

1 Introduction
Because of the strong implications of fluorinated molecules in many areas of research and science (medicinal chemistry, medical imaging, agrochemistry, materials, and so forth) [1], organofluorine chemistry has dramatically blossomed over the past decade. Among the organofluorinated compounds, fluorinated alkenes have recently emerged as an important class of molecules, which can notably be found in bioactive compounds [2]. Moreover, the fluoroolefin moiety can also be envisioned as a stable isosteric and isoelectronic mimic of the peptide bond and can therefore be used to overcome problems associated with the instability of peptides [3]. The metathesis reaction [4], which stands at the top of the class of olefin production and possesses enormous industrial potential, appears as an ideal solution to produce fluoroalkenes. Despite the tremendous developments reported over the past two decades for the development of remarkably efficient processes based on metathesis reactions to synthesize alkenes, only sporadic examples have been reported with fluoroalkenes [5], because of the inherent challenges associated with the use of these reluctant substrates. To date, the few publications describing the ring-closing metathesis (RCM) reactions of fluoroalkene derivatives are mainly dealing with carbon and nitrogen links between the two alkene moieties (Fig. 1) [6–10]. These reactions proved to be efficient mainly with Grubbs II catalyst (and more rarely with Hoveyda–Grubbs II catalyst) and mostly required long reaction time (up to 10 h), high catalyst loading (up to 20 mol %), and/or high temperature reaction (up to 100 °C). This RCM reaction has only been reported twice with O-linker (ester or ether) with low to good yields and has never been reported with other linkers.
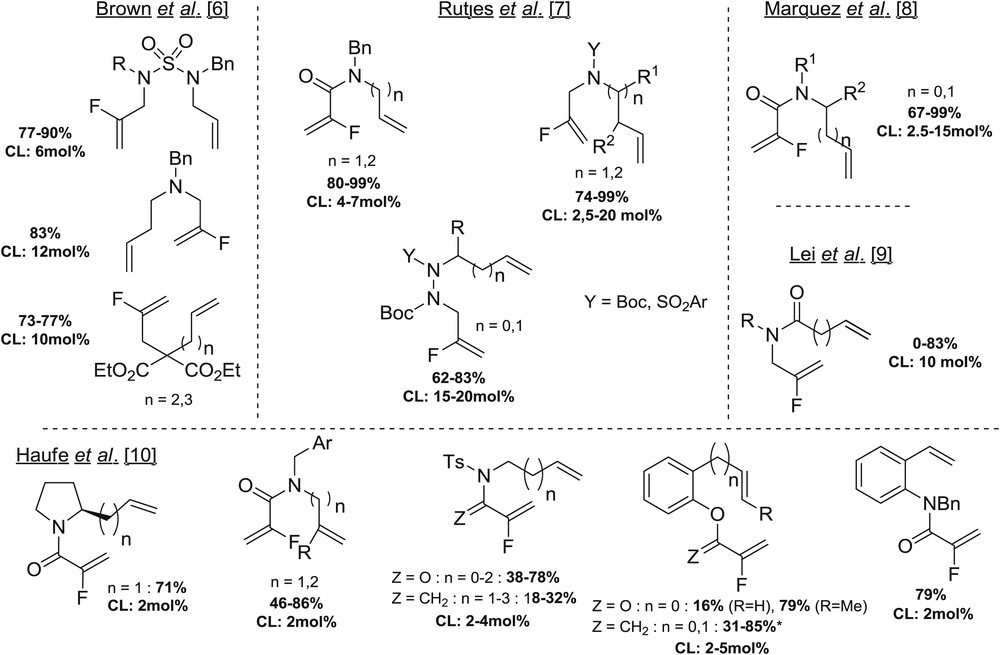
Main examples of fluoroalkenes involved in RCM reaction with Grubbs II catalyst (∗with Hoveyda–Grubbs II catalyst).
In our ongoing project devoted to the production of fluorinated heterocycles and biorelevant molecules [11], we recently published the synthesis of five- and six-membered ring lactam [12], as well as chiral lactam using the RCM reaction (Scheme 1) [13]. We based our strategy on the activation of the substrate by the introduction of appropriate substituent on the fluoroalkene. This strategy, proposed with nonfluorinated substrates by Grubbs et al. [14] and Dorta et al. [15], allows us to reach the best turnover number (TON), to our knowledge, reported with fluoroalkenes, as well as to work under milder experimental conditions than usual. Indeed, the use of fluorinated aromatic hydrocarbons (FAHs) [16] combined with activation of substrates allowed us to work at a temperature of 60 °C and to reach a TON of 100 [12].

Our previous work in the RCM reaction leading to fluorinated lactams.
Taking into account the few links existing between olefinic partners and applicable to the RCM reaction, it appears interesting to develop the synthesis of new heterocycles bearing a fluoroalkene moiety by the RCM reaction. In this context, we turned our attention to the synthesis of bisolefins linked by an oxaza moiety to produce the corresponding fluorinated heterocycles. These NO heterocycles could be valuable intermediates in the production of fluorinated analogs of biomolecules [17], agrochemical agents [18], fluoropseudopeptides [3,13], or other relevant fluorinated building blocks as amino-alcohol or 2,5-dihydro-2-furanylamines [19] (Scheme 2). It has to be noted that the synthesis of such heterocycles has already been reported in nonfluorinated series by Yang and Tae [20] via RCM reaction.
