

1 Introduction
1,5-Diketones are useful precursors to various substituted anilines [1] and heterocyclic compounds including pyridine derivatives [2–5], dihydropyridines [5,6], and pyrazolo[1,5-a] pyrimidines [7]. Moreover, 1,5-diketones with abstractable γ-hydrogen atoms were shown to be highly photoactive cage molecules for the release of fragrances [8]. These dicarbonyl compounds were also used for the synthesis of bicyclic ethers via their annulation reaction with bis (trimethylsilyl) enol ethers [9] and pinacol coupling through their reductive dimerization [10].
Michael addition of ketone enolates or their surrogates onto α,β-enones is one of the common methods for the synthesis of 1,5-diketones [11–13]. These compounds can also be prepared by a cross-coupling reaction of aryl methyl ketones with either aromatic aldehydes [14] or acetohydrazones [15], as well as by dimerization of α,β-enones in the presence of a quaternary ammonium salt as a phase-transfer catalyst [16]. In addition, the cross-metathesis of olefins was found to be a rapid access to such dicarbonyl derivatives [1,3]. In another synthetic protocol, Zard et al. reported a free radical approach for the synthesis of 1,5-diketones from alkenylacylphosphonates and keto-xanthates [2].
On the other hand, during the last decades, the nucleophilic behavior of enamines has been explored in palladium-catalyzed nucleophilic substitution of allylic compounds. Indeed, the reaction of allylic acetates with enamines worked well in the presence of Pd(II)/metallocene-based ligands, affording the corresponding allylation products [17]. Such a nucleophilic allylic substitution was used to prepare γ,δ-unsaturated ketones through the reaction of allylic benzotriazoles with enamines under the action of Pd(0)/PPh3 in the presence of ZnBr2 [18].
Furthermore, it was shown that the one-pot reaction of functionalized acyclic Morita–Baylis–Hillman (MBH) acetates with various enamino β-ketoesters in the presence of NaH afforded a series of trisubstituted 2-pyridones [19,20]. Nevertheless, it is worth noting that up to now, no Pd-catalyzed nucleophilic reaction of enamines with acyclic or cyclic MBH adducts has been reported. In our previous studies on the MBH chemistry, we have reported a Pd-free Tsuji-Trost–type reaction of MBH alcohols or acetates with β-dicarbonyl compounds, affording γ,δ-unsaturated 1,5-diketones [21,22]. In continuation of our interest in the allylic substitution of MBH derivatives with various nucleophiles [23,24], we report herein the first examples of a Pd-catalyzed allylic nucleophilic substitution of both cyclic and acyclic MBH adducts, as functionalized allylic substrates, with ketoenamines, providing a new series of saturated and γ,δ-unsaturated 1,5-dicarbonyl compounds.
2 Results and discussion
For this purpose, we have initially prepared the enamines 2a–c in 60–70% yields (Table 1) from the reaction of cyclic ketones 1a–c with morpholine using conventional reaction conditions [25].
Synthesis of enamines 2a–c from the reaction of ketones 1a–c with morpholine.a
| Entry | Enamine 2 | Reaction time (days) | Yield 2 (%)b | |
| 1 | 2a | Image 2 | 3 | 60 |
| 2 | 2b | Image 3 | 4 | 70 |
| 3 | 2c | Image 4 | 1 | 65 |
a The reaction was performed using ketones 1a–c (1 equiv), morpholine (1.5 equiv), p-toluenesulfonic acid (2 equiv) in toluene.
b Isolated yield.
Next, upon treatment of the enamine 2a with the MBH acetate 3a [21], at room temperature (rt), under solvent-free conditions, and without any additive, the starting materials were recovered within 48 h (Table 2, entry 1). The same result was also obtained when toluene was used as a solvent in the same reaction at rt or at reflux for 72 h (Table 2, entries 2 and 3). Interestingly, in dichloromethane at rt, the 1,5-dicarbonyl compound 4a was obtained, within 72 h, but in only 30% yield (Scheme 1, Table 2, entry 4). To improve the reaction yield and reduce the reaction time, our subsequent experiment was conducted in refluxing dichloromethane. Under these conditions, the 1,5-keto ester 4a was produced in the same yield within 72 h (Scheme 1, Table 2, entry 5).
