore whether TMPMgCl was the base of choice and to increase the scope of this chemistry.
The base screening was carried out with acetone as the electrophile and the nitrile (S)-6 to give the substituted product 15 (Scheme 4). The results to show the comparison of the different bases in regard to the yield of the isolated product 15 are shown in Table 1. We were concerned about optimising both the yield of the product 15 and its er. In each case the major enantiomer is that shown in Scheme 4, in which the reaction takes place with retention of configuration, as shown by preparation of the p-bromophenyl derivative and subsequent X-ray analysis [9].

Metallation of nitrile 6 and quench with acetone.
Effect of different bases and conditions for formation of product 15.
| Entry | Base | Solvent | Method | t (min) | Yield (%) 15 | er (S:R) |
| 1 | 1.2 equiv i-PrMgCl | Et2O | Normal | 10 | 44 | 82:18 |
| 2 | 1.2 equiv i-PrMgCl | Et2O | Inverse | 10 | 30 | 87:13 |
| 3 | 4 equiv i-PrMgCl | Et2O | Inverse | 10 | 45 | 91:9 |
| 4 | 4 equiv i-PrMgCl | Et2O | Inverse | In situ | 24 | 87:13 |
| 5 | 4 equiv i-PrMgCl | t-BuOMe/Et2O | Inverse | 10 | 50 | 71:29 |
| 6 | 4 equiv i-PrMgCl | THF–Et2O (1:1) | Inverse | 10 | 24 | 89:11 |
| 7 | 4 equiv n-Bu2Mg | Et2O | Inverse | 10 | 45 | 80:20 |
| 8 | 5 equiv TMPMgCl·LiCl | Et2O | Inverse | 10 | 73 | 70:30 |
| 9 | 4 equiv TMPMgCl | Et2O | Inverse | 2 | 55 | 85:15 |
| 10 | 3 equiv TMPMgCl | CPMEa/Et2O | Inverse | In situ | 79 | 60:40 |
| 11 | 4 equiv TMPMgCl | Et2O | Inverse (4 min) | 2 | 50 | 87:13 |
| 12 | 3 equiv TMPMgCl | Et2O | Inverse (4 min) | 0.1 | 48 | 86:14 |
a CPME = cyclopentyl methyl ether.
Initially we investigated the base i-PrMgCl, which was effective for the transformation (Table 1, entries 1–6). Using inverse addition, whereby the nitrile (S)-6 was added to the base gave similar results to normal addition of base to the nitrile, but perhaps with slightly improved enantioselectivity (compare entries 1 and 2). We therefore opted to continue further experiments with inverse addition. The yield was improved with excess base (compare entries 2 and 3). Remarkably, even using in situ acetone that we expected would simply form its enolate did in fact give some product (entry 4), although disappointingly the er was not improved. It therefore appears that there is a rapid partial loss of enantiopurity on metallation, although subsequently the magnesiated intermediate has reasonable configurational stability (for several minutes) at −107 °C. Two other solvents were tested but t-BuOMe gave reduced er (entry 5) and the THF–Et2O mixture gave reduced yield (entry 6). We then tried some other bases and found that Bu2Mg was effective and gave similar results to i-PrMgCl (entry 7). More promising in terms of the yield was the use of TMPMgCl·LiCl; however, the er was poorer (entry 8). This may be because of the presence of lithium cations that could coordinate to the nitrogen atom of the nitrile and start to favour a ketene-imine–metallated species. The best results were obtained by using TMPMgCl in the absence of LiCl. This base can be prepared readily from 2,2,6,6-tetramethylpiperidine (TMPH) and i-PrMgCl in THF or in Et2O [14]. Similar results were obtained by using an excess of this base in Et2O using inverse addition, either by rapid addition of nitrile (S)-6 (entry 9) or by addition of (S)-6 more slowly for more than about 4 min (entries 11 and 12). An attempt to use cyclopentyl methyl ether as the main solvent gave a good yield but poor selectivity (entry 10).
