1 Introduction
During the past three decades, many studies have been conducted on amino acids and peptides including those consisting of amino acids that have aromatic side chains. They have received a great experimental and theoretical attention because of their particular importance as chromophores to study the interactions within proteins. Their large faces π can be bound to metal ions such as Na+, Mg+, and Al+ [1,2], and they often take action to cause repulsive and attractive electrostatic forces, which lead to structural changes in their environments [3]. The presence of a UV chromophore in these biomolecules such as phenylalanine, tyrosine, tryptophan, and their peptides, respectively, has allowed the use of the different methods of spectroscopic analysis to determine their most stable structures in the gas phase. The identification of these structures experimentally detected requires the comparison with computed data on the energetics and vibrational frequencies to convert the observed spectra into structural assignments. This symbiotic relationship between experiment and theory has been used to study a wide range of biological molecules in the gas phase, including tryptamine neurotransmitters and serotonin [4–6], adrenergic neurotransmitters and their analogues [7–10], and more recently, small peptide chains [11–16]. Moreover, the experimental procedures performed on these molecules revealed a decay in the amount of conformations detected as the size of these systems increases [11,15,17]. In stark contrast with this small number of peptide conformations experimentally detected, there are a large number of conformations with low energies obtained through theoretical calculations. This poses extra requirements on computational techniques, in which the high-level quantum chemistry methods (e.g., which take into account a part of the electron correlation) will be necessary to accurately describe the intramolecular interactions within peptide, and in particular, the dispersion interactions involving the aromatic residues. Duan et al. [18] showed that the interaction between the aromatic cycle and the peptide backbone is surprisingly large (˜11 kJ mol−1). This indicates the methods that describe the adequate dispersion interactions will be necessary to obtain a correct energy order of conformers. However, the identification of the global minimum by investigating all possible conformers to a high level of theory (theory of perturbations Møller-Plesset, MP2, or beyond) quickly becomes an impossible task for peptides that consist of more than two or three amino acid residues.
A solution to the conformational problem of larger-size molecules (long peptide chains) consists in using a less rigorous method of calculation to generate the most stable structures to be used than as starting structures for an ab initio calculation. In this sense, a new mathematical optimization road knows a growing development. The stochastic methods gradually take precedence over the conventional deterministic technical: on the one hand, they help in locating the optimum of a function in the parameters' space without using the derivatives of the function with respect to these parameters, and on the other hand, they do not get trapped by a local optimum and usually manage to determine the global optimum of the function in question.
This work is part of the development framework of a class of evolutionary methods called “multiniche crowding” (MNC) to locate the different typical secondary folds of the protected amino acid N-formyl-l-tyrosinamide (HCO-l-Tyr-NH2). The combination of this technique, the basic operation of which is relevant to our desired objective to seek potential minima (global and local minimum) of molecular structures with the tools of semiempirical quantum calculation, should help afterward, on the one hand, to understand the conformational preference of this residue in the long polypeptide chains, and on the other hand, to provide vital data on the stacks problems of aromatic rings.
2 Calculation details
2.1 Numbering and abbreviations
Tyrosine can be derived from alanine amino acid by replacing one of its three hydrogen atoms of the methyl group by a phenol group constituting the side chain whose orientation is regulated by the three torsion angles: χ1 around C4C12, χ2 around C12C14, and χ3 around C17O24. The backbone of HCO-l-Tyr-NH2 has four torsion angles ω0, Φ, ψ, and ω1 and represent the rotations around the bonds C2N3, N3C4, C4C5, and C5N6, respectively, as shown in Fig. 1a, where ωi angles take 180° and correspond to trans-peptide bonds. The numbering of the different atoms of the compound HCO-l-Tyr-NH2 is given in Fig. 1b.