Optimization of the reaction conditions for the synthesis of 4a from the reaction of 2a with 3a.a
| Entry | T (°C) | Solvent | Time (h) | Yield 4ab (%) |
| 1 | rt | None | 48 | – |
| 2 | rt | Toluene | 72 | – |
| 3 | 110 | Toluene | 72 | – |
| 4 | rt | CH2Cl2 | 72 | 30 |
| 5 | 40 | CH2Cl2 | 72 | 30 |
a The reaction was performed using the acetate 3a (1 equiv) and enamine 2a (5 equiv) in CH2Cl2.
b Isolated yield.
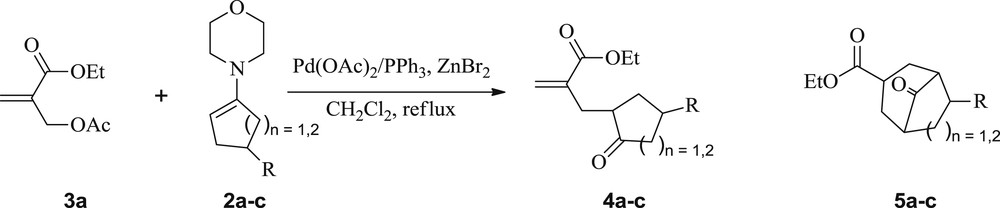
Synthesis of ketoesters 4a–c from acyclic MBH 3a and enamines 2a–c.
Taking into account the low yields of the compound 4a and long reaction times recorded in the previous preliminary attempts, we therefore decided to explore a new synthetic approach for 4a under catalytic conditions. In this context, we reacted, under the established conditions, the enamine 2a (5 equiv) with the MBH acetate 3a (1 equiv) in the presence of Pd(OAc)2 (0.04 equiv)/PPh3 (0.16 equiv) and ZnBr2 (2.66 equiv) as a Lewis acid in refluxing dichloromethane. The conversion of the substrate 3a was complete within 6 h, yielding exclusively, after hydrolysis of the reaction mixture, the unsaturated 1,5-keto ester 4a in 70% yield (Scheme 1, Table 3, entry 1). Under the same conditions, the allylic nucleophilic substitution of the substrate 3a was also successfully performed with enamines 2b and 2c, affording similarly the corresponding SN2 and/or SN2′-type products 4b and 4c in 80 and 68% yield, respectively (Scheme 1, Table 3, entries 2 and 3).
Synthesis of 1,5-dicarbonyl compounds 4 and 5 through the reaction of substrates 3a–e with enamines 2a–c.a
| Entry | 3 | 2 | Time (h) | Product 4 and/or 5b | Ratio 4:5 | |
| 1 | 3a | 2a | 6 | Image 5 4a, 70% | 4a/5a = 100:0 | |
| 2 | 3a | 2b | 4 | Image 6 4b, 80% | 4b/5b = 100:0 | |
| 3 | 3a | 2c | 4 | Image 7 4c, 68% | 4c/5c = 100:0 | |
| 4 | 3b | 2a | 24 | Image 8 5d, 80% | 4d/5d = 0:100 | |
| 5 | 3c | 2a | 48 | Image 9 5d, 75% | 4d/5d = 0:100 | |
| 6 | 3d | 2a | 24 | Image 10 5d, 75% | 4d/5d = 0:100 | |
| 7 | 3e | 2a | 37 | Image 11 5e, 60% | 4e/5e = 0:100 | |
| 8 | 3e | 2b | 32 | Image 12 5f, 64% | 4f/5f = 0:100 | |
| 9 | 3e | 2c | 40 | Image 13 5g, 57% | 4g/5g = 0:100 | |
| 10 | 3b | 2b | 7 | Image 14 4h, 27% | Image 20 5h, 60% | 4h/5h = 31:69 |
| 11 | 3c | 2b | 28 | Image 15 4h, 26% | Image 21 5h, 63% | 4h/5h = 29:71 |
| 12 | 3d | 2b | 12 | Image 16 4h, 35% | Image 22 5h, 57% | 4h/5h = 38:62 |
| 13 | 3b | 2c | 24 | Image 17 4i, 37% | Image 23 5i, 54% | 4i/5i = 40:60 |
| 14 | 3c | 2c | 28 | Image 18 4i, 28% | Image 24 5i, 65% | 4i/5i = 30:70 |
| 15 | 3d | 2c | 24 | Image 19 4i, 27% | Image 25 5i, 64% | 4i/5i = 30:70 |
a The reaction was performed using MBH adducts 3 (1 equiv), ketoenamine 2 (5 equiv), Pd(OAc)2 (0.04 equiv), PPh3 (0.16 equiv), and ZnBr2 (2.66 equiv) in refluxing CH2Cl2.
b Isolated yield.