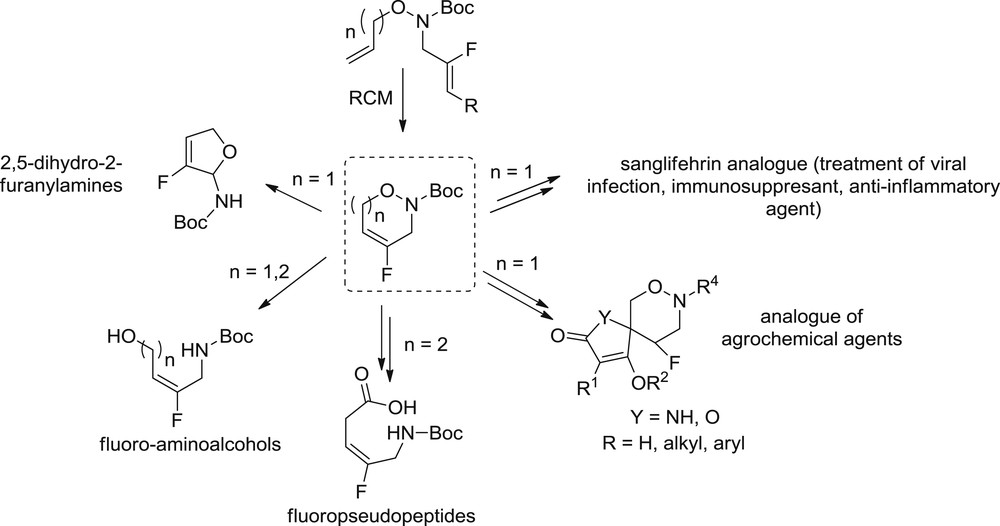
Fluorinated NO heterocycles and potential applications.
2 Results and discussion
2.1 Synthesis of substrates
First, we synthesized two sets of substrates. The first set was devoted to the synthesis of gem-disubstituted fluoroalkenes 2. Starting from commercially available N-Boc-hydroxylamine, a nucleophilic substitution with allyl or homoallyl bromides [20] followed by the reaction with commercially available 2-fluoro-3-chloroprop-1-ene gave access to two bisolefins 2a and 2b (Scheme 3).
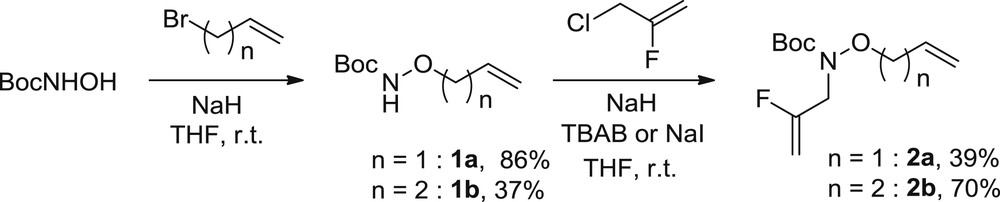
Synthesis of substrates 2.
The second set of substrates was designed to obtain “activated substrates” implying a trisubstituted fluoroalkenes bearing a phenyl group under the Z-configuration. First, we synthesized the ethyl-2-fluorocinnamate 3 by an olefination reaction in 60% yield [21]. The ester moiety was then reduced into alcohol 4 in quantitative yield before transformation into leaving tosylate group (compound 5) [12]. It has to be noted that the latter was used without purification because of its instability. Following the synthesis sequence described in Scheme 4, the N-Boc-hydroxylamine was submitted to a first alkylation with allyl bromide or derivatives followed by a second alkylation with compound 5 to give access to four fluorinated substrates 6 (Scheme 4).
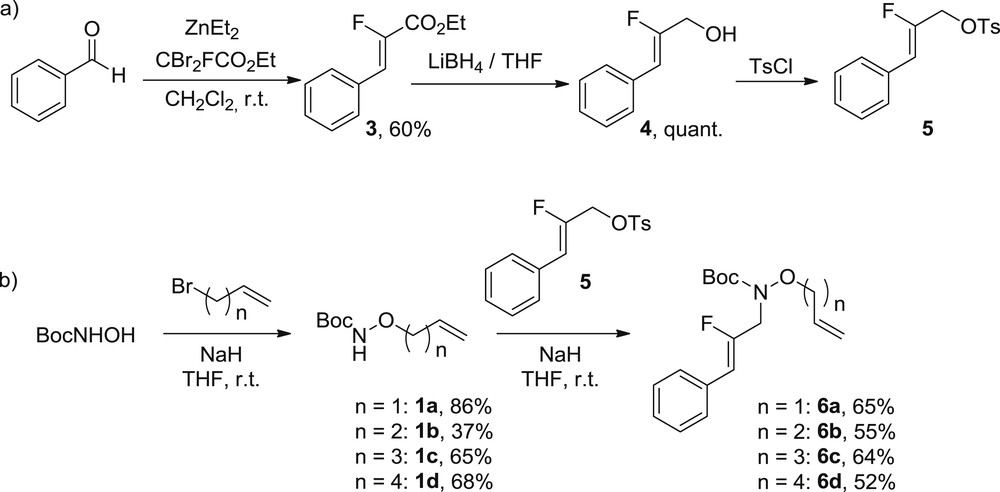
Synthesis of substrates 6. Eq. a) synthesis of fluorinated tosylate 5. Eq. b) synthesis of trisubstituted fluoroalkenes 6.
2.2 RCM
Before discussing the results we obtained concerning the formation of fluorinated NO heterocycles, it has to be noted that the use of experimental procedure described previously by Yang and Tae [20] with nonfluorinated substrates toward NO heterocycles totally failed in the fluorinated series [22].
2.2.1 Six-membered ring
The main results we obtained from synthesis of 4-fluoro-3,6-dihydro-1,2-oxazine 7 are summarized in Table 1.
