

1 Introduction
Hydrogen (H2) is expected to be the clean energy carrier and regarded as an ideal long-term solution for the future energy development [1–4]. However, many obstacles with respect to its generation, storage, and safety still need to be overcome. H2 could be produced directly by the environmentally friendly process of proton reduction using abundant and renewable water and sunlight using efficient low-cost photocatalysts, which may provide a technologically feasible basis for artificial photosynthesis [5]. Chemical energy in this process can be stored as HH bonds and used in the fuel cells to form water as a sole product. H2 can also be stored in hydrogenation products of unsaturated organic compounds that can release H2 under suitable conditions [6].
However, one of the crucial questions on proton reduction for a sustainable hydrogen technology is the electron source. Clearly, stoichiometric electron reservoirs from nonrenewable sources are not an option. In nature, conversion of solar energy to chemical energy takes place in the oxygen-evolving center of the photosystem II (PS II), which is present in green plants, algae, and cyanobacteria [5]. The green pigment chlorophyll assists in harvesting solar energy to oxidize water to dioxygen, whereas the generated electrons reduce carbon dioxide through a highly complicated multistep process [7].
Over the years, several catalysts containing precious and nonprecious metals with promising efficiency to split water have been demonstrated [8]. What is unclear at this point in time is the well-defined and commonly accepted protocol for the measurement, identical device, and reporting the new data for a direct comparison with the literature. For instance, when a new material is tested for the water splitting, the effectiveness of the catalyst in comparison to other literature reported catalytic systems in terms of activity and stability becomes seemingly impossible because of the different applied reaction conditions. The total amount of evolved gas from water photolysis and the rate of production cannot be practically compared with two materials from different laboratories. There is a strong need to correlate the literature data (benchmark systems) with own experimental results.
In recent years, few attempts have been made to benchmark the electrocatalytic water splitting [9]. Under similar reaction conditions and using the same experimental device and setup, several electrocatalysts have been tested for oxygen evolution reaction and the overpotential at a current density of 10 mA cm−2 has been compared [9a]. On the other hand, the electrocatalytic hydrogen and oxygen evolution activity and the stability of the catalysts have been measured and compared under identical conditions in acidic as well as in alkaline solutions [9b]. A standard protocol has been followed to report the catalytic activity considering the electrochemically active surface area and the short-term stability that indeed helps to get a clear understanding of the catalytic activities of different catalysts. In this respect, many of us have come out with the approach of comparing newly made catalysts with the commercially available catalysts like RuO2, IrO2, and Pt/C on various electrode substrates under the identical reaction conditions [10–15].
For the photocatalytic reaction with heterogeneous powder catalyst dispersed in a solution, the parameters like turnover number (TON) and turnover frequency (TOF) are often used like a thermal reaction [16]. Because the incident area of radiation, available active sites, and surface area of the dispersed photocatalyst are ambiguous, IUPAC has recommended the use of the term “quantum efficiency” with respect to the absorbed photons [17]. For the photocatalytic water splitting, the activity has been often described by photonic efficiency, that is, the initial rate of a photoreaction with respect to the incident photons irradiated inside the reactor [18]. Photonic efficiency neglects the intrinsic property of the photocatalyst without counting the adsorbed photon by the photocatalyst. Even the photonic yield has been reported by various groups when monochromatic light is used [19]. In this respect, the calculation of quantum efficiency is the most convenient way to compare the intrinsic properties of different photocatalysts [20]. It should be noted here that the energy of the incident photon also varies with the light source. Therefore, the specification of the light source along with the energy of the incident light is highly recommended. In an attempt to represent the photocatalytic activity of the solid catalysts dispersed in solution, Bahnemann and Kisch et al. [21] suggested to determine apparent optimal quantum yield using monochromatic light and compare the activities of different photocatalysts under identical conditions in a single photoreactor.
The situation becomes more complicated in the case of a photosensitized reaction, where multiple components come into play starting from the nature of the sensitizer, concentration, or stability [22–25]. The transfer of the energy from the sensitizer to the catalyst is the most important step of the whole process. The concentration of the dark catalyst, sacrificial agent, and their stability in the presence of light also controls the overall photochemical reaction. So far, no standard protocol has been followed to compare the activity of the catalyst in a photosensitized reaction.