Next, to establish the substrate scope, we selected the cyclic MBH derivatives 3b–d bearing three different leaving groups (LG = OAc, OMe, OEt), and we explored their behavior toward the nucleophilic enamines 2. First, upon treatment of allylic acetate 3b or allylic ethers 3c and 3d with enamine 2a, under the previous conditions (MBH adduct (1 equiv), ketoenamine (5 equiv), Pd(OAc)2 (0.04 equiv), PPh3 (0.16 equiv), and ZnBr2 (2.66 equiv) in CH2Cl2), the reaction worked well. Nevertheless, the expected SN2-type product 4d was not isolated but we exclusively obtained the same compound 5d, in 75–80% yields (entries 4–6). This derivative was presumably formed through an in situ intramolecular Michael addition on the enone moiety of its precursor 4d (Scheme 2, Table 3, entries 4–6). The best yield (80%) and the shorter reaction time (24 h) were achieved with the allylic acetate 3b that was found to be the most reactive.
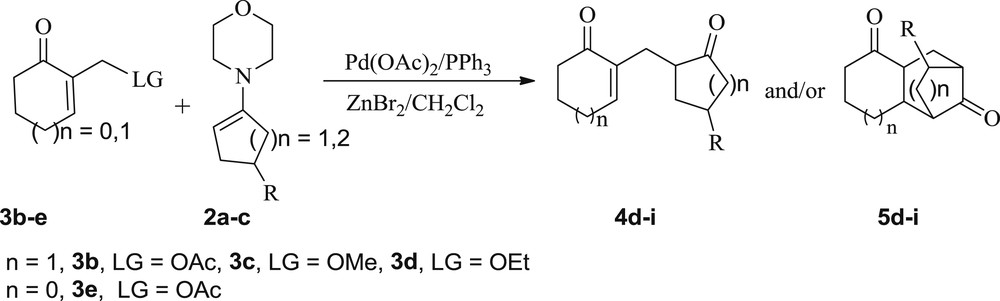
Allylic substitution of MBH adducts 3b–e with enamines 2a–c.
It is noteworthy that the monitoring of the reaction progress by thin layer chromatography, from the beginning of our experiment, showed only the formation of the compound 5d with no trace of its precursor 4d (Table 3, entries 4–6), suggesting that the conversion of this intermediate into 5d was kinetically faster than that of substrates 3b–d into the intermediate 4d.
Correlatively, an interesting example on Michael–Stork addition of enamine 2a to allenyl ketones and esters was previously reported by Lepore et al. [26]. They showed that a Michael addition onto the title substrates provided an iminium ion intermediate, which could further undergo hydrolysis to afford the 1,4-adducts 6. Alternatively, the corresponding enamines can undergo an intramolecular cyclization to yield the bicyclic compounds 7 (Scheme 3).
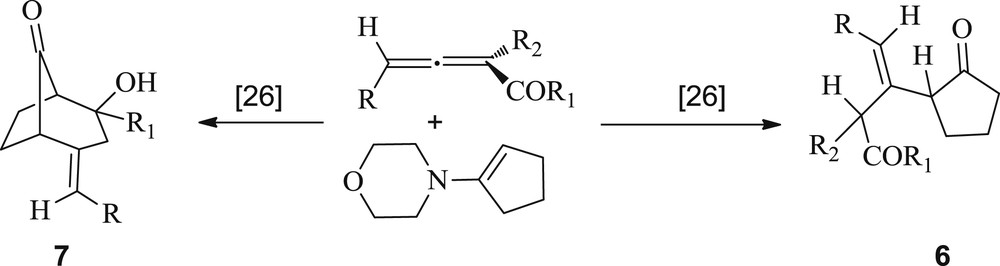
Michael–Stork addition of an enamine to allenyl ketones and esters.
Furthermore, we explored the scope of this allylic nucleophilic substitution reaction involving the five-membered MBH acetate 3e with enamines 2a–c. Under the previous conditions (Pd(OAc)2/ZnBr2), adducts 5e–g were exclusively obtained in 57–64% yields (Scheme 2, Table 3, entries 7–9).