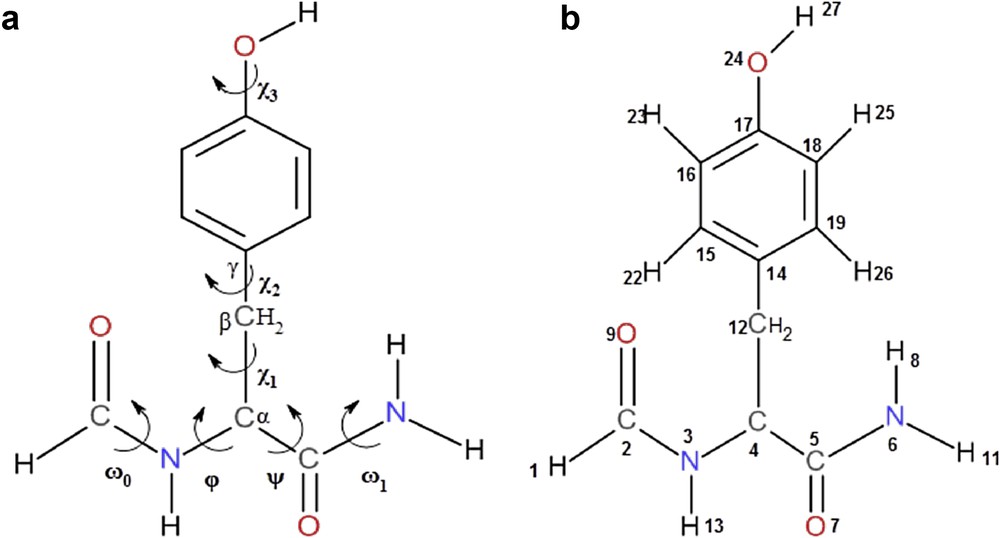
(a) Numbering of atoms and (b) defining dihedral angles for HCO-l-Tyr-NH2.
The multidimensional conformational analysis (MDCA) [19] predicted the existence of nine possible conformations for the backbone of any amino acid defined by two torsion angles Φ and ψ associated with the peptide bond. These nine conformations marked in Greek letters attached to both letters L or D (αD, αL, γD, γL, βL, δD, δL, εD, and εL) are often represented on a map called Ramachandran map E = f(Φ, ψ) (Fig. S1a). The side chain of tyrosine is defined by three angles of χ1, χ2, and χ3, whose two angles χ1 and χ2 can take three possible orientations, gauche+ (g+ = +60), anti (a = 180), and gauche− (g− = −60) leading in total to 3 × 3 = 9 possible orientations for this chain (Fig. S1b). Torsional angle χ3 may generally take both cys (s) and trans (a) positions corresponding to the values 0° and 180°, respectively.
In accordance with the IUPAC–IUB recommendation [20], dihedral or torsional angles were specified [21] within −180° and 180° for both backbone (Φ, ψ) and side chain (χ1, χ2) conformations (Fig. S1).
2.2 Molecular computations
The adopted strategy for the scanning of the potential energy surfaces (PESs) of the peptide systems is to vary systematically and simultaneously both torsional angles Φi and ψi of the ith polypeptide residue while maintaining other residues unchangeable attributing them to previously well-defined folds [22,23], a strategy that may show a great weakness in the exploration of the research space because it may neglect some conformations with low energies.
Our research team [24] published an approach that allows to explore the PES of an isolated molecular system and which may be of high molecular weight. The stochastic approach “genetic algorithm” based on the mechanisms of natural selection and genetic recombination, is used to create population generations from population point candidates called individuals or chromosomes that randomly product to optimize an objective function or fitness. Each chromosome is composed of a set of elements or characteristics called genes. In the study of the problem of conformational space of a given molecule, individuals correspond to conformations, the genes to dihedral angles, and the fitness function to the total energy of the system. This approach, which is coded into computer language and driven by scripts, automates the scan of all minima of molecular PES.