In this study, we will discuss the necessity to follow certain experimental standards such as the need for well-defined and commonly accepted standard reaction conditions, apparatus, instrumental setup, and detectors, which play a huge role while performing and optimizing water-splitting experiments. Furthermore, we emphasize on how one can correlate the experimental data of different laboratories to avoid misleading interpretations and to identify “real” benchmarks in the field. Therefore, the pinpoints of this essay are to refer to the predominantly experimental parameters that affect water photolysis. We believe that comparing data from various reaction conditions in a well-defined and most commonly accepted way will be important to facilitate progress in the field.
2 Basic principles of water splitting
Photocatalytic water splitting with semiconductor photocatalysts involves suitable band gap and band positions to overcome the demand of 1.23 eV energy [26–28]. In semiconductors, the photogenerated electrons reduce protons to form hydrogen, whereas the holes oxidize water to evolve oxygen [5,29,30]. Several kinetic phenomena affect the electron and hole transfer to split water or their recombination to hinder the process. Numerous semiconductors or composite materials have been comprehended so far for the two half-reactions of water reduction and oxygen formation by using sacrificial agents or even by applying Z-scheme systems [31–33].
As an alternative approach, so-called “dark materials” (nonphotocatalysts) such as metal oxides or metals have been studied systematically and used effectively in the presence of a photosensitizer that in turn have proven to be effective for water oxidation and hydrogen evolution reactions [34–37]. Special interest and maximum efforts have been devoted to developing highly abundant and low-cost transition metal oxides as the functional mimics of PS II [35]. For the photochemical water splitting with metal oxide–based dark catalysts, the photosensitizer is very essential. At the same time, the electron acceptor or donor must be added to carry out the process.
In overall water splitting using semiconductors, generally, a cocatalyst is required to generate catalytic centers, lowering activation energy, and facilitating electron–hole separation and transfer [38]. Either a single semiconductor is used or composites of materials could be organized. Heterojunction of p–n type materials often functions as catalytic centers [39]. In a Z-scheme mechanism, two photocatalysts are combined to form an overall system where the mediator transports charge transfer between two materials [31–33]. Various loading and dispersion of the cocatalyst on the photocatalyst surface may also affect the process to a large extent. Homogeneous distribution along with a strong contact of the cocatalyst and photocatalyst makes the system more effective for the long-term photocatalytic reaction. For the half-reactions, there is a need of sacrificial agent, which in fact supplies electrons for hydrogen evolution and accepts electrons in water oxidation to continue the process [40]. Since the concentration of the sacrificial agent, their photostability, and its interaction with the catalyst and the photosensitizer direct the reaction in multiple ways, respective analytic data should be provided in comparative investigations.
3 Influence of experimental conditions on the photocatalytic activity
Designing the setup for the photochemical water splitting requires a light source, a vessel for the reaction, and detectors for the generated gases. Most of the differences in measurements have arisen from the experimental conditions, as there is no guideline or rule in performing the photocatalytic experiments. Most of the groups have implemented their own setups for conducting the photocatalytic investigation [41,42]. Therefore, the experimental conditions and the used setup vary throughout the literature. We will discuss several experimental parameters and their influence in the determination of the photocatalytic and photochemical activities as well as a way to address the crucial factors influencing the activity in the following sections (Fig. 1).