Under the optimised conditions, we selected to add the nitrile (S)-6 slowly to 3 equiv of TMPMgCl in Et2O at −107 °C, followed by addition of the electrophile. Reasonable yields and er values were obtained on using alkyl cyanoformates as the electrophile to give the products 16a–c (Scheme 5). We also studied some new electrophiles. Benzoyl chloride gave the product 17 with reduced er. This may be because of slower reaction with the acid chloride that allows partial racemisation of the intermediate organomagnesium species, or possible reaction by a mixture of retention and inversion of configuration. However, we were pleased to find that the ketone cyclopentanone was successful to give the product 18 with high enantioselectivity (er 92:8). In addition, the electrophile benzaldehyde gave, as an inseparable mixture, the enantioenriched diastereomeric products 19a and 19b. We have determined that the absolute configuration of the product 15 demonstrated that reaction with acetone occurs with retention of configuration [9]. However, we have not determined the stereochemistry of the major enantiomers of the products 16–19. It is possible that these products are formed after reaction with retention of configuration, particularly using cyclopentanone that is similar to acetone. Despite this, reactions of metallated nitrile 5 with ethyl cyanoformate and with benzoyl chloride are known to occur with inversion of configuration [9,11a]. Regardless of the absolute configurations, the reactions occur with high enantioselectivity for a selection of electrophiles, as illustrated in Scheme 5.
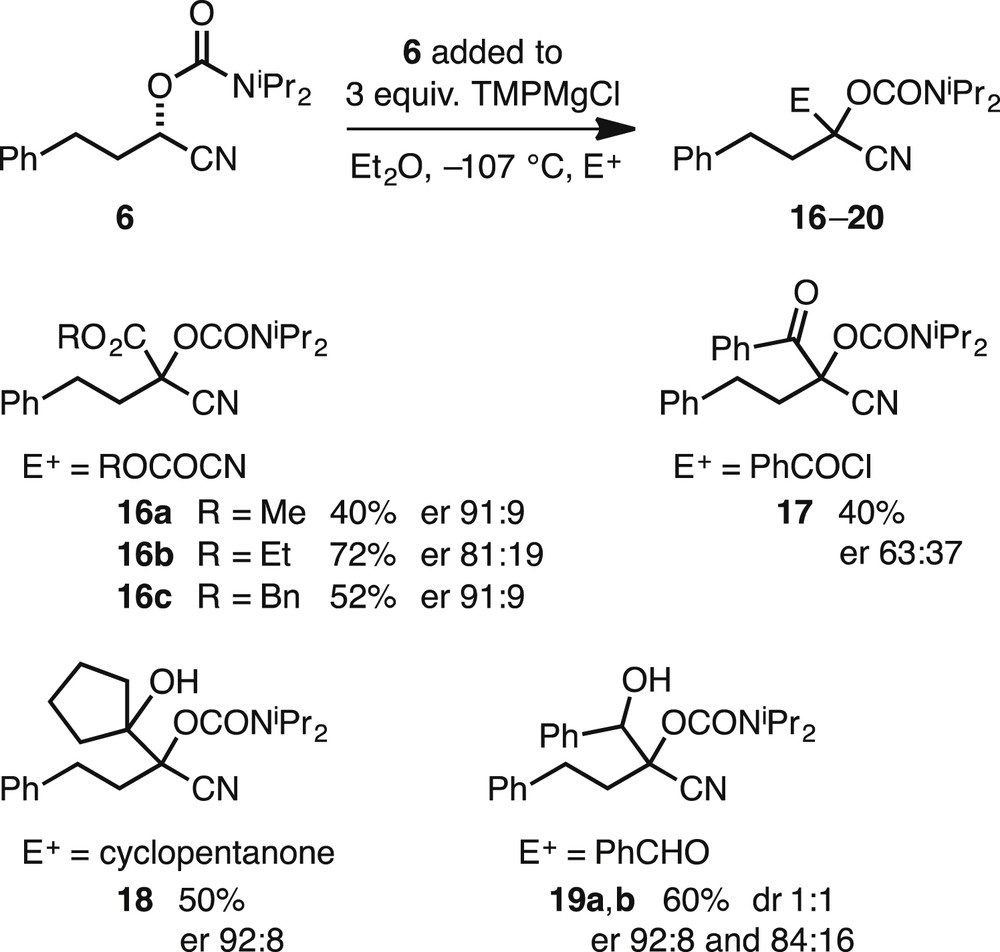
Metallation of nitrile (S)-6 and quench with various electrophiles E+.
3 Conclusions
The metallation of chiral nitriles with magnesium bases such as TMPMgCl at low temperature occurs with significant retention of enantiopurity. The magnesiated intermediates can be quenched with a variety of electrophiles. Better results were found with the carbamate 6 than the ether 10 or ester 11. This is likely because of the better coordinating ability of a carbamate that can stabilise the magnesiated intermediate through chelation of the carbonyl oxygen atom with the magnesium. This will then reduce the extent of loss of the metal from the stereocentre and help to retain high levels of enantiopurity in the substituted product after electrophilic quench.