MNC method, proposed by Cedeno et al. [25] based on the concept of crowding, is used not only during the insertion of offspring in the population but also during the selection of the individuals that are going to mate. In MNC method, selection by fitness proportionate reproduction used in simple genetic algorithm (SGA) is replaced by what the author calls “crowding selection”. For each individual Ii of the population, its mate Ij is selected from a group of individuals of crowding selection size (Cs), picked at random from the population. The mate Ij, thus chosen, must be the most similar one to Ii. During the replacement step, the algorithm uses a replacement policy called worst among most similar, as illustrated in Fig. 2. It consists first of all in choosing randomly Cf groups of s individuals per group from the population. These groups are called “crowding factor groups”. Second, one individual from each group who is the most similar to the offspring is identified. Finally, among these Cf individuals who are candidates for replacement, the less fitted one is replaced by the offspring.

Schematic illustration of worst among most similar technique.
The result is an algorithm that (1) maintains stable subpopulations within different niches, (2) maintains diversity throughout the search, and (3) converges to different local minima. Another major advantage of this method is that it requires no prior knowledge of the research space. Therefore, it is adopted in this study to locate the global minimum of a given molecular system on the one hand, and to find all local minima on its PES on the other hand.
The genetic algorithm based on the MNC technique is implemented in a package of program interfaced with MOPAC [26] (version 6.0) to evaluate the quality of the individual to insert into the population in each iteration. The evaluation criterion is the energy of the molecule (the heat of formation in our case). The semiempirical method AM1 is used separately to accomplish this task. The data file has been designed so that a constraint is imposed to the values of the dihedral angles defining the freedom degrees of the conformation randomly generated beforehand. This constraint permits indeed to calculate exactly the energy of the conformation generated during the construction of the initial population and after the application of the crossover and mutation operators eventually. Once the algorithm converges after the fixed maximum number of generation, an optimization without constraint is performed to release the structure so that the individuals of the same niche converge to the corresponding minimum. It has been discussed elsewhere [27] that real encoding is ideally suited to handle problems in a continuous research space. The encoding scheme is so that the genome of each individual (conformation) is composed of n dihedral angles (φ1, φ2,…, φn), in the case of a molecule of n degrees of freedom, whose values represent a location on the PES. The crossover operator used is called interval crossover [28]. In interval crossover, only one offspring is generated. For each pair of parent genes φ1 and φ2, the offspring's gene is selected at random from the interval [φ1 − ε/2, φ2 + ε/2], assuming without loss of generality that φ1 < φ2, if we use a real encoding as in our case. The parameter ε will be called in the following the parameter of interval crossover. The crossover operation is thus performed so that it generates an offspring close to the parents. The mutation is applied to the offspring, generated by the crossover operation, with a Pm probability (see Table 1), whereas permuting a couple of genes of the offspring selected at random.
Control parameters of the MNC genetic algorithm.
| Parameters | Values |
| Population size | 500 |
| Crowding selection size (Cs) | 25 |
| Crowding factor size (Cf) | 3 |
| Crowding size (s) | 25 |
| Interval crossover parameter (ε) | 10 |
| Crossover probability (Pc) | 1.0 |
| Mutation probability (Pm) | 0.06 |
With this approach, a large number of conformations are treated. The exploration of the PES system is conducted intelligently and according to its promising and productive areas. The conformations of the first population are generated randomly. The values of the torsion angles, freedom degrees of the PES that define a molecular conformation are produced by mutation. The data files used by MOPAC are built automatically and one file in dynamic access gathers conformations forming the population of the current generation.
3 Results and discussion
3.1 Conformational analysis
Gordon et al. [29] have conducted the first systematic mapping of the PES of the formyl-l-alaninamide (HCO-l-Ala-NH2) at Restricted Hartree-Fock (RHF)/3-21G and RHF/6-31+G* level of theory with a step size of 30° for φ and ψ angles successively, they found only seven minimum among the nine predicted by the MDCA. Therefore, the conformational building units of the right-handed helical structure αL and that of polyproline II (εL) did not turn out be stationary points on the PES of HCO-L-Ala-NH2.
Subsequently, the PES of the other amino acids, including glycine [30], phenylalanine [31], valine [32], and glutamine [33], have been explored and showed similar topologies to that of alanine. On the basis of these results, Császár and Perczel [34] suggested that the structural polyproline II (εL) element cannot be obtained for any of single amino acids using the diamide approximation. However, subsequent studies have reported the existence of εL for Trp [35], Ile [36], and Cys [37].