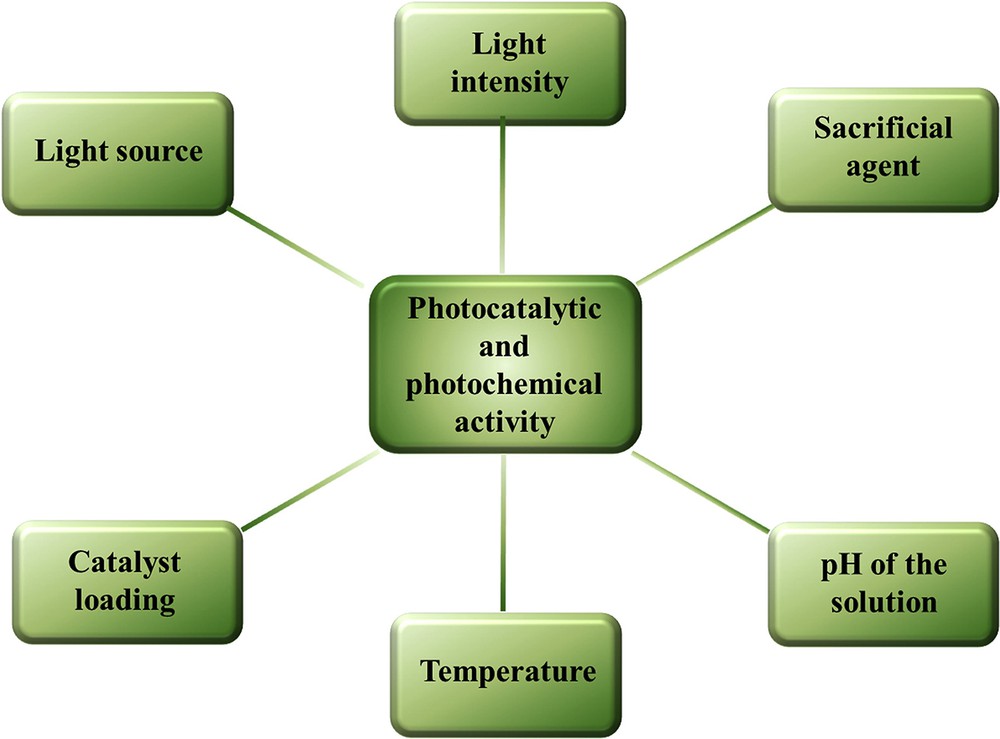
Experimental parameters that influence the photocatalytic and photochemical activities.
By combining the already known literature procedures and based on our own experiences, it is now clear that several physical parameters largely affect the photochemical water-splitting reactions and the quantification of the evolved gases. The key influencing parameters are discussed in the following sections.
3.1 Light source
For the light source, four features mainly come into play: nature of the light source, intensity, exposed area to light, and effective intensity. In the literature, sunlight simulator, Xe lamp, Hg lamp, and incandescence lamp with various intensities have been used as a source of UV and visible light [43]. A variation in the activity is expected with different light sources of similar intensity with the same catalyst under comparable reaction conditions. A recent study on intensity-based catalytic activity of blue light showed a strong dependency of the catalytic activity on light intensity [44]. An increase in activity with the increase in the intensity of the light has been shown to be effectual. Use of a filter to cut off the UV part of the light, usually 395 or 420 nm, often leads to a decrease in activity, whereas the presence of UV light fastens photodecomposition of the catalyst, sacrificial agent, and photosensitizer [45]. Effective intensity of the light at the reaction center depends on the distance from light source, transparency of the photowindow, and reaction mixture. The representation of the optimal rate should be independent of the photoreactor, and hence the activity should be reported against the number of photons at a particular wavelength at a particular time, that is, against photon flux [20].
At the same time, as the exposed area of the catalytic mixture to the light plays an essential role that must be mentioned in the experimental part so that obtained gas evolution rate can be normalized and given as micromoles per square meter per hour for better comparison. In photoreactors, the deposition of particles onto the window blocks the light resulting in lower activity (e.g., Ag deposition when AgNO3 was used as the sacrificial agent) [46]. We investigated the water oxidation activity of Ag3PO4 without using AgNO3 as the sacrificial agent. This helped in preventing the blocking of the quartz window but decomposition of Ag3PO4 to metallic Ag occurred at a faster rate [47,48].
3.2 Reaction temperature
Photocatalytic activity and the detection of the gas are largely affected by the temperature of the system. For the Clark-type electrode measurements, the detection of soluble oxygen varies with temperature [17]. In pressure-based reactor–detector system, the gas volume totally depends on the temperature [42]. Therefore, control of the reaction temperature is mandatory to produce precise data.
3.3 Influence of pH
The pH of a solution and the use of different buffers are also consequential and have a wide impact on the photocatalytic activity [2]. Several buffers have been reported in the literature for the effective water oxidation and reduction. In fact, no rule that has been followed but often slightly acidic buffer or neutral buffer produces an enhancement in the photosensitized water oxidation activity. The observed activity can vary significantly depending upon the type of catalyst and the reaction medium. In general, acetate (pH 5.8), silicate buffer (pH 4.7), and phosphate buffer (pH 7) have been well established to produce good results for the photosensitized water oxidation with transition metal oxide–based catalysts [2].