4 Experimental section
Procedures and data for compounds 6–9 have been reported [9,11a].
4.1 2-(Methoxymethoxy)-4-phenylbutanenitrile (10)
To a solution of alcohol (±)-8 (1.0 g, 6.2 mmol) in dimethoxymethane (12 mL) was added LiBr (285 mg, 3.3 mmol) and p-TsOH monohydrate (118 mg, 0.62 mmol), and the mixture was heated under reflux. After 3 d, the mixture was cooled to room temperature and the solvent was evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (9:1) gave the nitrile 10 (686 mg, 54%) as an oil; Rƒ 0.2 [petrol–Et2O (9:1)]; νmax (neat)/cm−1 2955, 2900, 1455, 1150, 1105, 1090, 1020; 1H NMR (400 MHz, CDCl3) δ = 7.39–7.20 (m, 5H, Ph), 4.87 (d, 1H, J = 7 Hz, CH), 4.69 (d, 1H, J = 7 Hz, CH), 4.35 (t, 1H, J = 7.5 Hz, CH), 3.45 (s, 3H, CH3), 2.87 (t, 2H, J = 7.5 Hz, CH2), 2.30–2.19 (m, 2H, CH2); 13C NMR (100 MHz, CDCl3) δ = 139.7, 128.7, 128.4, 126.5, 118.3, 95.8, 64.2, 56.3, 35.1, 30.9; HRMS (ES) found: M+, 205.1112. C12H15NO2 requires M+ 205.1103.
The enantiomers were resolved by CSP-HPLC using a Cellulose-2 column at 1 mL min−1 , ambient temperature and detection by UV absorbance at 254 nm. Injection volume (20 μL) of the sample was prepared in a 2 g L−1 solution of 0.5% i-PrOH in hexanes. Retention times were 14.5 and 15.4 min.
In the same way as mentioned previously, the alcohol (S)-8 (4.1 g, 25.5 mmol), dimethoxymethane (50 mL), LiBr (1.22 g, 14.1 mmol) and p-TsOH monohydrate (507 mg, 2.67 mmol) gave, after purification by column chromatography on silica and eluting with petrol–Et2O (9:1), the nitrile (S)-10 (1.8 g, 33%) as an oil; [α]D21 −57 (c 1.0, CHCl3); other data are as mentioned previously; er 98:2 (major peak was at 14.5 min).
4.2 1-Cyano-3-phenylpropyl 2,2-dimethylpropanoate (11)
To a solution of alcohol (±)-8 (2.0 g, 12.4 mmol) and DMAP (25 mgand 4-dimethylaminopyridine (DMAP) (25 mg, 2 mmol) in pyridine (126 mL) was added pivaloyl chloride (1.67 mL, 13.7 mmol) at room temperature. After 12 h, the solvent was evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (9:1) gave the nitrile 11 (1.0 g, 33%) as an oil; Rƒ 0.8 [petrol–Et2O (9:1)]; νmax (neat)/cm−1 2975, 1740, 1500, 1480, 1455, 1275, 1130, 1035; 1H NMR (400 MHz, CDCl3) δ = 7.39–7.17 (m, 5H, Ph), 5.28 (t, 1H, J = 7 Hz, CH), 2.85 (t, 2H, J = 8 Hz, CH2), 2.32–2.20 (m, 2H, CH2), 1.27 (s, 9H, t-Bu); 13C NMR (100 MHz, CDCl3) δ = 176.5, 139.1, 128.8, 128.4, 126.7, 116.9, 60.5, 38.8, 33.9, 30.8, 26.9; HRMS (ES) found: M+, 245.1408. C15H19NO2 requires M+ 245.1416.
In the same way as mentioned above, the alcohol (S)-8 (1.0 g, 6.2 mmol), DMAP (13 mg, 1.0 mmol), pyridine (64 mL) and pivaloyl chloride (0.83 mL, 6.8 mmol) gave, after purification by column chromatography on silica and eluting with petrol–Et2O (9:1), the nitrile (S)-11 (0.26 g, 17%) as an oil; [α]D21 −40 (c 1.0, CHCl3); other data are as mentioned above.