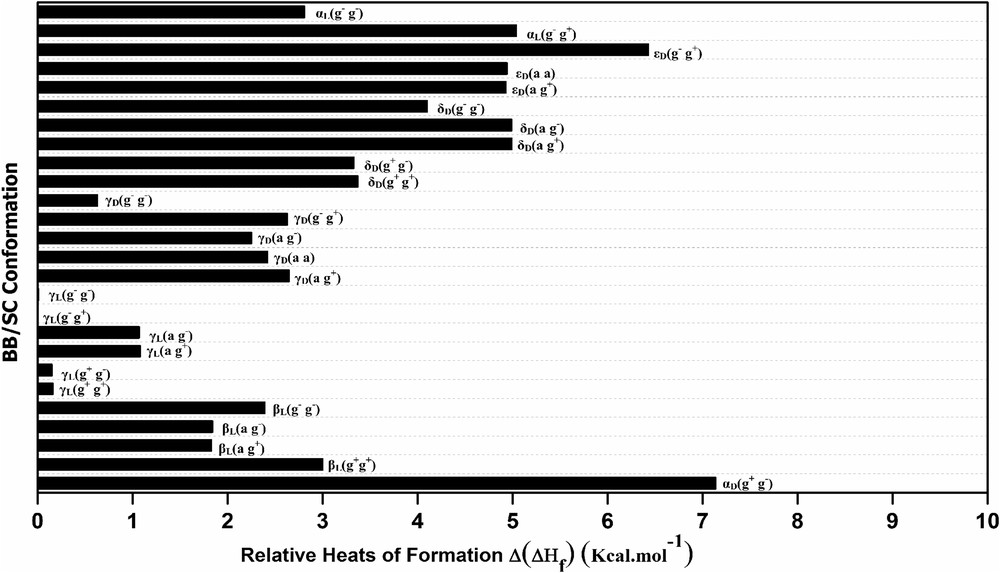
The calculations carried out using the MNC genetic algorithm coupled with the AM1 method on the diamide system HCO-l-Tyr-NH2 have revealed the existence of 26 different conformations (Table S1) spread over seven zones (αL, αD, βL, γL, γD, δD, and εD) among the nine legitimate predefined areas on the Ramachandran map, where the fold αL appears this time as a potential minimum, a fold which usually presents as a minimum on the PESs of diamide models whose side chains comprise the hydroxyl grouping OH [38,39], whereas no structure corresponding to the polyproline II εL (φ = −75 ± 30°, ψ = 160 ± 30°) was detected. The number of different side-chain orientations (g+ g+, … …,g− g−) varies according to each of the seven types of backbone folds found (αD, αL, γD, γL, βL, εD, and δD), as shown in Table 2.
Number of detected conformations of HCO-L-Tyr-NH2 according to the different areas of the Ramachandran map.
| Configuration of the backbone conformers | Number of orientations of the side chain |
| αD | 1 |
| βL | 4 |
| γL | 6 |
| γD | 5 |
| δD | 5 |
| εD | 3 |
| αL | 2 |
The graphical representation of relative heat of formation Δ(ΔHf) for the 26 located conformations of HCO-l-Tyr-NH2 diamide system (Fig. S2) shows that conformers with D subscript (αD, γD, εD, and δD) have energy values (formation heat) relatively higher, which makes them structurally less stable. This result is consistent with results obtained for other N- and C-protected l-amino acids containing trans-peptide bonds (ωi = 180°) treated at an ab initio level as glycine [30], phenylalanine [31], serine [39], proline [40], and aspartic acid [41].