Synthesis of heterocycles 7 by RCM reactions.
| Entry | Substrate | Solvent (concn) | Catalyst (mol %) | Conditions | Side productsa (%) | Yieldb (%) |
| 1 | 2a | C7F8 (0.02 M) | 10 | 120 °C, 15 min MW (200 W) | 0 | 9 |
| 2 | 2a | C7F8 (0.02 M) | 20 | 105 °C, 180 min, reflux | 77 | 23 |
| 3 | 2a | C7F8 (0.003 M) | 15 | 105 °C, 180 min, reflux | 0 | 71 |
| 4 | 6a | C7F8 (0.02 M) | 5 | 105 °C, 90 min, reflux | 43 | 57 |
| 5 | 6a | C7F8 (0.003 M) | 5 | 105 °C, 90 min, reflux | 0 | 96 |
| 6 | 6a | C7F8 (0.003 M) | 2 | 105 °C, 90 min, reflux | 16 | 76 |
| 7 | 6a | C7F8 (0.02 M) | 5 | 120 °C, 15 min MW (200 W) | 12 | 88 |
| 8 | 6a | C7F8 (0.003 M) | 5 | 120 °C, 30 min MW (200 W) | 6 | 94 |
| 9 | 6a | C7F8 (0.003 M) | 2 | 120 °C, 30 min MW (200 W) | 0 | 10 |
a Percentage of side products generated during the RCM process.
b 1H NMR yield with 2,4-dinitrofluorobenzene as an internal standard.
After testing various experimental conditions (precatalysts and solvents) [22], the best results were obtained with M2 catalyst in perfluorotoluene [16] as a solvent. With substrate 2a, we were able to reach complete conversion only using high catalyst loading (20 mol %) to give 23% yield in the desired heterocycle 7 along with most side products including mainly the homodimer of the nonfluorinated alkene (entry 2). Working under high dilution conditions allowed us to minimize the homodimerization side reaction and to reach 71% yield in 7 albeit with high (15 mol %) catalyst loading (entry 3). As recently reported for the synthesis of fluorinated lactams via RCM [11,12], the use of modified substrate 6 proved to be relevant and more efficient than the nonsubstituted substrate 2. Indeed, the complete conversion of 6 was obtained with a quarter amount of the catalyst as compared to substrate 2, giving 57% yield of the desired heterocycle 7 (entry 4). Nevertheless, the side products were still present in high quantity (43%) at the end of the reaction. Diluting the system to 0.003 M allowed us to get 96% yield under reflux (entry 5) or 94% yield under microwave activation (entry 8) of the desired product 7 with 5 mol % catalyst loading. The use of 2 mol % catalyst loading led to lower yields (entries 6 and 9). The substitution of fluoroalkene with a phenyl moiety proved, as for the production of fluorinated lactam, to be an efficient strategy to activate the substrate for the formation of the six-membered ring heterocycle 7.
2.2.2 Seven-membered ring
The main results obtained from synthesis of 4-fluoro-3,6,7-trihydro-1,2-oxazepine 8 are summarized in Table 2.
Synthesis of heterocycle 8 by RCM reactions.
| Entry | Substrate | Solvent (concn) | Catalyst (mol %) | Conditions | Side productsa (%) | Yieldb (%) |
| 1 | 2b | C7F8 (0.003 M) | 10 | 110 °C, 30 min MW (200 W) | 0 | 30 |
| 2 | 2b | C7F8 (0.003 M) | 20 | 105 °C, 210 min, reflux | 0 | 79 |
| 3 | 2b | C7F8 (0.003 M) | 30 | 105 °C, 1500 min, reflux | 0 | 90 |
| 4 | 6b | C7F8 (0.003 M) | 5 | 120 °C, 30 min MW (200 W) | 0 | 98 |
| 5 | 6b | C7F8 (0.003 M) | 2 | 120 °C, 30 min MW (200 W) | 0 | 75 |
| 6 | 6b | C7F8 (0.003 M) | 2 | 105 °C, 30 min, reflux | <5 | 81 |
a Percentage of side products generated during the RCM process.
b 1H NMR yield with 2,4-dinitrofluorobenzene as an internal standard.
For the seven-membered ring, the reaction with substrate 2b gave only 30% yield of the desired product 8 with 10 mol % catalyst loading (entry 1). Increasing the catalyst loading to 20 mol % led to 79% yield in 8 (entry 2), whereas a high catalyst loading of 30 mol % allowed us to obtain excellent 90% yield without side products (entry 3). The use of the modified substrate 6b proved once again to be highly beneficial for the reaction giving, with 5 mol % catalyst, the best result obtained in product 8 with almost quantitative yield (entry 4). Decreasing the catalyst loading to 2 mol % gave fair results under reflux or microwave irradiation with 81% and 75% yields, respectively (entries 5 and 6).
2.2.3 Eight- and nine-membered ring
Unfortunately, different attempts to get the cyclized compound from 6c (eight-membered ring) and 6d (nine-membered ring) proved to be unsuccessful yielding only side products and mainly the homodimers resulting in Cross-Metathesis (CM) reaction of nonfluorinated double bond.
2.3 Improved experimental procedure
The RCM reaction of substrates 2 and 6 required to work under high dilution conditions, which can be problematic because of the high quantity of a solvent used during the reaction. Moreover, the reaction also required to use perfluorotoluene, FAH solvent, which is quite expensive, even if we can recover a major quantity by distillation at the end of the reaction. Inspired by previous report by Grela et al. [16c], to minimize the quantity of FAH solvent used, we decided to carry out the reaction in a mixture of solvents, using less expensive toluene or dichloromethane, two common solvents for the metathesis reaction. To test the relevancy of the mixture of solvents we carried out the RCM reaction with amide substrate 9 requiring only a concentration of 0.02 M in the media.