4.3 3-Hydroxy-2-(methoxymethoxy)-3-methyl-2-(2-phenylethyl)butanenitrile (12)
n-BuLi (254 μL, 0.63 mmol, 2.5 M in hexanes) was added to i-Pr2NH (83 μL, 0.63 mmol) in THF (2 mL) at −78 °C. After 10 min, nitrile (±)-10 (92 mg, 0.45 mmol) in THF (1 mL) was added. Then, after 10 min, acetone (72 μL, 0.97 mmol) was added. Next, after 30 min, the mixture was warmed to room temperature and saturated NH4Cl(aq) (4 mL) was added. The mixture was extracted with Et2O (3 × 10 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (95:5) gave the nitrile 12 (90 mg, 70%) as an oil; Rƒ 0.2 [petrol–Et2O (95:5)]; νmax (neat)/cm−1 3055, 2985, 1420, 1265, 1015; 1H NMR (400 MHz, CDCl3) δ = 7.37–7.15 (m, 5H, Ph), 5.18 (d, 1H, J = 7.5 Hz, CH), 4.94 (d, 1H, J = 7.5 Hz, CH), 3.79 (br s, 1H, OH), 3.55 (s, 3H, CH3), 2.99–2.82 (m, 2H, CH2), 2.10–1.96 (m, 2H, CH2), 1.37 (s, 3H, CH3), 1.29 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ = 140.8, 128.6, 128.5, 126.3, 117.3, 95.7, 86.6, 74.4, 56.7, 37.7, 31.3, 25.7, 23.8; HRMS (ES) found: MH+, 264.1589. C15H22NO3 requires MH+ 264.1600.
The enantiomers were resolved by CSP-HPLC using a Cellulose-1 column at 1 mL min−1, ambient temperature and detection by UV absorbance at 254 nm. Injection volume (20 μL) of the sample was prepared in a 2 g L−1 solution of 2% i-PrOH in hexanes. Retention times were 19.6 and 22.2 min.
4.4 Methyl 2-cyano-2-(methoxymethoxy)-4-phenylbutanoate (13)
In the same way as nitrile 12, n-BuLi (254 μL, 0.63 mmol, 2.5 M in hexanes), i-Pr2NH (83 μL, 0.63 mmol), nitrile 10 (100 mg, 0.49 mmol) and MeOCOCN (77 μL, 0.97 mmol) gave, after purification by column chromatography on silica and eluting with petrol–Et2O (9:1), the nitrile 13 (84 mg, 66%) as an oil; Rƒ 0.1 [petrol–Et2O (9:1)]; νmax (neat)/cm−1 3055, 2985, 1760, 1420, 1265, 1160, 1105, 1050; 1H NMR (400 MHz, CDCl3) δ = 7.37–7.18 (m, 5H, Ph), 5.10 (d, 1H, J = 7 Hz, CH), 4.83 (d, 1H, J = 7 Hz, CH), 3.81 (s, 3H, CH3), 3.43 (s, 3H, CH3), 3.08–2.72 (m, 2H, CH2), 2.46–2.28 (m, 2H, CH2); 13C NMR (100 MHz, CDCl3) δ = 167.1, 139.4, 128.7, 128.6, 128.5, 126.7, 126.5, 115.6, 95.5, 76.4, 57.2, 53.8, 39.9, 30.0; HRMS (ES) found: M+, 263.1170. C14H17NO4 requires M+ 263.1158.
The enantiomers were resolved by CSP-HPLC using a Cellulose-1 column at 1 mL min−1, ambient temperature and detection by UV absorbance at 254 nm. Injection volume (20 μL) of the sample was prepared in a 2 g L−1 solution of 0.2% i-PrOH in hexanes. Retention times were 35.0 and 40.3 min.
The same reaction was conducted at −107 °C with nitrile 10 (100 mg, 0.49 mmol) to give the nitrile 13 (41 mg, 34%) as an oil; data are as given above; er 52:48.
4.5 1-Cyano-3-phenylpropyl 2,2-dimethylpropanoate (14)
The nitrile (±)-10 (42 mg, 0.17 mmol) in Et2O–THF (0.5 mL, 1:1) was added to TMPMgCl (1.56 mL, 0.51 mmol, 0.33 M in THF) in Et2O–THF (2.5 mL, 1:1) at −107 °C. After 10 min, MeOCOCN (41 μL, 0.51 mmol) was added. Then, after 10 min, the mixture was allowed to warm to room temperature and then saturated NH4Cl(aq) (4 mL) was added. The mixture was extracted with Et2O (3 × 10 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (95:5) gave the nitrile 14 (45 mg, 90%) as an oil; Rƒ 0.1 [petrol–Et2O (9:1)]; νmax (neat)/cm−1 2975, 2935, 1750, 1455, 1280, 1255, 1125, 1105, 1085, 1055, 1030; 1H NMR (400 MHz, CDCl3) δ = 7.38–7.19 (m, 5H, Ph), 3.87 (s, 3H, CH3), 3.00–2.87 (m, 2H, CH2), 2.50–2.39 (m, 2H, CH2), 1.32 (s, 9H, t-Bu); 13C NMR (100 MHz, CDCl3) δ = 176.5, 165.4, 138.9, 128.7, 128.4, 126.7, 115.1, 72.5, 54.0, 39.2, 38.3, 30.0, 26.7; HRMS (ES) found: MH+, 304.1536. C17H22NO4 requires MH+ 304.1549.