Note that the relative heat of formation Δ(ΔHf)X of each conformation X, is calculated with regard to the global minimum γL(g− g+) according to Eq. 1:
| (1) |
We can easily notice that the first four most stable conformers adopt the inverse gamma turn folding γL with a low energy gap (≈0.16 kcal), this shows that the orientation of the side chain –CH2-(C6H5) phenol has only a very modest effect on the backbone, which is quickly found by comparing the values of the angles (φ, ψ) that appear similar [φ = −83.6 ± 1°, ψ = 60.1 ± 5.2°]. This rigidity of the backbone is because of the strong hydrogen bond CO9⋯HN6 forming a ring with seven atoms (Table S1).
3.2 Molecular interactions
The studies conducted on N- and C-protected l-amino acids (HCO-X-NH2 and CH3CO-X-NHCH3), which carry polar or nonpolar side chains, showed that the latter causes attractive electrostatic forces that result from hydrogen bondings, van der walls interactions, and other repulsive caused by the steric hindrance leading to structural changes in their environment, namely at the level of adjacent main chains, thus the length and nature of these side chains in addition to the interactions within the backbone itself play a key role in the adopted folds. The different types of hydrogen bonds, which may occur in the various conformers of HCO-l-Tyr-NH2, are shown in Fig. 3.

Schematic representation of different types of hydrogen bonds for HCO-l-Tyr-NH2.
The corresponding distances to these potential hydrogen bonds within each conformation of HCO-l-Tyr-NH2 detected by the MNC/AM1 approach are reported in Table 3.
Relative distances of potential hydrogen bonds and electrostatic interactions within the 26 conformations of the HCO-l-Tyr-NH2 obtained by MNC algorithm.
| Conformers | N6H⋯N3C | N6H⋯O9CN (C7) | N3H13⋯O7CN (C5) |
| αD(g+ g−) | 2.64 | – | – |
| βL(g+ g+) | – | – | 2.84 |
| βL(a g+) | – | – | 2.45 |
| βL(a g−) | – | – | 2.46 |
| βL(g− g−) | – | – | 2.77 |
| γL(g+ g+) | – | 1.81 | – |
| γL(g+ g−) | – | 1.87 | – |
| γL(a g+) | – | 1.74 | – |
| γL(a g−) | – | 1.80 | – |
| γL(g− g+) | – | 1.81 | – |
| γL(g− g−) | – | 1.85 | – |
| γD(a g+) | – | 1.54 | – |
| γD(a a) | – | 1.64 | – |
| γD(a g−) | – | 1.65 | – |
| γD(g− g+) | – | 1.55 | – |
| γD(g− g−) | – | 1.54 | – |
| δD(g+ g+) | – | – | – |
| δD(g+ g−) | – | – | – |
| δD(a g+) | – | – | – |
| δD(a g−) | – | – | – |
| δD(g− g−) | – | – | – |
| εD(a g+) | – | – | – |
| εD(a a) | – | – | – |
| εD(g− g+) | – | – | – |
| αL(g− g+) | 2.20 | – | – |
| αL(g− g−) | 2.20 | – | – |
3.3 Comparison with HCO-l-phenylalanine-NH2
3.3.1 Structural analysis
The amino acid tyrosine differs from that of phenylalanine per hydroxyl substituent on its benzene ring. A recent study carried out on N-formyl-l-phenylalaninamide (HCO-l-Phe-NH2) using genetic algorithm based on the MNC technique and coupled with the semiempirical method AM1 [42] allowed us to locate 28 different conformations for this diamide system (Table S2). The results obtained from this study will serve to make a comparison on the structural changes between these diamide systems. Then, if the OH hydroxyl substitution in the benzene ring of the diamide system HCO-l-Tyr-NH2 has no effect on the foldings adopted, then all stereochemical and energetic characteristics observed from tyrosyl residue must be identical to those of the phenylalanyl residue. However, comparing the two diamide systems revealed remarkable differences:
- • The number of folds (αD, γD, γL, …) detected for the two systems is different. In the case of the protected tyrosine, two conformations whose dihedral angles are: (φ1 = −102.5°; ψ1 = −0.1°) and (φ2 = −103.7°; ψ2 = −1.2°) corresponding to the right-handed helical structure αL are detected. However, no conformation that corresponds to this fold was located by either the stochastic approach MNC [42] or by ab initio calculations for the protected phenylalanine [31,43].