The results obtained from the reaction of amide 9 in a mixture of C7F8/solvent are shown in Fig. 2.

Study of mixture of solvents to achieve RCM reaction in good yields.
With toluene as a cosolvent, the results were not interesting because the reaction yield dropped as soon as toluene was introduced in the media. On the contrary, dichloromethane gave relevant results using a 50:50 mixture of solvents. The reaction yield dropped when less than 50% of octafluorotoluene was used. So to carry out the reaction on larger quantity of substrate 6, we decided to use a mixture of solvent C7F8/CH2Cl2 (50:50) to isolate the heterocycles 7 and 8 (Scheme 5).

Synthesis of heterocycles 7 and 8 in a mixture of solvents.
In these conditions, we could save half quantity of the FAH solvent and the heterocycles 7 and 8 were isolated in 40% and 89% yields, respectively.
3 Conclusions
We developed the RCM reaction on new substrates bearing a fluoroalkene moiety with olefins linked for the first time by an oxaza link. We showed that the substitution of fluoroalkene by a phenyl ring is, as for the formation of lactam, an astonishing strategy to improve the reaction conversion. The heterocycles could be obtained in very good yields, albeit in high diluting system. A modified experimental procedure with a mixture of solvents allowed saving half quantity of the FAH solvent although still furnishing good yields of the reaction.
4 Experimental section
4.1 General
All reactions have been carried out under inert atmosphere in predried glassware. The dry common solvents used were purified by an Innovative Technology PureSolv purification system. Octafluorotoluene (perfluorinated solvent; FluoroChem) was directly used from a commercial bottle and kept under argon atmosphere. Commercially available chemicals were obtained from Acros Organics, Aldrich Chemical, Strem Chemicals, Alfa Aesar, TCI, and used as received unless otherwise stated. The M2 catalyst was purchased from Aldrich. All commercial chemicals were used without further purification. NMR spectra were recorded on a Bruker AC 400 spectrometer. Chemical shifts (δ) are quoted in ppm relative to TMS (1H) and CFCl3 (19F). Coupling constants (J) are quoted in Hz. Microwave-assisted reactions were carried out into CEM Microwave Discover (200 W). High-resolution mass spectra (HRMS) were recorded on Q-TOF Micro WATERS. Infrared spectra were recorded on a PerkinElmer Spectrum One Spectrophotometer. The wave numbers (ν) of recorded IR signals are quoted in cm−1. Analytical thin layer chromatography was performed on precoated 200 μm layer thickness silica gel 60 Alumigram Xtra SIL G/UV254. Visualization was accomplished with short wave UV light, potassium permanganate, or phosphomolybdic acid staining solution followed by heating. Flash chromatography was performed on Merck silica gel (40–63 mesh, solid deposit) by a standard technique. Yields refer to chromatographically and spectroscopically pure compounds. The products 1a–d [20,23], 3 [21], 4 [24], 5 [12], 9 [12], and 10 [12] have previously been described and were identified by comparison of their physical and spectroscopic data (1H, 19F, and 13C NMR) with those in the cited references.
4.2 Synthesis of substrates
4.2.1 tert-Butyl allyloxy(2-fluoroallyl)carbamate (2a)
A 100 mL two-neck flask equipped with a stir bar and a reflux condenser was flushed with argon. A solution of tert-butyl allyloxycarbamate (1 equiv, 400 mg, 2.31 mmol) in THF (20 mL) was prepared. NaH (2 equiv, 110.8 mg, 4.62 mmol) was then added. At the end of the bubbling, 3-chloro-2-fluoropropene (2 equiv, 273 μL, 4.62 mmol) was added slowly followed by TBAB (2 equiv, 4.62 mmol, 1.49 g). The mixture was heated to 40 °C and stirred for 24 h. The reaction was stopped by adding NH4Cl (20 mL) and extracted with DCM (3 × 25 mL). The organic layers were combined and dried over anhydrous MgSO4. After filtration and evaporation of the volatile materials under reduced pressure, the crude material was purified by flash chromatography on silica gel (cyclohexane/DCM = 7:3, Rf = 0.41) to afford 2a (208 mg, 39%) as a colorless oil.
1H NMR (400.13 MHz, CDCl3): δ 5.98 (ddt, 3JH-H = 17 Hz, 3JH-H = 10 Hz, 3JH-H = 6 Hz, 1H, HC=CH2), 5.31 (m, 2H, HC=CH2), 4.74 (dd, 3JF-H = 16 Hz, 3JF-H = 3 Hz, 1H, FC=CH2), 4.52 (dd, 3JF-H = 48 Hz, 3JF-H = 3 Hz, 1H, FC=CH2), 4.38 (d, 3JH-H = 6 Hz, 2H, O–CH2), 4.14 (d, 3JH-F = 13 Hz, 2H, N–CH2), 1.51 (s, 9H, C(CH3)3); 19F NMR (376.50 MHz, CDCl3): δ −103.53 (ddt, 3JF-H = 48 Hz, 3JF-H = 16 Hz, 3JF-H = 13 Hz, 1F); 13C NMR (75.47 MHz, CDCl3): δ 160.8 (d, 1JC-F = 261 Hz, 1C, CF), 156.3 (s, 1C, N–C=O), 132.4 (s, 1C, HC=CH2), 119.9 (s, 1C, HC=CH2), 93.1 (d, 2JC-F = 18 Hz, 1C, FC=CH2), 82.1 (s, 1C, C(CH3)3), 76.5 (s, 1C, O–CH2), 50.6 (d, 2JC-F = 32 Hz, 1C, N–CH2), 28.2 (3C, C(CH3)3).
IR (neat) ν 2951, 2929, 1710, 1489, 1466, 1450, 1363, 1224, 1191, 1117, 1086, 972, 925, 884, 838, 693 cm−1; HRMS (ESI+): m/z = 254,1172 [M+Na]+, calcd for C11H18FNO3Na: 254.1168.
4.2.2 tert-Butyl but-3-en-1-yloxy(2-fluoroallyl)carbamate (2b)
A 100 mL two-neck flask equipped with a stir bar and a reflux condenser was flushed with argon. A solution of tert-butyl but-3-en-1-yloxycarbamate (1 equiv, 230 mg, 1.23 mmol) in THF (13 mL) was prepared. NaH (2 equiv, 368 mg, 2.46 mmol) was added. At the end of the bubbling, 3-chloro-2-fluoropropene (2 equiv, 145 μL, 2.46 mmol) was added slowly followed by NaI (2 equiv, 2.46 mmol, 368 mg). The mixture was heated to 40 °C and stirred for 24 h. The reaction was stopped by adding NH4Cl (20 mL) and extracted with DCM (3 × 25 mL). The organic layers were combined and dried over anhydrous MgSO4. After filtration and evaporation of the volatile materials under reduced pressure, the crude material was purified by flash chromatography on silica gel (cyclohexane/DCM = 7:3, Rf = 0.44) to afford 2b (211 mg, 70%) as a colorless oil.
1H NMR (400.13 MHz, CDCl3): δ 5.84 (ddt, 3JH-H = 17 Hz, 3JH-H = 10 Hz, 3JH-H = 7 Hz, 1H, HC=CH2), 5.10 (m, 2H, HC=CH2), 4.74 (dd, 3JF-H = 16 Hz, 3JF-H = 3 Hz, 1H, FC=CH2), 4.52 (dd, 3JF-H = 48 Hz, 3JF-H = 3 Hz, 1H, FC=CH2), 4.13 (d, 3JH-F = 13 Hz, 2H, N–CH2), 3.95 (t, 3JH-H = 7 Hz, 2H, O–CH2), 2.38 (dt, 3JH-H = 7 Hz, 3JH-H = 7 Hz, 2H, CH2) 1.51 (s, 9H, C(CH3)3); 19F NMR (376.50 MHz, CDCl3): δ −103.54 (ddt, 3JF-H = 48 Hz, 3JF-H = 16 Hz, 3JF-H = 13 Hz, 1F); 13C NMR (75.47 MHz, CDCl3): δ 160.6 (d, 1JC-F = 260 Hz, 1C, CF), 156.3 (s, 1C, N–C=O), 134.4 (s, 1C, HC=CH2), 116.5 (s, 1C, HC=CH2), 93.4 (d, 3JC-F = 17 Hz, 1C, FC=CH2), 81.9 (s, 1C, C(CH3)3), 74.4 (s, 1C, O–CH2), 50.6 (d, 2JC-F = 31 Hz, 1C, N–CH2), 32.3 (s, 1C, CH2)28.1 (3C, C(CH3)3).
IR (neat) ν 2955, 2936, 1699, 1495, 1461, 1445, 1421, 1371, 1226, 1152, 1105, 1073, 989, 854, 827 cm−1; HRMS (ESI+): m/z = 268.1333 [M+Na]+, calcd for C12H20FNO3Na: 268.1325.
4.2.3 (Z)-tert-Butyl allyloxy(2-fluoro-3-phenylallyl)carbamate (6a)
A 100 mL two-neck flask equipped with a stir bar and a reflux condenser was flushed with argon. A solution of tert-butyl allyloxycarbamate (1 equiv, 400 mg, 2.31 mmol) in THF (20 mL) was prepared. NaH (2 equiv, 110.8 mg, 4.62 mmol) was added. At the end of the bubbling, the mixture was cooled down to 0 °C and the tosylate (1.2 equiv, 849 mg, 2.77 mmol) in a solution of THF was added slowly. The mixture was stirred for 5 h. The reaction was monitored by TLC and 19F NMR. When full consumption of the starting material was observed, the reaction was stopped by adding NH4Cl (20 mL) and extracted with DCM (3 × 25 mL). The organic layers were combined and dried over anhydrous MgSO4. After filtration and evaporation of the volatile materials under reduced pressure, the crude material was purified by flash chromatography on silica gel (cyclohexane/DCM = 7:3, Rf = 0.39) to afford 6a (461 mg, 65%) as a colorless oil.
1H NMR (400.13 MHz, CDCl3): δ 7.27 (m, 5H, ArH), 5.90 (ddt, 3JH-H = 17 Hz, 3JH-H = 10 Hz, 3JH-H = 6 Hz, 1H, HC=CH2), 5.65 (d, 3JH-F = 38 Hz, 1H, FC=CH), 5.21 (m, 2H, HC=CH2), 4.33 (d, 3JH-H = 6 Hz, 2H, O–CH2), 4.20 (d, 3JH-F = 16 Hz, 2H, N–CH2), 1.45 (s, 9H, C(CH3)3); 19F NMR (376.50 MHz, CDCl3): δ −108.84 (dt, 3JF-H = 38 Hz, 3JF-H = 16 Hz, 1F); 13C NMR (75.47 MHz, CDCl3): δ 156.3 (s, 1C, N–C=O), 156.0 (d, 1JC-F = 269 Hz, 1C, CF), 132.7 (d, 3JC-F = 3 Hz, 1C, ArC), 132.3 (s, 1C, HC=CH2), 128.6 (2C, ArCH), 128.4 (2C, ArCH), 127.3 (d, 6JC-F = 2 Hz, 1C, ArCH), 119.8 (s, 1C, HC=CH2), 109.2 (d, 2JC-F = 7 Hz, 1C, FC=CH), 81.9 (s, 1C, C(CH3)3), 76.6 (s, 1C, O–CH2), 51.8 (d, 2JC-F = 30 Hz, 1C, N–CH2), 28.1 (3C, C(CH3)3).
IR (neat) ν 2979, 2933, 1702, 1495, 1476, 1451, 1367, 1232, 1155, 1089, 991, 932, 876, 851, 754, 693 cm−1; HRMS (ESI+): m/z = 330.1484 [M+Na]+, calcd for C17H22FNO3Na: 330.1481.
4.2.4 (Z)-tert-Butyl but-3-en-1-yloxy(2-fluoro-3-phenylallyl)carbamate (6b)
A 100 mL two-neck flask equipped with a stir bar and a reflux condenser was flushed with argon. A solution of tert-butyl but-3-en-1-yloxycarbamate (1 equiv, 300 mg, 1.6 mmol) in THF (10 mL) was prepared. NaH (2 equiv, 76.9 mg, 3.20 mmol) was added. At the end of the bubbling, the mixture was cooled down to 0 °C and the tosylate (1.2 equiv, 589 mg, 1.92 mmol) in a solution of THF was added slowly. The mixture was stirred for 5 h. The reaction was monitored by TLC and 19F NMR. When full consumption of starting material was observed, the reaction was stopped by adding NH4Cl (10 mL) and extracted with DCM (3 × 15 mL). The organic layers were combined and dried over anhydrous MgSO4. After filtration and evaporation of the volatile materials under reduced pressure, the crude material was purified by flash chromatography on silica gel (cyclohexane/DCM = 7:3, Rf = 0.43) to afford 6b (283 mg, 55%) as a colorless oil.
1H NMR (400.13 MHz, CDCl3): δ 7.27 (m, 5H, ArH), 5.74 (ddt, 3JH-H = 17 Hz, 3JH-H = 10 Hz, 3JH-H = 7 Hz, 1H, HC=CH2), 5.63 (d, 3JH-F = 38 Hz, 1H, FC=CH), 5.00 (m, 2H, HC=CH2), 4.18 (d, 3JH-F = 16 Hz, 2H, N–CH2), 3.89 (t, 3JH-H = 7 Hz, 2H, O–CH2), 2.30 (q, 3JH-H = 7 Hz, 2H, O–CH2–CH2), 1.44 (s, 9H, C(CH3)3); 19F NMR (376.50 MHz, CDCl3): δ −108.95 (dt, 3JF-H = 38 Hz, 3JF-H = 16 Hz, 1F); 13C NMR (75.47 MHz, CDCl3): δ 156.3 (s, 1C, N–C=O), 153.5 (d, 1JC-F = 269 Hz, 1C, CF), 134.6 (s, 1C, HC=CH2), 132.8 (d, 3JC-F = 3 Hz, 1C, ArC), 128.7 (2C, ArCH), 128.5 (2C, ArCH), 127.5 (d, 6JC-F = 2 Hz, 1C, ArCH), 116.7 (s, 1C, HC=CH2), 109.4 (d, 2JC-F = 7 Hz, 1C, FC=CH), 82.1 (s, 1C, C(CH3)3), 74.6 (s, 1C, O–CH2), 51.8 (d, 2JC-F = 30 Hz, 1C, N–CH2), 32.6 (s, 1C, O–CH2–CH2), 28.3 (3C, C(CH3)3).
IR (neat) ν 2978, 1702, 1642, 1495, 1476, 1451, 1418, 1367, 1232, 1156, 1090, 1028, 991, 915, 876, 852, 753, 693 cm−1; HRMS (ESI+): m/z = 344.1646 [M+Na]+, calcd for C18H24FNO3Na: 344.1638.
4.2.5 (Z)-tert-Butyl (2-fluoro-3-phenylallyl)(pent-4-en-1-yloxy)carbamate (6c)
A 50 mL two-neck flask equipped with a stir bar and a reflux condenser was flushed with argon. A solution of tert-butyl but-3-en-1-yloxycarbamate (1 equiv, 300 mg, 1.49 mmol) in THF (5 mL) was prepared. NaH (2 equiv, 71.54 mg, 2.98 mmol) was added. At the end of the bubbling, the mixture was cooled down to 0 °C and the tosylate (1.2 equiv, 548 mg, 1.79 mmol) in a solution of THF was added slowly. The mixture was stirred for 5 h. The reaction was monitored by TLC and 19F NMR. When full consumption of the starting material was observed, the reaction was stopped by adding NH4Cl (5 mL) and extracted with DCM (3 × 5 mL). The organic layers were combined and dried over anhydrous MgSO4. After filtration and evaporation of the volatile materials under reduced pressure, the crude material was purified by flash chromatography on silica gel (cyclohexane/CH2Cl2 = 7:3, Rf = 0.43) to afford 6c (320 mg, 64%) as a colorless oil.
1H NMR (400.13 MHz, CDCl3): δ 7.51–7.23 (m, 5H, ArH), 5.78 (ddt, 3JH-H = 17 Hz, 3JH-H = 10 Hz, 3JH-H = 7 Hz, 1H, HC=CH2), 5.70 (d, 3JH-F = 38 Hz, 1H, FC=CH), 4.97 (m, 2H, HC=CH2), 4.26 (d, 3JH-F = 16 Hz, 2H, N–CH2), 3.92 (t, 3JH-H = 7 Hz, 2H, O–CH2), 2.14 (m, 2H, O–CH2–CH2), 1.70 (m, 2H, O–CH2–CH2–CH2), 1.52 (s, 9H, C(CH3)3); 19F NMR (376.50 MHz, CDCl3): δ −108.85 (dt, 3JF-H = 37 Hz, 3JF-H = 16 Hz, 1F); 13C NMR (75.47 MHz, CDCl3): δ 156.3 (s, 1C, N–C=O), 154.9 (d, 1JC-F = 269 Hz, 1C, CF), 137.9 (s, 1C, HC=CH2), 132.8 (d, 3JC-F = 2 Hz, 1C, ArC), 128.7 (2C, ArCH), 128.5 (2C, ArCH), 127.5 (d, 6JC-F = 2 Hz, 1C, ArCH), 114.9 (s, 1C, HC=CH2), 109.3 (d, 2JC-F = 7 Hz, 1C, FC=CH), 82.0 (s, 1C, C(CH3)3), 74.8 (s, 1C, O–CH2), 51.6 (d, 2JC-F = 30 Hz, 1C, N–CH2), 30.1 (s, 1C, O–CH2–CH2–CH2), 28.3 (3C, C(CH3)3), 27.4 (s, 1C, O–CH2–CH2).
IR (neat) ν 2977, 2933, 1704, 1641, 1495, 1475, 1450, 1417, 1367, 1231, 1157, 1091, 1033, 994, 959, 913, 877, 851, 753, 692 cm−1; HRMS (ESI+): m/z = 358.1810 [M+Na]+, calcd for C19H26FNO3Na: 358.1794.
4.2.6 (Z)-tert-Butyl (2-fluoro-3-phenylallyl)(hex-5-en-1-yloxy)carbamate (6d)
A 50 mL two-neck flask equipped with a stir bar and a reflux condenser was flushed with argon. A solution of tert-butyl but-3-en-1-yloxycarbamate (1 equiv, 300 mg, 1.4 mmol) in THF (5 mL) was prepared. NaH (2 equiv, 66.9 mg, 2.8 mmol) was added. At the end of the bubbling, the mixture was cooled down to 0 °C and the tosylate (1.2 equiv, 589 mg, 1.92 mmol) in a solution of THF was added slowly. The mixture was stirred for 5 h. The reaction was monitored by TLC and 19F NMR. When full consumption of starting material was observed, the reaction was stopped by adding NH4Cl (5 mL) and extracted with DCM (3 × 5 mL). The organic layers were combined and dried over anhydrous MgSO4. After filtration and evaporation of the volatile materials under reduced pressure, the crude material was purified by flash chromatography on silica gel (cyclohexane/CH2Cl2 = 70/30, Rf = 0.43) to afford 6d (250 mg, 52%) as a colorless oil.
1H NMR (400.13 MHz, CDCl3): δ 7.53–7.26 (m, 5H, ArH), 5.78 (ddt, 3JH-H = 17 Hz, 3JH-H = 10 Hz, 3JH-H = 7 Hz, 1H, HC=CH2), 5.73 (d, 3JH-F = 38 Hz, 1H, FC=CH), 4.97 (m, 2H, HC=CH2), 4.28 (d, 3JH-F = 16 Hz, 2H, N–CH2), 3.94 (t, 3JH-H = 7 Hz, 2H, O–CH2), 2.07 (m, 2H, OCH2CH2CH2–CH2), 1.63 (m, 2H, O–CH2–CH2), 1.54 (s, 9H, C(CH3)3), 1.50 (m, 2H, O–CH2–CH2–CH2); 19F NMR (376.50 MHz, CDCl3): δ −108.85 (dt, 3JF-H = 38 Hz, 3JF-H = 16 Hz, 1F); 13C NMR (75.47 MHz, CDCl3): δ 156.4 (s, 1C, N–C=O), 154.9 (d, 1JC-F = 271 Hz, 1C, CF), 138.5 (s, 1C, HC=CH2), 132.9 (s, 1C, ArC), 128.6 (2C, ArCH), 128.5 (2C, ArCH), 127.4 (s, 1C, ArCH), 114.7 (s, 1C, HC=CH2), 109.3 (d, 2JC-F = 7 Hz, 1C, FC=CH), 81.9 (s, 1C, C(CH3)3), 75.3 (s, 1C, O–CH2), 51.6 (d, 2JC-F = 30 Hz, 1C, N–CH2), 33.5 (s, 1C, O–CH2–CH2–CH2–CH2), 28.3 (3C, C(CH3)3), 27.6 (s, 1C, O–CH2–CH2), 25.3 (s, 1C, O–CH2–CH2–CH2).
IR (neat) ν 2977, 2935, 1704, 1640, 1495, 1450, 1416, 1367, 1231, 1158, 1091, 1032, 994, 911, 875, 853, 753, 692 cm−1; HRMS (ESI+): m/z = 372.1959 [M+H]+, calcd for C20H28FNO3Na: 372.1951.
4.3 RCM reaction
4.3.1 Under reflux
An oven-dried two-neck flask, equipped with a reflux condenser, was filled under argon with diene (1 equiv, 33 μmol) and solvent before the addition of the ruthenium precatalyst. Progress of the reaction could be monitored by TLC and 1H NMR. The solvent was removed under vacuum or distilled and recovered. The internal standard 2,4-dinitrofluorobenzol was then added to the crude residue and the yield was calculated by both 1H and 19F NMR integrations. The crude residue could be purified by flash column chromatography to reach the analytically pure product.