The enantiomers were resolved by CSP-HPLC using a Cellulose-1 column at 1 mL min−1, ambient temperature and detection by UV absorbance at 254 nm. Injection volume (20 μL) of the sample was prepared in a 2 g L−1 solution of 1% i-PrOH in hexanes. Retention times were 11.8 and 13.8 min.
In the same way as mentioned above, the nitrile (S)-10 (50 mg, 0.2 mmol), TMPMgCl (2.6 mL, 0.44 mmol, 0.17 M in THF) and MeOCOCN (38 μL, 0.48 mmol) gave, after purification by column chromatography on silica and eluting with petrol–Et2O (9:1), the nitrile 14 (57 mg, 92%) as an oil; data are as given above; er 51:49.
4.6 [1-Cyano-2-hydroxy-2-methyl-1-(2-phenylethyl)]propyl N,N-bis(propan-2-yl)carbamate (15) [9]
Method for Table 1, entry 10: a solution of nitrile (S)-6 (100 mg, 0.35 mmol) in dry Et2O (2 mL) was added dropwise for more than 4 min using a syringe pump to a solution of TMPMgCl (3.5 mL, 1.4 mmol, 0.4 M solution in Et2O) in dry Et2O (1 mL) at −107 °C. After 2 min, dry acetone (0.12 mL, 1.75 mmol) was added. Then, after 30 min, saturated NH4Cl(aq) (2 mL) was added and the mixture was allowed to warm to room temperature. The mixture was extracted with Et2O (3 × 5 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (9:1) gave the nitrile 15 (60 mg, 50%) as an oil; [α]D21 −8.0 (c 1.0, CHCl3); other data are as reported [9]; er 87:13 (major peak at 15 min) was determined by CSP-HPLC using a Cellulose-1 column at 1 mL min−1, ambient temperature and detection by UV absorbance at 254 nm. Injection volume (20 μL) of the sample was prepared in a 2 g L−1 solution of 1% i-PrOH in hexanes. Retention times were 15 and 17 min.
4.7 Methyl 2-{[bis(propan-2-yl)carbamoyl]oxy}-2-cyano-4-phenylbutanoate (16a) [9]
A solution of nitrile (S)-6 (50 mg, 0.17 mmol) and methyl cyanoformate (0.05 mL, 0.52 mmol) in dry Et2O–THF (1:1) (0.5 mL) was added dropwise for more than 4 min using a syringe pump to a solution of TMPMgCl (1.3 mL, 0.52 mmol, 0.4 M solution in Et2O–THF) in dry Et2O–THF (1:1) (2.5 mL) at −107 °C. After 30 min, saturated NH4Cl(aq) (2 mL) was added and the mixture was allowed to warm to room temperature. The mixture was extracted with Et2O (3 × 5 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (9:1) gave the ester 16a (25 mg, 40%) as needles; mp 70–73 °С; other data are as reported [9]; [α]D23 +3.0 (c 1.0, CHCl3); er 91:9 (major peak at 16.8 min) was determined by CSP-HPLC using a Cellulose-1 column at 1 mL min−1, ambient temperature and detection by UV absorbance at 254 nm. Injection volume (20 μL) of the sample was prepared in a 2 g L−1 solution of 1% i-PrOH in hexanes. Retention times were 16.8 and 20.9 min.
4.8 Ethyl 2-{[bis(propan-2-yl)carbamoyl]oxy}-2-cyano-4-phenylbutanoate (16b) [11a]
From racemic nitrile 6: n-butyllithium (0.4 mL, 0.95 mmol, 2.5 M solution in hexanes) was added to diisopropylamine (0.15 mL, 1.02 mmol) in Et2O (2.5 mL) at −78 °C. After 10 min, nitrile (±)-6 (50 mg, 0.17 mmol) in Et2O (0.5 mL) was added. Then, after 10 min, ethyl cyanoformate (0.10 mL, 1.02 mmol) was added. Next, after 30 min, the mixture was allowed to warm to room temperature and saturated NH4Cl(aq) (2 mL) was added. The mixture was extracted with Et2O (3 × 5 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (9:1) gave the ester 16b (50 mg, 82%) as an oil; 1H NMR (400 MHz, CDCl3) δ = 7.36–7.32 (m, 2H, Ph), 7.28–7.23 (m, 3H, Ph), 4.39–4.26 (m, 2H, CH2), 4.09–4.00 (m, 1H, CH), 3.80–3.73 (m, 1H, CH), 3.03–2.90 (m, 2H, CH2), 2.48–2.36 (m, 2H, CH2), 1.35 (t, 3H, J = 7 Hz, CH3), 1.32–1.25 (m, 12H, 4 × Me); 13C NMR (100 MHz, CDCl3) δ = 165.6, 152.7, 139.2, 128.7, 128.3, 126.6, 116.0, 73.5, 62.9, 47.4, 46.1, 38.6, 30.4, 21.6, 21.3, 20.3, 20.2, 13.9; HRMS (ES) found: MH+, 361.2132. C20H29N2O4 requires MH+, 361.2127; data are as reported [11a].
The enantiomers were resolved by CSP-HPLC using a CHIRALPAK® AD column at 1 mL min−1, ambient temperature and detection by UV absorbance at 254 nm. Injection volume (20 μL) of the sample was prepared in a 2 g L−1 solution of 1% i-PrOH in hexanes. Retention times were 8.1 and 9.1 min.
From nitrile (S)-6: the nitrile (S)-6 (100 mg, 0.35 mmol) and dry ethyl cyanoformate (0.10 mL, 1.02 mmol) in dry Et2O–THF (1:1) (0.5 mL) was added dropwise for more than 4 min to TMPMgCl (3.3 mL, 1.04 mmol, 0.4 M in THF) in dry Et2O–THF (1:1) (2.5 mL) at −107 °C. After 30 min, saturated NH4Cl(aq) (2 mL) was added and the mixture was allowed to warm to room temperature. The mixture was extracted with Et2O (3 × 5 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (9:1) gave the ester 16b (90 mg, 72%) as an oil; [α]D23 +27.0 (c 1.0, CHCl3); er 80:10 (major peak at 9.1 min) was determined by CSP-HPLC; other data are as mentioned above or as reported (no specific rotation data is given in the literature) [11a].
4.9 Benzyl 2-{[bis(propan-2-yl)carbamoyl]oxy}-2-cyano-4-phenylbutanoate (16c) [9]
A solution of nitrile (S)-6 (50 mg, 0.17 mmol) and benzyl cyanoformate (0.08 mL, 0.52 mmol) in dry Et2O–THF (1:1) (0.5 mL) was added dropwise for more than 4 min using a syringe pump to a solution of TMPMgCl (1.3 mL, 0.52 mmol, 0.4 M solution in Et2O–THF) in dry Et2O–THF (1:1) (2.5 mL) at −107 °C. After 30 min, saturated NH4Cl(aq) (2 mL) was added and the mixture was allowed to warm to room temperature. The mixture was extracted with Et2O (3 × 5 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (19:1) gave the ester 16a (40 mg, 52%) as an oil; data are as reported [9]; [α]D23 +12.0 (c 1.0, CHCl3); er 91:9 (major peak at 21.4 min) was determined by CSP-HPLC using a Cellulose-1 column at 1 mL min−1, ambient temperature and detection by UV absorbance at 254 nm. Injection volume (20 μL) of the sample was prepared in a 2 g L−1 solution of 1% i-PrOH in hexanes. Retention times were 21.4 and 26.9 min.
4.10 1-Benzoyl-1-cyano-3-phenylpropyl N,N-bis(propan-2-yl)carbamate (17)
From racemic nitrile 6: n-butyllithium (0.79 mL, 1.9 mmol, 2.5 M solution in hexanes) was added to diisopropylamine (0.29 mL, 2.08 mmol) in Et2O (2.5 mL) at −78 °C. After 10 min, nitrile (±)-6 (100 mg, 0.35 mmol) in Et2O (0.5 mL) was added. Then, after 10 min, benzoyl chloride (0.25 mL, 2.0 mmol) was added. Next, after 30 min, the mixture was allowed to warm to room temperature and saturated NH4Cl(aq) (2 mL) was added. The mixture was extracted with Et2O (3 × 5 mL). The combined organics layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (9:1) gave the ketone 17 (100 mg, 72%) as plates; mp 127–130 °C; Rƒ 0.65 [petrol–Et2O (4:1)]; νmax (neat)/cm−1 2970, 2940, 1735, 1705, 1690, 1430; 1H NMR (400 MHz, CDCl3) δ = 8.03 (d, 2H, J = 7.5 Hz, Ph), 7.58 (t, 1H, J = 7.5 Hz, Ph), 7.46 (t, 2H, J = 7.5 Hz, Ph), 7.36–7.31 (m, 2H, Ph), 7.26–7.24 (m, 3H, Ph), 3.92–3.85 (m, 1H, CH), 3.75–3.69 (m, 1H, CH), 3.14–3.01 (m, 2H, CH2), 2.71–2.64 (m, 1H, CH), 2.59–2.51 (m, 1H, CH), 1.31–1.15 (m, 12H, 4 × Me); 13C NMR (100 MHz, CDCl3) δ = 190.4, 151.9, 139.4, 133.3, 128.8, 128.7, 128.5, 128.3, 128.2, 126.6, 116.8, 79.6, 46.8, 46.7, 38.5, 30.8, 21.4, 21.3, 20.3, 19.7; HRMS (ES) found: MH+, 393.2161. C24H29N2O3 requires MH+, 393.2178.
The enantiomers were resolved by CSP-HPLC using a Cellulose1 column at 1 mL min−1, ambient temperature and detection by UV absorbance at 254 nm. Injection volume (20 μL) of the sample was prepared in a 2 g L−1 solution of 1% i-PrOH in hexanes. Retention times were 15.0 and 18.7 min.
From nitrile (S)-6: the nitrile (S)-6 (100 mg, 0.35 mmol) and dry benzoyl chloride (0.12 mL, 1.04 mmol) in dry Et2O–THF (1:1) (0.5 mL) was added dropwise for more than 4 min to TMPMgCl (3.1 mL, 1.0 mmol, 0.4 M in THF) in dry Et2O–THF (1:1) (2.5 mL) at −107 °C. After 30 min, saturated NH4Cl(aq) (2 mL) was added and the mixture was allowed to warm to room temperature. The mixture was extracted with Et2O (3 × 5 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (9:1) gave the ester 17 (40 mg, 40%) as plates; [α]D23 +2.0 (c 1.0, CHCl3); er 63:37 (major peak at 14.9 min) was determined by CSP-HPLC; other data are as mentioned above.
4.11 1-Cyano-1-(1-hydroxycyclopentyl)-3-phenylpropyl N,N-bis(propan-2-yl)carbamate (18)
From racemic nitrile 6: TMPMgCl (3.80 mL, 1.4 mmol, 0.4 M solution in Et2O) was added to the nitrile (±)-6 (100 mg, 0.35 mmol) in Et2O (3 mL) at −78 °C. After 10 min, dry cyclopentanone (0.15 mL, 1.75 mmol) was added. Then, after 30 min, the mixture was allowed to warm to room temperature and saturated NH4Cl(aq) (2 mL) was added. The mixture was extracted with Et2O (3 × 5 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (9:1) gave the alcohol 18 (70 mg, 54%) as needles; mp 135–137 °С; Rƒ 0.75 [petrol–Et2O (4:1)]; νmax (neat)/cm−1 3465, 2970, 2940, 1700; 1H NMR (400 MHz, CDCl3) δ = 7.34–7.30 (m, 2H, Ph), 7.25–7.22 (m, 3H, Ph), 4.47 (s, 1H, OH), 4.09–3.97 (m, 1H, CH), 3.91–3.79 (m, 1H, CH), 3.02–2.95 (m, 1H, CH), 2.88–2.79 (m, 2H, CH2), 2.31–2.23 (m, 1H, CH), 2.12–1.99 (m, 2H, 2 × CH), 1.97–1.87 (m, 2H, 2 × CH), 1.79–1.71 (m, 4H, 2 × CH2), 1.28 (d, 12H, J = 7 Hz, 4 × Me); 13C NMR (100 MHz, CDCl3) δ = 153.3, 140.3, 128.6, 128.3, 126.3, 118.3, 85.8, 85.7, 47.1, 46.5, 38.0, 37.9, 35.3, 31.3, 24.7, 24.0, 21.4, 20.3; HRMS (ES) found: MH+, 373.2475. C22H33N2O3 requires MH+, 373.2491.
The enantiomers were resolved by CSP-HPLC using a Cellulose1 column at 1 mL min−1, ambient temperature and detection by UV absorbance at 254 nm. Injection volume (20 μL) of the sample was prepared in a 2 g L−1 solution of 1% i-PrOH in hexanes. Retention times were 14.4 and 20.3 min.