- • Among the 26 and 28 conformations detected for both systems HCO-l-Tyr-NH2 and HCO-l-Phe-NH2, respectively, only 19 have similar geometric structures (identical backbones and side-chain orientations). The differences between the dihedral angles of 19 common structures of the two diamide systems HCO-l-Tyr-NH2 and HCO-l-Phe-NH2 (Table S3) are calculated by using Eq. 2:
| (2) |
These differences observed between the dihedral angles can be explained by the influence of the tyrosyl residue (C6H4OH) on some intramolecular interactions, which are responsible for the structures' rigidity in question.
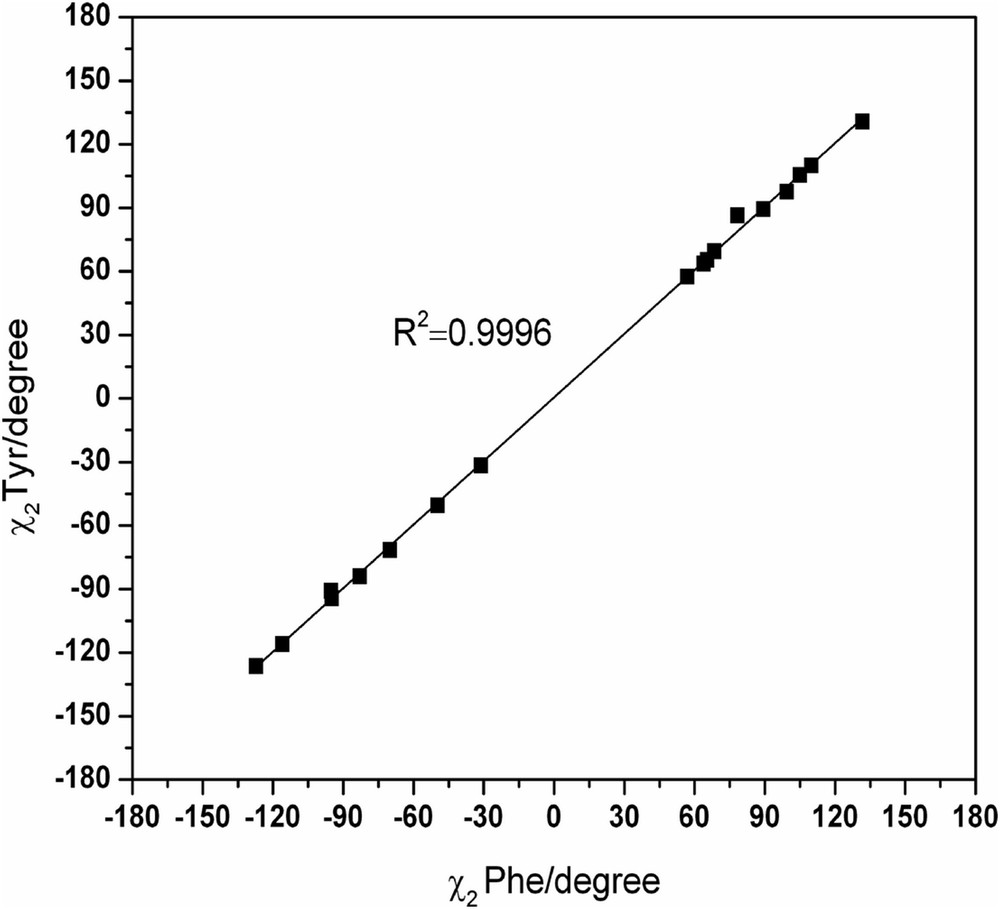
The dihedral angles φ, ψ, χ1, and χ2 of HCO-l-Tyr-NH2 can be represented in function of those of HCO-l-Phe-NH2 to assess the concordance quality between the common conformers of the two systems (Figs. S3–S6).