3.4 Concentration of the reactants
Except all of the physical parameters, the concentration of the photocatalyst, sacrificial agent, cocatalyst, and the photosensitizer controls the activity in different ways. At this point, determination of the optimum rate by varying the amount of the photocatalyst to attain the maximum rate is necessary [20]. On the other hand, with the increasing concentration of a photosensitizer, the catalytic activity reaches to a maximum and then drops subsequently [49]. Increase in the catalytic activity was observed with increasing concentration of a sacrificial agent up to a certain limit. In general, the electron donor sacrificial agents (e.g., ascorbic acid, triethanolamine, alcohols, etc.) are photostable, whereas electron acceptors such as persulfate or silver nitrate undergo photodecomposition under irradiation [2,19,49]. This causes a serious problem to the photochemical water oxidation as the sacrificial concentration decreases with time. The reaction becomes slower and ceases because of the consumption of the sacrificial reagent. In fact, the water oxidation has mostly been studied only for few minutes in the presence of a photosensitizer [24].
3.5 Source of evolved H2 and O2
For the overall water splitting, the amount of produced H2 must be stoichiometrically double of generated O2. This indeed proves once more that evolved hydrogen and oxygen originate from the water. For half-reactions, the detection of the source of evolved H2 or O2 is required. Because sacrificial agents like alcohols, biomass, and acids or catalysts like organic polymer graphitic carbon nitride can also produce H2 under photochemical conditions to show an incremental effect. Labeling study with D2O and detection of peak only at m/z = 4 for D2 confirms water as the source of H2 [50]. In experimental conditions, low-intensity peak at m/z = 3 is often observed because of the presence of some water in D2O and adsorbed water in the catalyst or other reagents.
In photoinduced water oxidation using persulfate, there is a possibility of O2 originating from the electron acceptor. Although the combination of persulfate and Ru(bpy)32+ produces a negligible amount of O2 under photochemical conditions, the presence of a catalyst can still change the situation [2]. However, the H218O labeling study is apt for the detection of the O2 source (Fig. 2).
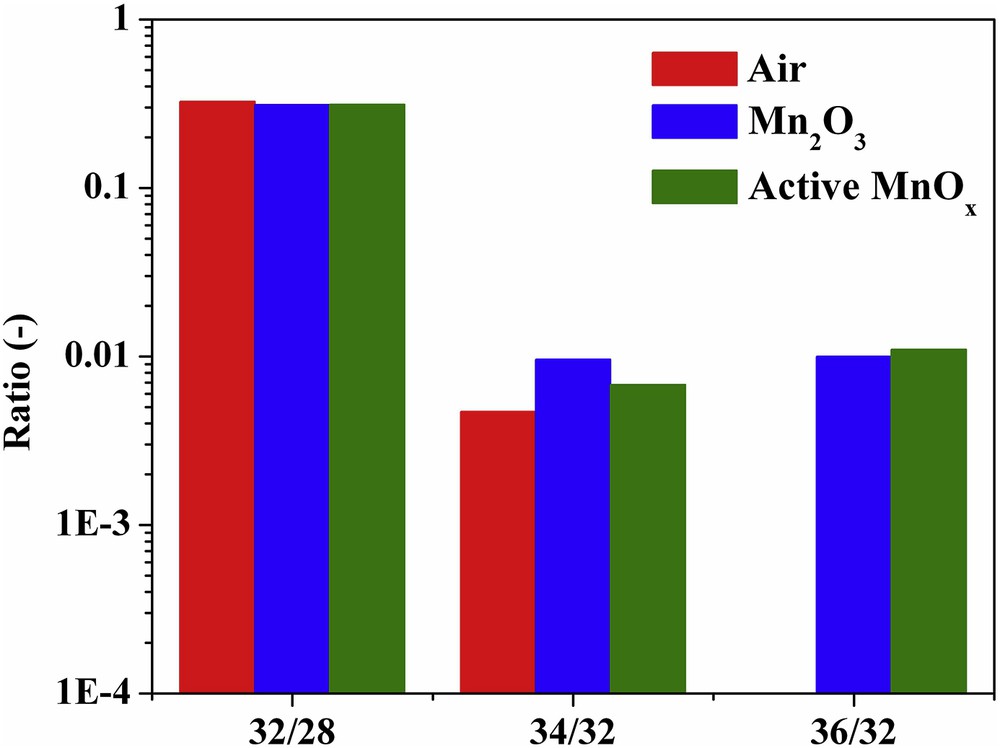
Using H218O in the presence of small amounts of H216O for the identification of the source of dioxygen by photoinduced water oxidation.
The presence of peak at m/z = 36 is expected to arise from labeled water. In fact, highly pure H218O (more than 99%) was not commercially available, and therefore peaks with m/z = 34 (18O16O) and 32 (16O16O) could also be observed in minor quantities along with major m/z = 36 (18O18O) [22,23]. These peaks could also be generated from the combination of the adsorbed water in the catalysts or other reagents and labeled water [2].
3.6 Detection of evolved gas
The amount of the evolved gas is detected either by gas chromatographic techniques, increase in the pressure/volumes, or by gas detectors [42]. For the oxygen evolution reaction, the Clark-type electrode is often used to quantify the amount of dissolved oxygen [23,51]. Because detection limit of the Clark-type electrode with the marginal error is up to ∼260 μmol/L of O2 at 25 °C (just below the water saturation level), higher concentrations of O2 cannot be measured precisely that indeed gives rise to an inadequate data set. In pressure or volume monitored systems, control of the reactor temperature with high precision as well as specific total reactor volume is vital.
4 Benchmarking catalyst system
The catalytic activity in water splitting is often expressed by the following terms: “highly active”, “effective”, “high performance”, or “efficient”. This ambiguity can be excluded from the quantitative comparison of the produced data with respect to a benchmark catalyst. The benchmark catalyst should be widely acceptable in terms of availability, ease of synthesis, cost-effective, and reproducibility. In this respect, a commercially available photocatalyst (e.g., TiO2, WO3, and BiVO4) or dark catalyst (e.g., Mn2O3, Co3O4, and NiO) from a defined company with a specific product number should be checked in different laboratory conditions to ensure the reproducibility of the data. Comparing the data (H2 or O2) produced by a new catalyst system with the benchmarking catalyst will certainly enhance the clarity of the system. In addition, control experiments must be carried out in dark under identical reaction conditions to make sure that the evolved gas is generating only by the photocatalytic reaction. Control experiments with different amounts of the photocatalyst, cocatalyst loading, and sacrificial agent should also be performed to find out the optimal condition and optimal rate of the reaction.
5 Representation of activity
The amount of evolved gas is expressed as micromoles or millimoles and the rate is expressed as micromoles per hour or millimoles per hour. To calculate the TON (TON = moles of H2 or O2 evolved per mole of metal), the molar concentration of a catalyst needs to be known. It is easy to calculate the TON of pure crystalline materials with known molecular formula. With doped material, molecular substitution, nonstoichiometric materials, or amorphous catalysts the difficulties arise. In that case, millimoles of metal in a certain amount of catalyst could be determined by techniques such as inductively coupled plasma atomic emission spectroscopic studies, X-ray photoelectron spectroscopy, or energy dispersive X-ray studies. For bimetallic or polymetallic materials, the situation becomes much more complicated to detect the active metal center of the catalyst. More experiments are required to understand the role of all metal centers and rule out the direct participation of some of the metal centers. This complex situation can be handled accepting all relevant metals as active.
After the generation of the data from overall water splitting or half-reactions thereof, representation of the data is also critical. The main apprehension involves whether the catalytic reaction is a surface reaction or only a bulk reaction. In bulk reaction, the availability of the active sites for the reaction is again a matter of debate. In that situation, the representation of mass activity and the surface activity of the reaction is also complex to interpret. In addition, many transition metal oxides, in fact, show higher activities when normalized with surface area as compared to the mass of catalyst. Normalization against the number of atoms present in the molecule is needed during the representation.
Still, the debate persists that reaction is taking place on the surface of the catalyst or in the bulk. For the highly crystalline material, it was often considered that the surface catalyzed reaction and the surface normalized data are represented. For the layered and amorphous materials, photochemical water oxidation cannot be explained only by the surface reaction. For example, bioinspired PS II catalysts based on manganese oxides have a layered structure with channels where water can easily pass through the layers to have a higher active area for the water oxidation. Again, all metal centers are also not available for the reaction. In this case, an overall idea about the number of active sites can be drawn by the number of reducible or oxidizable sites in the catalyst. Temperature-programmed hydrogen reduction or oxygen oxidation is the common techniques for this purpose. Therefore, a surface area determined by N2 adsorption further could be corrected against the available active sites.
For the Clark-type electrode measurements, the generated data are represented as micromoles per liter or milliliter. Therefore, the solution volume is extremely important in addition to the catalyst amount, concentration of sacrificial agent, and photosensitizer. TOF (TOF = TON/time) needs to be addressed to show how fast the reaction proceeds. In the case of Clark-type electrode data, determination of TOF is tricky because of the uncertainty in total oxygen evolution for the possible leakage with higher O2 content and solubility limit of O2. Often the rate of the oxygen evolution is reported for the first 60 s and compared between different catalysts to confirm the efficiency of the catalyst [52]. It is noteworthy that for the photosensitized reaction, the activation of the sensitizer is needed for the first few seconds and different rates could be observed without the activation when compared to preactivation (Figs. 3 and 4).
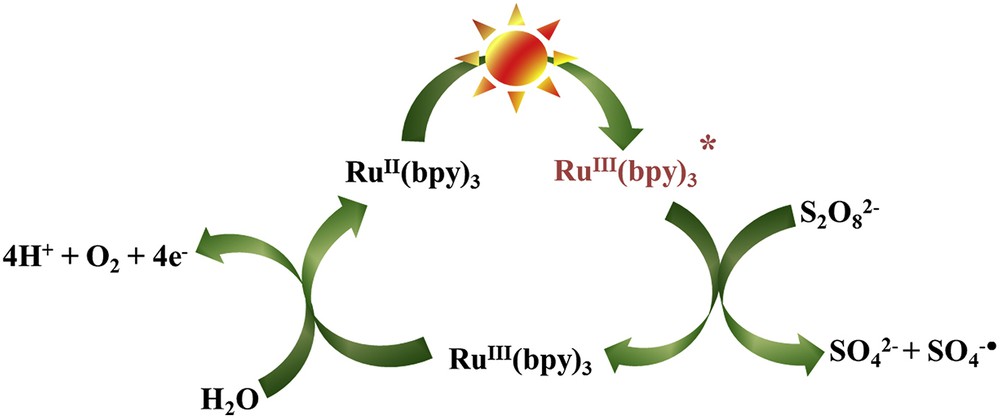
Catalytic cycle for photocatalytic water oxidation using Ru(bpy)32+ as a photosensitizer and persulfate as an electron acceptor.
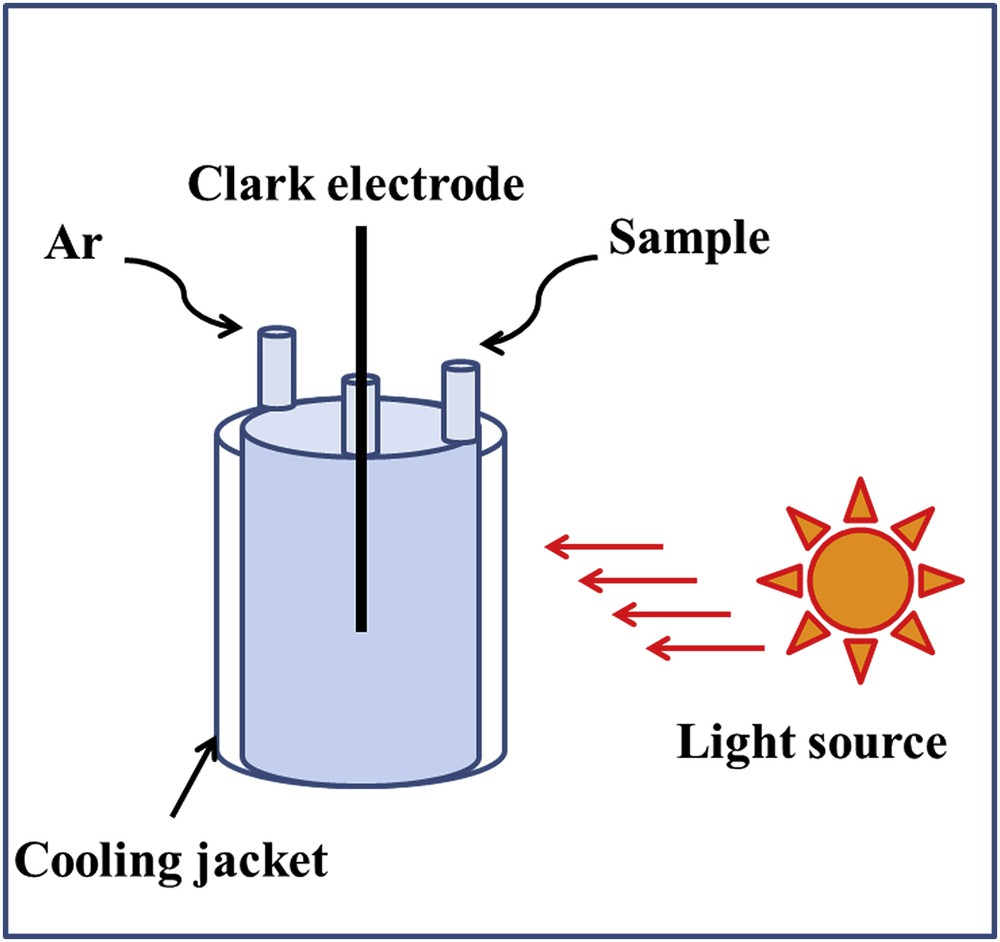
Setup for a Clark-type electrode for photocatalytic water oxidation.
6 Optimization of standards: our approach