From nitrile (S)-6: the nitrile (S)-6 (100 mg, 0.35 mmol) in dry Et2O (2 mL) was added dropwise for more than 4 min to TMPMgCl (3.8 mL, 1.4 mmol, 0.4 M in Et2O) in dry Et2O (1 mL) at −107 °C. After 2 min, dry cyclopentanone (0.15 mL, 1.75 mmol) was added. Then, after 30 min, saturated NH4Cl(aq) (2 mL) was added and the mixture was allowed to warm to room temperature. The mixture was extracted with Et2O (3 × 5 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (9:1) gave the alcohol 18 (65 mg, 50%) as needles; [α]D23 −2.0 (c 1.0, CHCl3); er 92:8 (major peak at 14.4 min) was determined by CSP-HPLC; other data are as mentioned above.
4.12 1-Cyano-1-(1-hydroxybenzyl)-3-phenylpropyl N,N-bis(propan-2-yl)carbamate (19a and 19b)
From racemic nitrile 6: isopropyl magnesium chloride (1.20 mol, 1.4 mmol, 1.15 M solution in Et2O) was added to nitrile (±)-6 (100 mg, 0.35 mmol) in Et2O (3 mL) at −78 °C. After 10 min, dry benzaldehyde (0.20 mL, 1.75 mmol) was added. Then, after 30 min, the mixture was allowed to warm to room temperature and saturated NH4Cl(aq) (2 mL) was added. The mixture was extracted with Et2O (3 × 5 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (9:1) gave an inseparable mixture of diastereomers 19a and 19b (dr 1:1) (100 mg, 77%) as a solid; Rƒ 0.3 [petrol–Et2O (4:1)]; νmax (neat)/cm−1 3445, 2965, 2940, 2255, 1685, 1455; 1H NMR (400 MHz, CDCl3) δ = 7.47–7.17 (m, 10H, Ph), 5.50 (d, 0.5H, J = 5 Hz, CH), 5.24 (d, 0.5H, J = 6.5 Hz, CH), 4.83 (d, 0.5H, J = 6.5 Hz, OH), 4.38 (d, 0.5H, J = 5 Hz, OH), 4.02–3.92 (m, 1H, CH), 3.75–3.63 (m, 1H, CH), 3.02–2.77 (m, 2H, CH2), 2.59–2.23 (m, 2H, CH2), 1.32–1.27 (m, 6H, 2 × Me), 1.13–0.97 (m, 6H, 2 × Me); 13C NMR (100 MHz, CDCl3) δ = 153.7, 153.6, 140.1, 140.0, 137.5, 137.2, 128.7, 128.6, 128.4, 128.35, 128.3, 127.4, 126.5, 126.4, 126.35, 126.3, 117.9, 117.1, 80.7, 76.5, 75.5, 46.9, 46.5, 37.2, 35.2, 30.8, 30.7, 21.1, 21.0, 20.8, 20.3; HRMS (ES) found: MH+, 395.2334. C24H31N2O3 requires MH+, 395.2335.
The diastereomers and enantiomers were resolved by CSP-HPLC using a Cellulose1 column at 1 mL min−1, ambient temperature and detection by UV absorbance at 254 nm. Injection volume (20 μL) of the sample was prepared in a 2 g L−1 solution of 1% i-PrOH in hexanes. Retention times were 30.3, 38.2, 51.4 and 60.0 min.
From nitrile (S)-6: the nitrile (S)-6 (100 mg, 0.35 mmol) in dry Et2O (2 mL) was added dropwise for more than 4 min to TMPMgCl (3.8 mL, 1.4 mmol, 0.4 M in Et2O) in dry Et2O (1 mL) at −107 °C. After 2 min, dry benzaldehyde (0.20 mL, 1.75 mmol) was added. Then, after 30 min, saturated NH4Cl(aq) (2 mL) was added and the mixture was allowed to warm to room temperature. The mixture was extracted with Et2O (3 × 5 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. Purification by column chromatography on silica and eluting with petrol–Et2O (19:1) gave an inseparable mixture of diastereomers 19a and 19b (dr 1:1) (75 mg, 60%) as a solid; [α]D23 +6.0 (c 1.0, CHCl3); er values 92:8 and 84:16 (major peaks at 30.3 and 38.2 min) were determined by CSP-HPLC; other data are as mentioned above.
Acknowledgements
We thank the Libyan Ministry of Higher Education, the Leverhulme Trust (grant RPG-056) and the University of Sheffield for funding. We thank Miss Ruaa Talk for carrying out the reaction (Table 1, entry 10) in cyclopentyl methyl ether.