The dihedral angles φ and χ1 for the conformations of the two diamide systems show a very good correlation (R2 = 0.9997 and R2 = 0.9998, respectively), which may show that the intervention of the grouping OH is limited in a manner that did not disrupt certain intramolecular bonds responsible for the rigidity of these structures (Figs. S3 and S5). However, this correlation decreases slightly for both dihedral angles ψ and χ2 (R2 = 0.9996 and R2 = 0.9996, respectively) (Figs. S4 and S6).
We can observe (from the spread of points in the figures) that the dihedral angles ψ and χ2 are less ideal in terms of the population of their respective conformational space. These can be contrasted to the dihedral angles φ and χ1, which have most of the stable conformers populating ideal states predicted by MDCA. The points representing the values of dihedral angles φ and χ1 are intensively grouped in the probable areas predicted by MDCA (g+, a, g−) (Figs. S3 and S5). However, the points representing both dihedral angles ψ and χ2 are distributed in a random manner, which indicates the flexible nature of each of those angles (Figs. S4 and S6).
3.3.2 Energetic analysis
The calculations carried out on the HCO-l-Tyr-NH2 system (Table S1) predicted the folded conformation γL(g− g+) as being the global minimum (see Fig. 4). The relative energy between the latter and the relative next higher minimum in energy γL(g− g−) amounts to 0.01 kcal mol−1, whereas the third and the fourth minimums are the folds γL(g+ g−) and γL(g+ g+) with relative energies of 0.15 and 0.16 kcal mol−1, respectively, above the global minimum γL(g− g+).
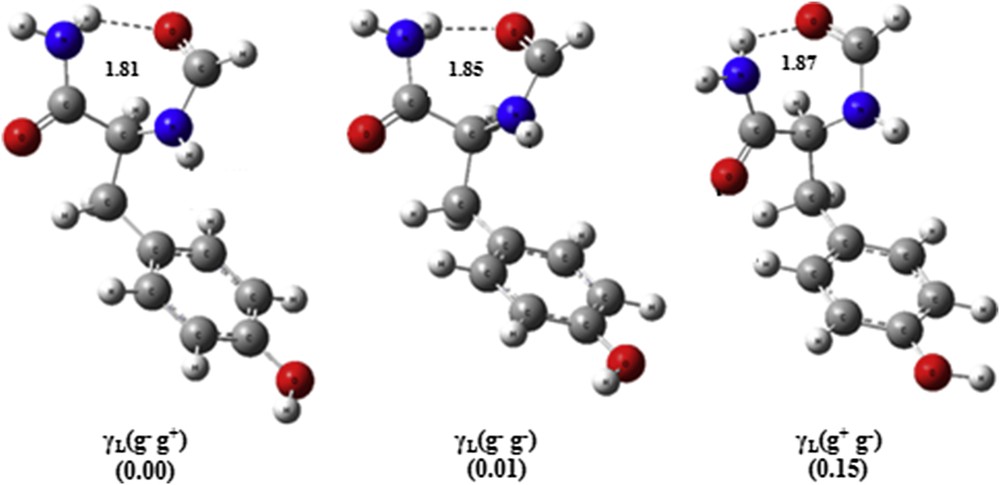
Geometric representation of the three most stable conformers of the HCO-l-Tyr-NH2 obtained by MNC/AM1 (bond lengths are in Å).
It is worth noting that these four most stable conformations of the HCO-l-Tyr-NH2 system are also the preferred ones for HCO-l-Phe-NH2 and in the same stability order but with a different energy gap (Fig. S7(a–e)).
Depicted 4D Ramachandran map based on four dihedral angles φ, ψ, and χ1, χ2 for located conformations of both HCO-l-Phe-NH2 and HCO-l-Tyr-NH2 diamide systems through the MNC/AM1 approach (Fig. S8) allows us a panoramic and simultaneously vision of adopted folds, relative orientations of the side chains, and relative heats of formation.