Finally, the nucleophilic allylic substitution of substrates 3b–d with six-membered enamines 2b and 2c has been investigated (Scheme 2, Table 3, entries 10–15). We observed that these substrates did not have the same behavior toward the five-membered enamine 2a (vide supra) and the six-membered enamines 2b and 2c (Scheme 2, Table 3, entries 10–15). Indeed, the reaction with enamine 2a gave exclusively the adduct 5d (Scheme 2, Table 3, entries 4–6), whereas the use of 2b and 2c gave predominantly adducts 5h and 5i along with their corresponding precursors 4h and 4i (Scheme 2, Table 3, entries 10–15) in high overall yields.
Compounds 5h and 5i and the corresponding precursors 4h and 4i were easily separated by column chromatography (petroleum ether/ether = 60:40) and fully characterized. Polycyclic compounds 5d–i were unequivocally elucidated by 1H NMR and high-resolution mass spectra (HRMS).
It is notable that, under the catalysis of both Pd(0) and ZnBr2, we did not observe any SN2′ product along with the SN2-type products 4d–i using cyclic MBH adducts 3b–e and enamines 2a–c. We believe that this allylic substitution is under thermodynamic control, affording exclusively the α-products [27,28].
To explain the formation of the 1,5-dicarbonyl compounds 4 and 5, we propose an initial departure of the leaving group on adducts 3, which would be assisted by the Lewis acid ZnBr2 [29–31]. Indeed, the resulting intermediate 7 undergoes an oxidative addition to Pd(0) to form a cationic Π–allyl Pd(II) complex 8 (i) that is further attacked by the enamines 2, yielding an iminium intermediate 9 (ii). After that, there is either the hydrolysis of the intermediate 9 to give the δ-diketones 4 (iii), or alternatively the carbanion derived from the tautomeric form 9′ of 9 undergoes an intramolecular Michael addition on the enone moiety to finally provide, after hydrolysis, the δ-diketones 5 (iv) (Scheme 4).
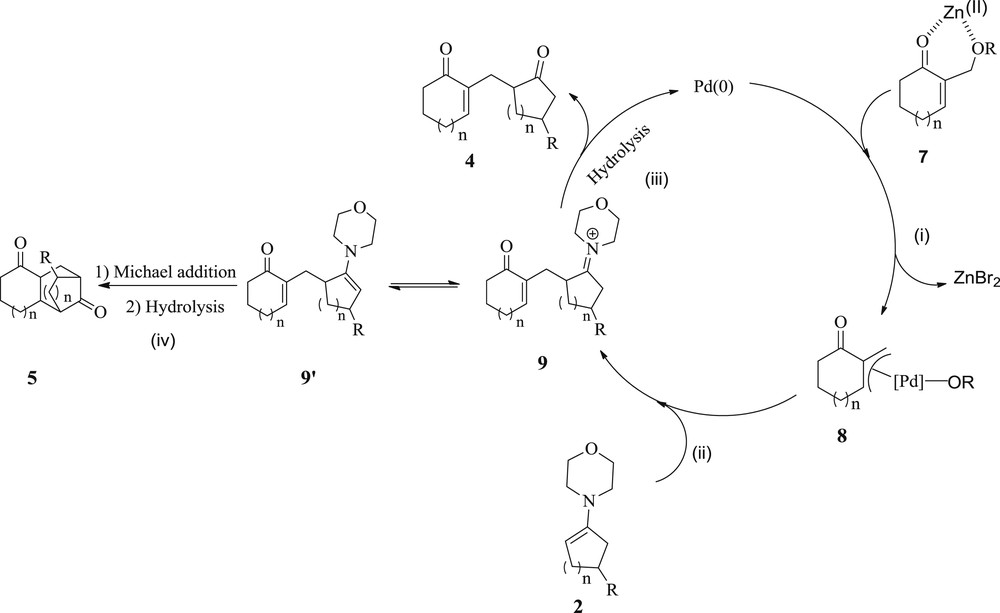
Proposed reaction mechanism for 1,5-dicarbonyl compounds 4 and 5.
3 Conclusion
We have reported the first examples of an efficient ZnBr2-promoted Pd-catalyzed allylic substitution reactions of cyclic and acyclic MBH adducts with cyclic ketoenamines in refluxing dichloromethane. These reactions afford a series of SN2-type unsaturated 1,5-dicarbonyl compounds, which can further undergo an intramolecular Michael addition on the enone moiety, yielding the corresponding 1,4-adducts. All the newly synthesized compounds were isolated in moderate to good yields and fully characterized.
4 Experimental section
4.1 General comments
1H NMR and 13C NMR spectra were recorded either on a 300 MHz Bruker for 1H or 75 MHz in CDCl3, using tetramethylsilane as an internal standard (chemical shifts in δ values and J in Hz). HRMS were recorded as ESI-HRMS on an Auto Spec Ultima/micromass mass spectrometer. Mass spectra (EI) were obtained at 70 eV on a Hewlett–Packard HP-5890. Analytical thin layer chromatography was performed using Fluka Kieselgel 60 F254 precoated silica gel plates. Visualization was achieved by UV light (254 nm). Flash chromatography was performed using silica gel 60 and a gradient solvent system (petroleum ether/ether). Compounds 3 and enamines 2 were prepared according to the literature methods [21,25].
4.2 General procedure for the synthesis of 1,5-dicarbonyl compounds 4a–i and 5a–i
Under nitrogen, a mixture of MBH acetate 3 (1.5 mmol), Pd(OAc)2 (13 mg, 0.06 mmol), PPh3 (63 mg, 0.24 mmol), and ZnBr2 (0.90 g, 4 mmol) was refluxed in CH2Cl2 (10 mL) for 15 min. Then the solution of enamine 2 (7.5 mmol) in CH2Cl2 (5 mL) was added, and the reaction mixture was refluxed for 4–48 h. After evaporation of CH2Cl2, water (10 mL) was added with NaOH (1 M, 30 mL), and the mixture was extracted with CH2Cl2 (3 × 30 mL). The organic phase was washed with a saturated solution of ammonium chloride (10 mL) and dried over magnesium sulfate. After removal of the solvent, the residue was subjected to column chromatography (petroleum ether/ether = 60:40) to produce pure unsaturated 1,5-dicarbonyl compounds 4a–i and 5a–i.
4.3 Ethyl 2-((2-oxocyclopentyl)methyl)acrylate (4a)
Yield 70%; colorless oil. 1H NMR (CDCl3, 300 MHz): δ 6.19 (d, J = 3 Hz, 1H), 5.59 (d, J = 3 Hz, 1H), 4.21 (q, J = 7.5 Hz, 2H), 2.91–2.85 (m, 1H), 2.38–2.34 (m, 2H), 2.33–2.21 (m, 4H), 1.99–1.86 (m, 1H), 1.54–1.50 (m, 1H), 1.32 (t, J = 7.5 Hz, 3H). 13C NMR (CDCl3, 75 MHz): δ 219.5, 166.9, 138.9, 126.1, 60.7, 48.1, 37.8, 32.1, 29.7, 20.6, 14.2. MS (m/z), 55 (53), 68 (50), 96 (100), 151 (21), 168 (51), 196 (M+, 39); HRMS calcd for C11H16O3+ [M−H]+ 195.1021, found 195.1020.
4.4 Ethyl 2-((2-oxocyclohexyl)methyl)acrylate (4b)
Yield 80%; colorless oil. 1H NMR (CDCl3, 300 MHz): δ 6.19 (d, J = 3 Hz, 1H), 5.57 (d, J = 3 Hz, 1H), 4.20 (q, J = 7.5 Hz, 2H), 2.36–2.32 (m, 4H), 2.12–2.08 (m, 2H), 1.89–1.85 (m, 4H), 1.74–1.68 (m, 1H), 1.30 (t, J = 7.5 Hz, 3H). 13C NMR (CDCl3, 75 MHz): δ 212.1, 166.9, 138.4, 126.6, 60.5, 49.1, 41.8, 33.5, 31.8, 27.9, 26.9, 14.0. MS (m/z), 55 (39), 67 (34), 97 (41), 136 (100), 165 (32), 210 (M+, 8); HRMS calcd for C12H18O3+ [M+H]+ 211.1327, found 211.1325.