To optimize the reaction conditions and to represent the catalytic activity in a meaningful way, we measured some of the commercial (MnO, Mn2O3, Mn3O4, MnO2, Co3O4, etc.) [2,46] dark materials purchased from Sigma-Aldrich that were then compared with respective as-synthesized catalysts (prepared solvothermally or via a single source precursor route) for photochemical water splitting in a similar way. The water oxidation experiments of the dark catalysts (Clark electrode setup) were first optimized by changing the concentration of the sensitizer, the sacrificial electron acceptor, and varying buffer solution. In our laboratory conditions, the best results were achieved while choosing 14 mM of the electron acceptor, 1 mg of the catalyst, and 2 mL of phosphate buffer (pH 7) in a 10 mL quartz reactor fitted with a water jacket by maintaining the temperature at 20 ± 1 °C. The quartz reactor was then illuminated with a continuous-output xenon lamp (illumination area of 1.5 cm × 3.5 cm; spot size of 16 cm2) of 300 W with a cutoff filter 395 nm. The light intensity was adjusted to 1.5 Wcm−2 and the dissolved oxygen concentration of the reaction was measured by a Clark electrode. As an example, the rate of oxygen evolution calculated for the commercial CoOx was 0.08 ± 0.001 , whereas for the as-synthesized CoOx with a similar surface area, 1.56 ± 0.021 was achieved for the first 60 s. A set of control experiments were carried out under similar conditions as above to quantify the evolved oxygen gas (for CoOx, ∼0.1 mL/h), and in addition, the labeled-oxygen study was also accomplished and the resulting values of m/z=36 (18O18O), 34 (16O18O), and 32 (16O16O) were systematically monitored using a GC–MS [2]. Furthermore, for the photocatalysts such as commercial and as-prepared Ag3PO4, BiVO4, and WO3, the best results could be obtained using similar conditions in water or phosphate buffer except using the sensitizer.
The water reduction experiments were conducted in a pressure or volume monitored systems where a photoreactor with defined irradiation geometry was developed for the photocatalytic experiments in our laboratory. The testing was carried out using graphitic carbon nitride systems, CdS and organic polymer materials by cocatalyst loading, and using triethanolamine as a hole scavenger. As an example, 50 mg graphitic carbon nitride was placed in the photoreactor of volume 73.55 mL (illumination area of 20 cm2) and then 60 mL of an aqueous triethanolamine solution (10 vol %) was added. The reactor was illuminated by a 300 W Xe lamp with a cutoff filter 395 nm (intensity 1.5 Wcm−2) at 25 ± 1 °C [48]. The hydrogen production rate was about 0.5 mL/h using Pt as a cocatalyst and under the same conditions, the efficiency was 0.09%. In addition to this, recently, non-noble metal cocatalytic systems with carbon nitride have also been demonstrated by our group [49]. We have tested the long-term hydrogen evolution for several days to establish the practical applicability of the catalyst system (Fig. 5).
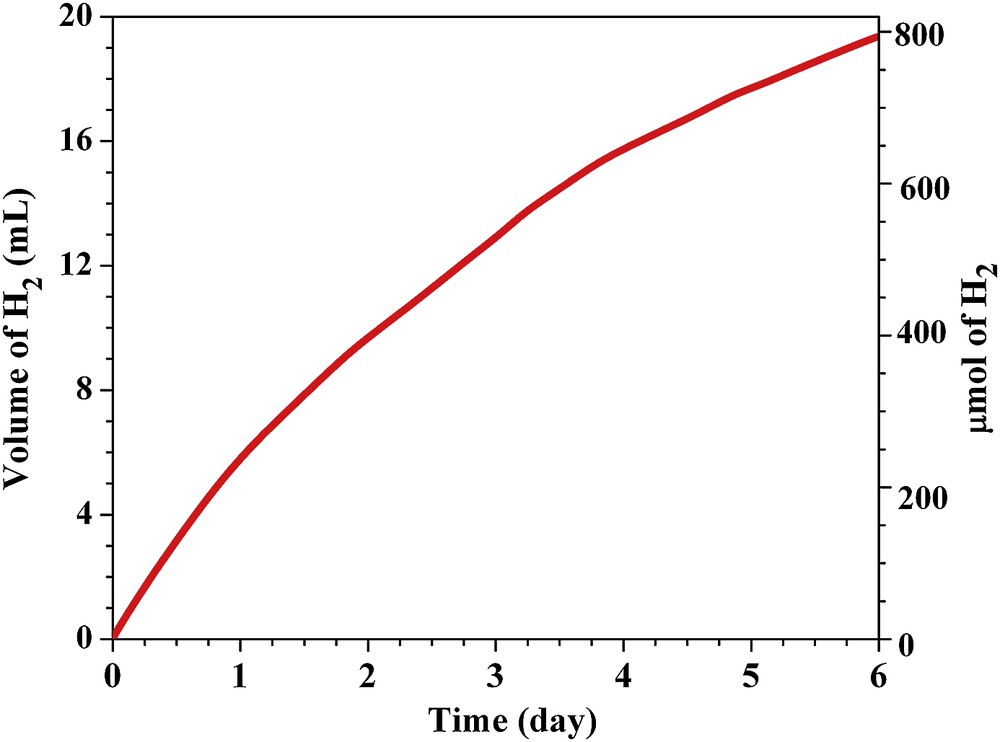
Measurement of the long-term stability of photocatalytic water reduction (hydrogen evolution reaction) using graphitic carbon nitride, CdS and organic polymer materials by cocatalyst loading, and aqueous triethanolamine solution as a hole scavenger under defined irradiation geometry.
Even the recovered catalyst from the long run showed photocatalytic hydrogen evolution efficiently. Comparison of the catalytic activity with the reported catalysts has been carried out by determining apparent quantum efficiency. The activity of the photocatalytic reaction with semiconductor material was represented by the optimal rate where the maximum rate is produced against the amount of a photocatalyst. In addition, incident photon flux with the light source and cutoff filter should be described to have a complete comparison of the photocatalytic activities of the different catalysts from different laboratories.
7 Conclusions
We plead to follow certain experimental standards such as the need for well-defined and commonly accepted standard reaction conditions, apparatus, instrumental setup, and detectors, which play a huge role while performing and optimizing water-splitting experiments. We strongly propose that for comparison purposes, all parameters involved in photochemical water splitting should be referenced clearly and precisely to avoid the “Babylonian confusion” in water splitting. The importance of surface area on the catalytic activity and the determination of TON values in terms of a number of electron transfer over the number of molecules of a photocatalyst during the photocatalytic reaction could possibly resolve the problems and difficulties in the representation of data of water photolysis.
Acknowledgments
Financial support from the BMBF (project MANGAN) and the Deutsche Forschungsgemeinschaft (Cluster of Excellence UniCat, EXC 314-2) is gratefully acknowledged.