4.3.2 Under microwave irradiation
An oven-dried cap sealed microwave vial was filled with diene (1equiv, 33 μmol) and solvent before the addition of the ruthenium precatalyst at the reported loading. The vial was flushed with argon and then placed in a microwave reactor and heated for the reported time. The vial content was then transferred to a round-bottom flask where the solvent was removed under vacuum or distilled and recovered. The internal standard 2,4-dinitrofluorobenzol was then added to the crude residue and the yield was calculated by both 1H and 19F NMR integrations. The crude residue could be purified by flash column chromatography to obtain the analytically pure product.
4.3.3 Under sealed vials (mixture of solvent)
An oven-dried cap sealed vial, equipped with a stir bar under argon atmosphere, was filled with diene, dichloromethane, and perfluorotoluene before the addition of the ruthenium precatalyst. The vial was placed in an oil bath and heated to 120 °C for 30 min. The vial content was then cooled down to room temperature and transferred to a round-bottom flask where the solvent was removed under vacuum. The crude residue could be purified by flash column chromatography to obtain the analytically pure product.
4.3.3.1 tert-Butyl 4-fluoro-3,6-dihydro-2H-1,2-oxazine-2-carboxylate (7)
An oven-dried cap sealed vial, equipped with a stir bar under argon atmosphere, was filled with bisolefin 6a (1equiv, 244 μmol, 75 mg), dichloromethane (40 mL), and perfluorotoluene (40 mL) before the addition of the precatalyst M2 (0.05 equiv, 12.2 μmol, 11.57 mg). The vial was placed in an oil bath and heated to 120 °C for 30 min. The vial content was then cooled down to room temperature and transferred to a round-bottom flask where the solvent was removed under vacuum. The crude material is then purified through silica gel column chromatography (pentane/diethyl ether = 85:15, Rf = 0.25) affording the desired heterocycle 7 (19.8 mg, 40%) as a colorless oil.
1H NMR (400.13 MHz, CDCl3): δ 5.40 (d, 3JH-F = 16 Hz, 1H, HC=CF), 4.46 (m, 2H, N–CH2), 4.15 (m, 2H, O–CH2), 1.52 (s, 9H, S–C(CH3)3); 19F NMR (376.50 MHz, CDCl3): δ −113.40 (m, 1F); 13C NMR (75.47 MHz, CDCl3): δ 154.7 (d, 4JC-F = 2 Hz, 1C, N–C=O), 154.2 (d, 1JC-F = 261 Hz, 1C, C–F), 99.8 (d, 2JC-F = 11 Hz, 1C, HC=CF), 82.4 (s, 1C, C(CH3)3), 66.3 (d, 2JC-F = 6 Hz, 1C, O–CH2), 45.1 (d, 2JC-F = 36 Hz, 1C, N–CH2–CF), 28.2 (s, 3C, C(CH3)3).
IR (neat) ν 2924, 2854, 1707, 1682, 1456, 1393, 1368, 1346, 1236, 1213, 1150, 1087, 1012, 992, 935, 851, 769 cm−1; HRMS (ESI+): m/z = 226.0861 [M+Na]+, calcd for C9H14FNO3Na: 226.0855.
4.3.3.2 tert-Butyl 4-fluoro-3,6,7-trihydro-3H-1,2-oxazepine-2-carboxylate (8)
An oven-dried cap sealed vial, equipped with a stir bar under argon atmosphere, was filled with bisolefin 6b (1equiv, 389 μmol, 125 mg), dichloromethane (65 mL), and perfluorotoluene (65 mL) before the addition of the precatalyst M2 (0.05 equiv, 19.45 μmol, 18.46 mg). The vial was placed in an oil bath and heated to 120 °C for 30 min. The vial content was then cooled down to room temperature and transferred to a round-bottom flask where the solvent was removed under vacuum. The crude material is then purified through silica gel column chromatography (pentane/diethyl ether = 85:15, Rf = 0.31) affording the desired heterocycle 8 (75 mg, 89%) as a colorless oil.
1H NMR (400.13 MHz, CDCl3): δ 5.40 (dt, 3JH-F = 21 Hz, 3JH-H = 6 Hz, 1H, HC=CF), 4.37 (m, 2H, N–CH2), 4.19 (t, 3JH-H = 6 Hz, 2H, O–CH2–CH2), 2.37 (m, 2H, O–CH2–CH2), 1.51 (s, 9H, C(CH3)3); 19F NMR (376.50 MHz, CDCl3): δ −107.92 (d, 3JF-H = 21 Hz, 1F); 13C NMR (75.47 MHz, CDCl3): δ 157.3 (d, 1JC-F = 251 Hz, 1C, C–F), 154.7 (d, 4JC-F = 2 Hz, 1C, N–C=O), 102.3 (d, 2JC-F = 20 Hz, 1C, HC=CF), 82.0 (s, 1C, C(CH3)3), 74.7 (s, 1C, O–CH2–CH2), 50.2 (d, 2JC-F = 46 Hz, 1C, N–CH2–CF), 28.3 (s, 3C, C(CH3)3), 22.1 (d, 3JC-F = 10 Hz, 1C, CH2–CH=CF);
IR (neat) ν 2929, 2856, 1708, 1681, 1458, 1371, 1346, 1235, 1217, 1140, 1091, 1008, 955, 932, 850 cm−1; HRMS (ESI+): m/z = 240.1021 [M+Na]+, calcd for C10H16FNO3Na: 240.1012.
Acknowledgments
This work was promoted by the Interregional Norman Chemistry Network (CRUNCh). The “Régions Basse et Haute−Normandie” are gratefully thanked for their financial support. ENSICAEN and University of Caen, INSA Rouen, Rouen University, CNRS, and Labex SynOrg (ANR-11-LABX-0029) are also thanked for their support.