4.5 Ethyl 2-((5-methyl-2-oxocyclohexyl)methyl)acrylate (4c)
Yield 68%; colorless oil. 1H NMR (CDCl3, 300 MHz): δ 6.20 (s, 1H), 5.56 (d, J = 3 Hz, 1H), 4.20 (q, J = 8 Hz, 2H), 2.87–2.80 (m, 1H), 2.71–2.67 (m, 1H), 2.42–2.27 (m, 3H), 2.14–2.04 (m, 2H), 1.70 (t, J = 6 Hz, 2H), 1.65–1.55 (m, 1H), 1.30 (t, J = 8 Hz, 3H), 1.08 (d, J = 6 Hz, 3H). 13C NMR (CDCl3, 75 MHz): δ 213.3, 166.8, 138.3, 126.4, 60.7, 46.5, 39.3, 37.9, 34.2, 32.9, 26.6, 19.5, 14.1. MS (m/z), 55 (89), 67 (42), 136 (28), 151 (100), 179 (62), 224 (M+, 10); HRMS calcd for C13H20O3+ [M+H]+ 225.1484, found 225.1486.
4.6 Decahydro-1H-5,8-methanobenzo[7]annulene-1,10-dione (5d)
Yield 80% from (6-oxocyclohex-1-en-1-yl)methyl acetate 3b, 75% from 2-(methoxymethyl)cyclohex-2-enone 3c, and 75% from 2-(ethoxymethyl)cyclohex-2-enone 3d; white solid; mp 128–130 °C. 1H NMR (CDCl3, 300 MHz): δ 2.66–2.57 (m, 1H), 2.40–2.32 (m, 3H), 2.19–1.92 (m, 7H), 180–1.60 (m, 4H), 1.25–1.16 (m, 1H). 13C NMR (CDCl3, 75 MHz): δ 219.4, 210.6, 51.4, 49.2, 47.1, 43.4, 41.3, 34.7, 29.7, 26.0, 23.1, 18.5. MS (m/z), 96 (48), 110 (40), 164 (31), 192 (M+, 100); HRMS calcd for C12H16O2+ [M+H]+ 193.1229, found 193.1230.
4.7 Octahydro-4,7-methanoazulene-1,9(2H)-dione (5e)
Yield 60%; white solid; mp 122–126 °C. 1H NMR (CDCl3, 300 MHz): δ 2.37–2.00 (m, 4H), 1.81–1.43 (m, 4H), 1.39–1.23 (m, 4H), 0.91–0.83 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 220.8, 220.7, 47.1, 46.8, 38.0, 30.2, 30.1, 29.9, 29.7, 29.6, 20.7. MS (m/z), 150 (100), 178 (M+, 60); HRMS calcd for C11H14O2+ [M+H]+ 179.1072, found 179.1071.
4.8 Decahydro-1H-4,8-methanocyclopenta[8]annulene-1,10-dione (5f)
Yield 64%; white solid; mp 119–121 °C. 1H NMR (CDCl3, 300 MHz): δ 3.04–2.95 (m, 1H), 2.82–2.74 (m, 1H), 2.43–2.31 (m, 3H), 2.19–2.05 (m, 9H), 1.77–1.63 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 219.3, 219.2, 50.3, 47.4, 45.7, 45.2, 36.9, 35.1, 34.3, 32.2, 27.0, 20.1. MS (m/z), 96 (48), 110 (40), 164 (31), 192 (M+, 100); HRMS calcd for C12H16O2+ [M+H]+ 193.1229, found 193.1241.
4.9 6-Methyldecahydro-1H-4,8-methanocyclopenta[8]annulene-1,10-dione (5g)
Yield 57%; white solid; mp 120–123 °C. 1H NMR (CDCl3, 300 MHz): δ 2.47–2.42 (m, 1H), 2.26–1.96 (m, 4H), 1.94–1.83 (m, 4H), 1.76–1.27 (m, 6H), 0.96 (d, J = 9 Hz, 3H). 13C NMR (CDCl3, 75 MHz): δ 219.2, 213.0, 51.8, 47.8, 47.5, 46.8, 37.9, 34.5, 34.1, 33.6, 27.9, 24.5, 20.0. MS (m/z) 96 (90), 110 (88), 164 (89), 206 (M+, 98); HRMS calcd for C13H18O2+ [M+H]+ 207.1385, found 207.1389.
4.10 2-((2-Oxocyclohexyl)methyl)cyclohex-2-enone (4h)
Yield 27% from (6-oxocyclohex-1-en-1-yl)methyl acetate 3b, 26% from 2-(methoxymethyl)cyclohex-2-enone 3c, and 35% from 2-(ethoxymethyl)cyclohex-2-enone 3d; colorless oil. 1H NMR (CDCl3, 300 MHz): δ 6.79 (t, J = 4.5 Hz, 1H), 1.76–1.70 (m, 1H), 2.60–2.50 (m, 1H), 2.43–2.28 (m, 6H), 2.08–1.95 (m, 5H), 1.86–1.81 (m, 1H), 1.68–1.59 (m, 2H), 1.21–1.36 (m, 1H). 13C NMR (CDCl3, 75 MHz): δ 212.9, 199.7, 147.8, 137.5, 49.2, 42.2, 38.5, 34.0, 30.3, 28.1, 26.3, 25.0, 23.0. MS (m/z) 55 (100), 67 (71), 95 (52), 110 (31), 122 (83), 206 (M+, 92); HRMS calcd for C13H18O2+ [M+H]+ 207.1385, found 207.1389.
4.11 Decahydro-5,9-methanobenzo[8]annulene-1,11(2H)-dione (5h)
Yield 60% from (6-oxocyclohex-1-en-1-yl)methyl acetate 3b, 57% from 2-(methoxymethyl)cyclohex-2-enone 3c, and 63% from 2-(ethoxymethyl)cyclohex-2-enone 3d; white solid; mp 113–116 °C. 1H NMR (CDCl3, 300 MHz): δ 3.44–3.35 (m, 1H), 2.58–2.53 (m, 2H), 2.46–2.22 (m, 4H), 2.15–1.98 (m, 7H), 1.74–1.54 (m, 4H). 13C NMR (CDCl3, 75 MHz): δ 219.2, 213.0, 51.8, 47.8, 47.5, 46.8, 37.9, 34.5, 34.1, 33.6, 27.9, 24.5, 20.0. MS (m/z): 55 (77), 95 (52), 110 (34), 150 (34), 178 (21), 206 (M+, 100); HRMS calcd for C13H18O2+ [M+H]+ 207.1385, found 207.1383.
4.12 2-((5-Methyl-2-oxocyclohexyl)methyl)cyclohex-2-enone (4i)
Yield 37% from (6-oxocyclohex-1-en-1-yl)methyl acetate 3b, 28% from 2-(methoxymethyl)cyclohex-2-enone 3c, and 27% from 2-(ethoxymethyl)cyclohex-2-enone 3d; colorless oil. 1H NMR (CDCl3, 300 MHz): δ 6.80 (t, J = 4.5 Hz, 1H), 2.77–2.37 (m, 2H), 2.34–2.31 (m, 6H), 2.02–1.92 (m, 7H), 1.90–1.28 (m, 1H), 0.96 (d, J = 6 Hz, 3H). 13C NMR (CDCl3, 75 MHz): δ 220.9, 199.7, 147.8, 137.5, 47.9, 42.1, 41.6, 38.5, 36.1, 31.9, 30.2, 26.2, 23.0, 21.2. MS (m/z), 55 (79), 110 (47), 203 (88), 220 (M+, 100); HRMS calcd for C14H20O2+ [M+H]+ 221.1542, found 221.1544.
4.13 7-Methyldecahydro-5,9-methanobenzo[8]annulene-1,11(2H)-dione (5i)
Yield 54% from (6-oxocyclohex-1-en-1-yl)methyl acetate 3b, 65% from 2-(methoxymethyl)cyclohex-2-enone 3c, and 64% from 2-(ethoxymethyl)cyclohex-2-enone 3d; white solid; mp 106–108 °C. 1H NMR (CDCl3, 300 MHz): δ 3.46–3.38 (m, 1H), 2.56–2.23 (m, 7H), 2.18–2.12 (m, 4H), 1.69–1.40 (m, 4H), 1.35–1.17 (m, 1H), 0.89 (d, J = 6 Hz, 3H). 13C NMR (CDCl3, 75 MHz): δ 219.4, 212.9, 51.9, 48.4, 47.8, 45.8, 43.2, 42.8, 37.4, 33.6, 27.6, 26.4, 24.4, 22.4. MS (m/z), 55 (100), 110 (77), 163 (42), 220 (M+, 59); HRMS calcd for C14H20O2+ [M+H]+ 221.1542, found 221.1546.