We easily notice that the conformations γD(g+ g+), βL(a g+), αD(g+ g+), αD(a g+), and αD(g− g−) are presented as potential minima in the PES of HCO-l-Phe-NH2 when they are absent from that of HCO-l-Tyr-NH2. A probable reason for this loss can be attributed to their relatively high energy values (heats of formation) (Table S1), which makes them structurally less stable. Consequently, the intervention of the hydroxyl OH grouping caused some structural changes (dihedral angles, hydrogen bonds, and so forth) that have led to the migration of these conformations to other more stable.
3.4 Correlation theory experience
The validity of the calculations presented in this work can be assessed by comparing, at the structural level, the conformations of the tyrosine predicted by the MNC approach with those derived experimentally either by X-ray crystallography, which will provide the technology needed to solve the three-dimensional structure of the proteins, or by nuclear magnetic resonance (NMR) spectroscopy.
The backbones of amino acids and consequently of proteins can be described by the two torsion angles φ and Ψ defining any secondary refolding. In this sense, a map showing the distribution of pairs (φ, Ψ) of 410 tyrosine residues, collected from 131 proteins was generated (Fig. 5a) by exploiting recent experimental protein data (May 2016) from protein data bank [44]. To conduct a comparison between the secondary folds observed experimentally and those predicted by the MNC approach, an additional map was drawn on the basis of the obtained results showing the pairs (φ, Ψ) corresponding to 500 conformations predicted by our calculations (Fig. 5b).

(a) Couple distribution (φ, Ψ) of 410 tyrosine residues, collected from 131 proteins. (b) Couple distribution (φ, Ψ) corresponding to 500 HCO-l-Tyr-NH2 conformations predicted by the MNC algorithm.
The comparison of these data shows a promising similarity. The map representing the experimental data (X-ray and NMR) has five zones, two of which are highly populated. The first zone (I) corresponds to the βL (extended β-strand) and εL (polyproline II) areas; the second zone (II) represents the γL (inverse gamma turn), δL, and αL (right-hand α-helix) areas; the third zone (III) corresponds to the δD; the fourth zone (IV) is the αD, which corresponds to the left-hand α-helix; and the fifth zone (V) corresponds to εD (inverse polyproline II). The five zones are shown by dotted lines in Fig. 5a.
However, the map representing the folds theoretically detected through the MNC algorithm (Fig. 5b) also showed five zones but with the following remarkable differences:
- • The absence of the conformation εL (polyproline II). The same result was already mentioned by Chass et al. [45] by using the protected diamide system N-acetyl-tyrosyl-N-methylamide (CH3CO-l-Tyrosine-NHCH3) at B3LYP/6-31G(d) level of theory.
- • The area designated by a dotted yellow line Fig. 5b corresponding to the folding γD (right gamma turn) does not appear in the experimental map.
The zones III, IV, and V are less populated and experimentally correspond to the δD, αD, and εD folds, respectively. These folds are less favorites energetically by calculations carried out using the MNC genetic algorithm—as detailed in Section 3.1—and therefore far from being the preferred conformations of the compound HCO-l-Tyr-NH2.
Such a correlation permits us to assume that if the diamide model is relevant to the description of main-chain folding of proteins, then the most stable conformers should have the lowest energy.
4 Conclusions
The exploration of the PES of HCO-l-Tyr-NH2 through the genetic algorithm based on the MNC technique revealed the existence of 26 different conformations. The lowest energy structure corresponds to a γL backbone conformation with (g−, g+) orientations in the side chain. A comparative study made between HCO-l-Tyr-NH2 and HCO-l-Phe-NH2 systems suggests that the OH substitution effect is indeed present. From the 19 common structures, dihedral angles φ and χ1 showed perfect linear fits with R2 values of 0.9997 and 0.9998, respectively. This correlation decreases slightly for dihedral angles ψ and χ2 with R2 values of 0.9996 for both angles.
The comparative study among theoretical calculations and experimental (NMR and X-ray) results revealed a promising similarity, which reflects the ability of the MNC genetic algorithm used to locate the different minima on the PES of HCO-l-Tyr-NH2.
Appendix A Supplementary data
The following are the supplementary data related to this article: