

1 Introduction
Many years ago, the photochemistry of o-divinylbenzene (1) and its alkyl derivatives was studied [1–6] to investigate the photochemical behavior of the hexatriene system in which the middle double bond is a part of the aromatic ring. The main product in this photochemical reaction was the tricyclic compound 3 with benzobicyclo[3.1.0]hexene skeleton, formed via a [4 + 2] cycloaddition followed by a vinylcyclopropane–cyclopentene rearrangement from 2 (Scheme 1).
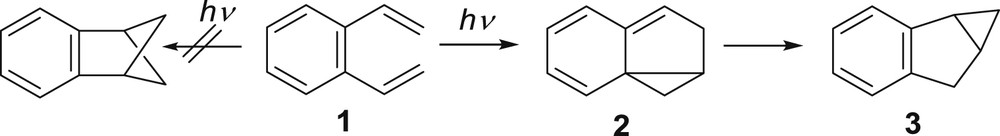
Photoproduct of intramolecular cycloaddition of o-divinylbenzene (1).
Laarhoven and co-workers, studying the photochemistry of stilbenes, have introduced the aryl substituent into the o-divinylbenzene and these diverse o-vinylstilbene derivatives were studied in detail [7–17]. The first studied compound was 2-vinylstilbene (4) [7]. Its photoexcitation afforded, besides 1-vinylphenanthrene (6, 15%), traces of unidentified compounds and polymeric compounds as well as endo- (2%) and exo-5-phenylbenzobicyclo[2.1.1]hex-2-ene (5, 70%) (Scheme 2).
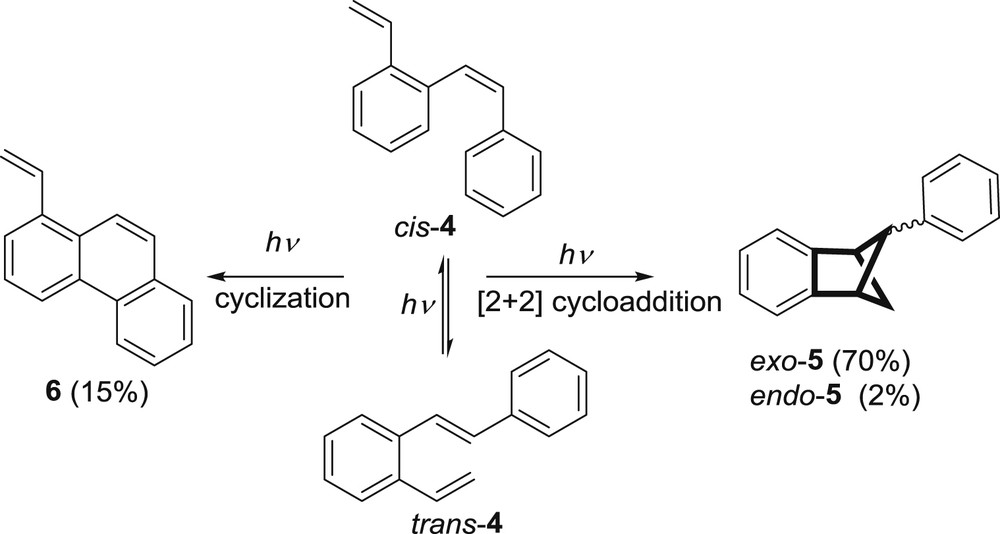
Excited state reactions of 2-vinylstilbene (4).
When a substituent is introduced into the 2-vinylstilbene, thus changing the conformation of the molecule in the ground state, excitation of those compounds afforded electrocyclization products and the products with benzobicyclo[3.1.0]hexene structure, besides formation of the products with benzobicyclo[2.1.1]hexene structure (Fig. 1) [15–17].
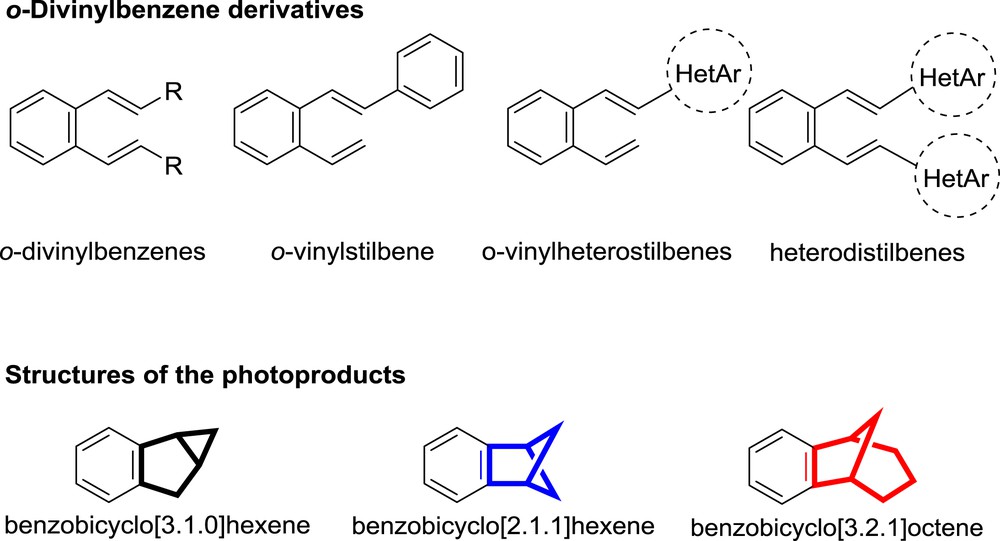
Investigated structures.
2 Photochemistry of o-vinyl-heterostilbenes
Foregoing research paved the way and opened a whole new area of investigations on ortho-vinyl–substituted heterostilbenes. The heterocycles that have been incorporated into the framework and investigated, up until now, were furan, thiophene, pyrrole, sydnone, oxazole, and most recently pyridine. The synthesis of the furan derivative, applying the method for the synthesis of 2-vinylstilbene [8], was not acceptable. New approach was chosen and the “one-pot” synthesis was developed utilizing a double Wittig reaction (Scheme 3) [18].
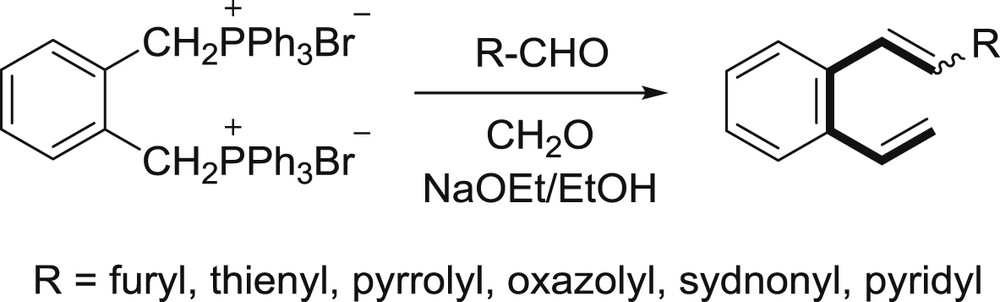
“One-pot” synthesis of β-heteroaryl-o-divinylbenzenes.
This simple and inexpensive synthetic path was applied for the preparation of almost all investigated o-vinyl–substituted heterostilbene compounds.
2.1 Furan and thiophene analogues of o-vinylstilbenes
Heterocycles are generally considered aromatic in character but this aromaticity varies depending on the heteroatom incorporated into the ring, so the photochemical behavior of heterocycle-containing compounds could also be different from the behavior of the corresponding compounds with the phenyl ring. The study of photochemical reactions of heterocyclic o-vinylstilbene analogues was primarily extended to β-(2-furyl)–substituted-o-divinylbenzenes 7 (Scheme 4) [19–21].
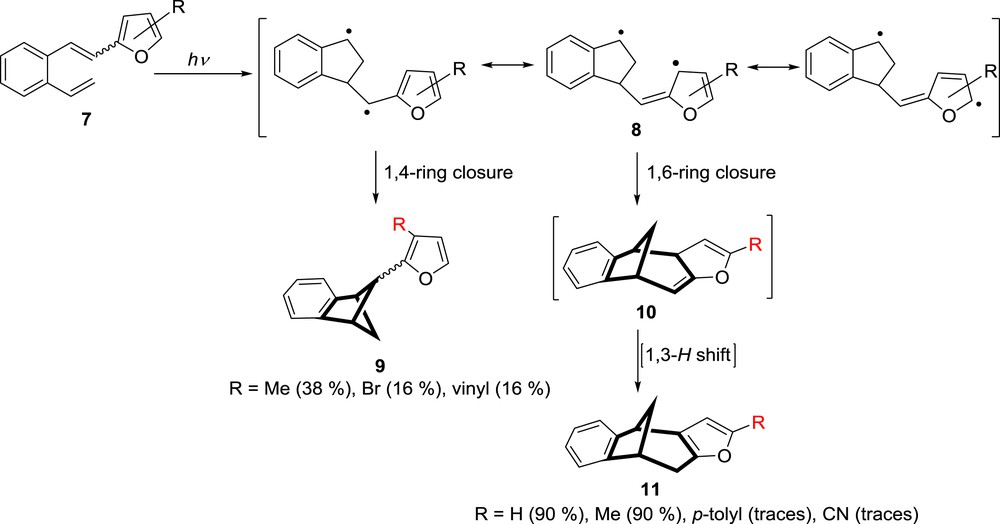
Photochemistry of β-(2-furyl)–substituted-o-divinylbenzenes 7.
In comparison with the photochemical behavior of 2-vinylstilbene (4) [7], which undergoes [2 + 2] cycloaddition and formation of the benzobicyclo[2.1.1]hexene derivatives 9 as the main product, unsubstituted β-(2-furyl)-o-divinylbenzene 7 is a system in which the β-substituent is involved in the intramolecular cycloaddition giving benzobicyclo[3.2.1]octadiene structure 11 as the main product in very good yield [19]. The formation of this tetracyclic product 11 was explained by formation of the stabilized biradical 8, followed by the preferred 1,6-ring closure to 10. Product 11 was isolated as the final product after the 1,3-H shift. A study of furan derivatives with different substituents at position 5 or 3 on the furan ring showed a profound steric and electronic effects on the intramolecular cycloaddition reactions [20]. Although the 5-methyl-furyl derivative 7 reacted equal as unsubstituted 7 and produced methyl-substituted benzobicyclo[3.2.1]octadiene derivative 11, 5-(p-tolyl)furyl or 5-(p-cyano)furyl derivatives 7 gave the substituted 11 only in traces (Scheme 4), besides isomerization of the double bond. When the substituents were placed at position 3 on the furan ring [21], the main photoproducts were substituted benzobicyclo[2.1.1]hexene derivatives 5. They are also formed via biradical 8, but because of the competitive steric hindrances, the more favorable was the 1,4-ring closure.
In all of the cases [20,21], unexpected substituted phenanthrenes 14 were also isolated and their formation explained as photoinduced [4 + 2] cycloaddition via biradical 12, followed by a ring closure to 2,5-dihydrofuran intermediate 13, which can lose water during the workup procedure and aromatize to product 14 (Scheme 5).
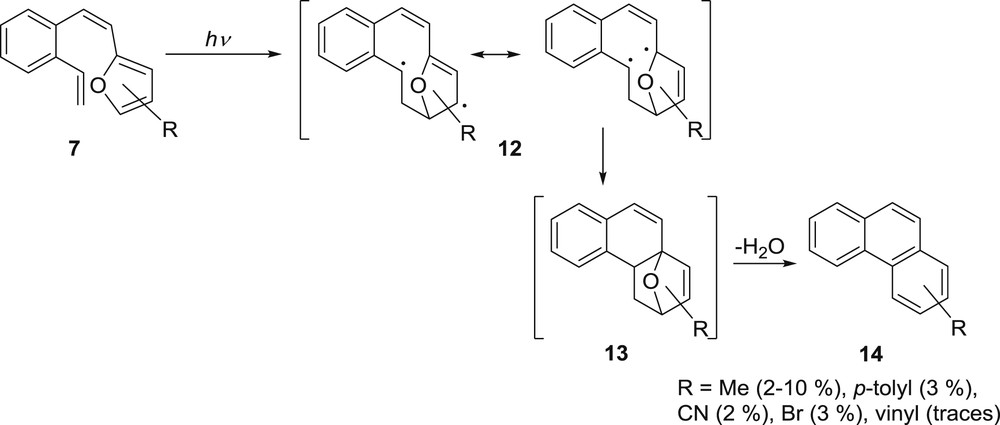
The proposed mechanism for the formation of the phenanthrenes 14.
The proposed mechanism was supported by the correct position of the substituents in all of the isolated phenanthrenes, obtained on irradiation of substituted β-(2-furyl)-o-divinylbenzenes 7. This process is more pronounced with the 3-substituted furyl derivatives in which, due to the steric effects, the presence of more suitable conformation in cis-configuration allows better overlap of the vinyl and the furan moieties and consequently formation of phenanthrenes 14 [21].
In continuation of the work the investigation was extended with the introduction of more substituents to the furan ring incorporated into the o-divinylbenzene molecule (Scheme 6).
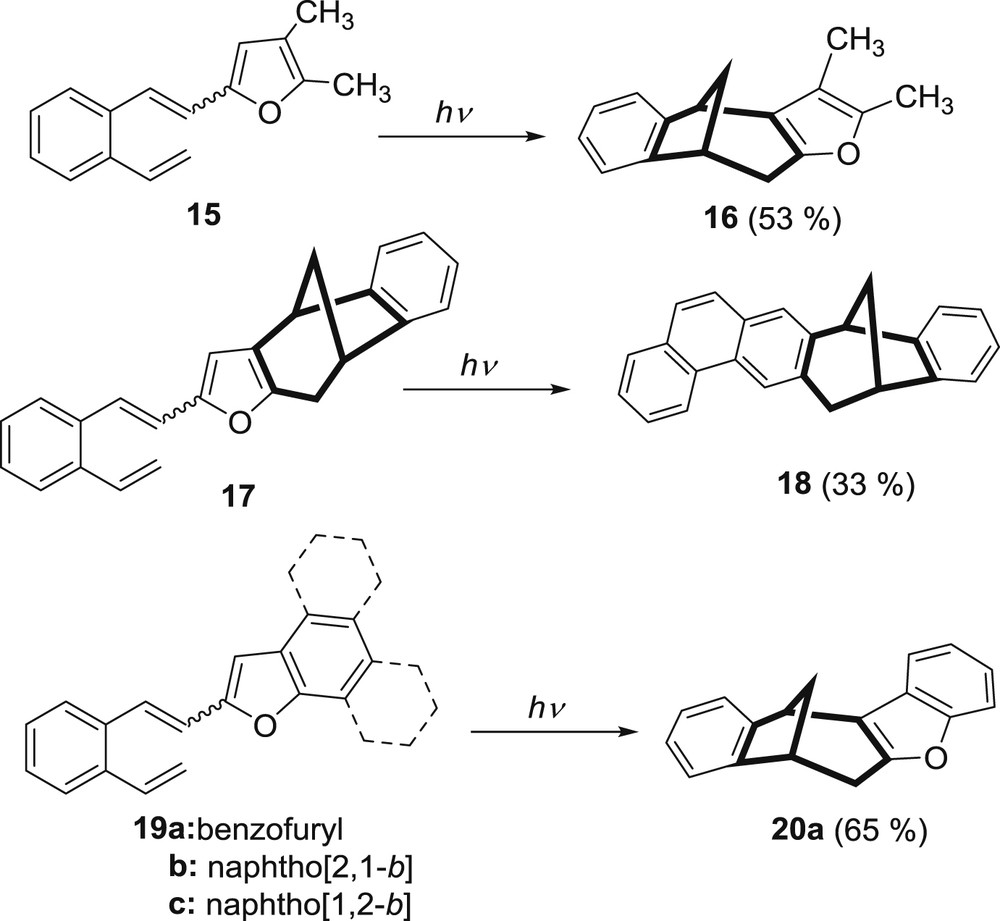
Photochemistry of various furan derivatives of β-substituted-o-divinylbenzene.
These derivatives included 2-(4,5-dimethyl)furyl compound 15 [22], the vinylstyryl-benzobicyclo[3.2.1]octadiene compound 17 [22], and condensed heteroaromatic compounds 19 [23,24]. The 2-(4,5-dimethyl)furyl o-divinylbenzene 15 afforded benzobicyclo[3.2.1]octadiene derivative 16 as a main product, beside traces of phenanthrene derivative. These results were consistent with the photochemistry of o-vinylstyryl furans that were studied earlier [19–21]. Unexpectedly, irradiation of the vinyl derivative 17, which could be observed as 2-(4,5-dialkyl)furyl derivative, resulted in fused phenanthrene bicyclic derivative 18 in moderate yield, besides high-molecular-weight products [22]. Formation of the bicyclo-phenanthrene photoproduct 18 is explained by the photoinduced [4 + 2] cycloaddition reaction (Scheme 5). This unique behavior of the o-vinylstyryl-furan derivative 17 is ascribed to conformational changes and π–π intramolecular interactions. Double bicyclic structure, which we have been striving for, has not been isolated. The benzofuryl derivative 19a upon irradiation gave the benzobicyclo[3.2.1]octadiene product 20a. Naphthofuryl derivatives 19b and c, regardless of concentration, underwent cis–trans isomerization and intermolecular cycloaddition [25,26] but do not form intramolecular photocycloaddition products. Benzofuryl-derivative 19a underwent intra- and intermolecular cycloaddition reactions depending on concentration.
In continuation of interest for the effect of the heteroatom and its position in the heteroaromatic ring on the formation of heteropolycyclic compounds, it was anticipated that introduction of sulfur as the heteroatom, by replacing the furan moiety with the thiophene, might influence the excited state properties of this new hexatriene system and have an impact on the formation of diverse photoproducts. Thiophene has a more aromatic character than furan, and it could be expected that thiophene derivative 21 might resemble the photochemical behavior of 2-vinylstilbene (1) (Scheme 1). Irradiation of 2-thienyl–substituted o-divinylbenzene 21 gave equal bicyclic structure as furan derivative 7, the thieno-fused benzobicyclo[3.2.1]octadiene 22 as the main product (Scheme 7) [27].
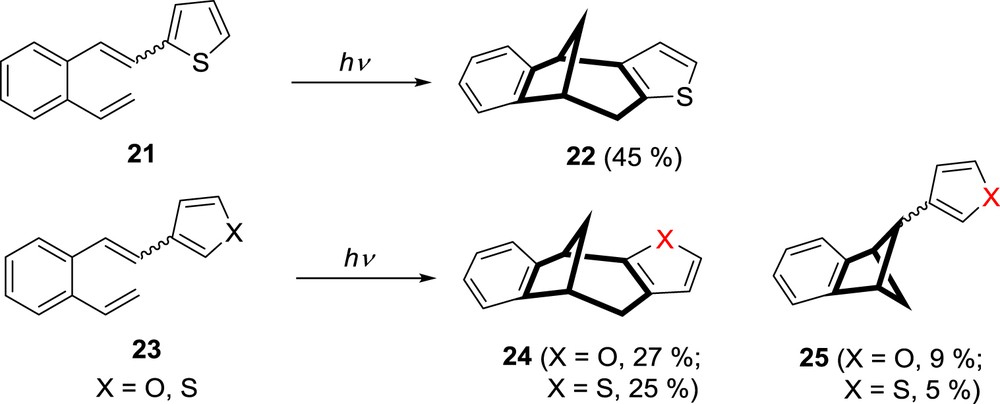
Photochemistry of β-2/3-thienyl and 3-furyl derivatives of o-divinylbenzene.
Furthermore, 3-thienyl and 3-furyl–substituted o-divinylbenzenes 23 (Scheme 7) showed comparable photochemical behavior as they gave the bicyclo[3.2.1]octadiene structures 24 and bicyclo[2.1.1]hexene structures 25 by 1,6- and 1,4-ring closure, respectively [27].
2.2 Pyrrole analogues of o-vinylstilbenes
Pyrrole moiety was introduced into the β-position of the o-divinylbenzene to investigate the photochemistry of nitrogen-bearing compound. The pyrrolyl derivative 26 did not behave by analogy to the furan/thiophene derivatives and it gave dimeric product 27 by regiospecific intermolecular addition of pyrrole to the double bond (Scheme 8) [28].
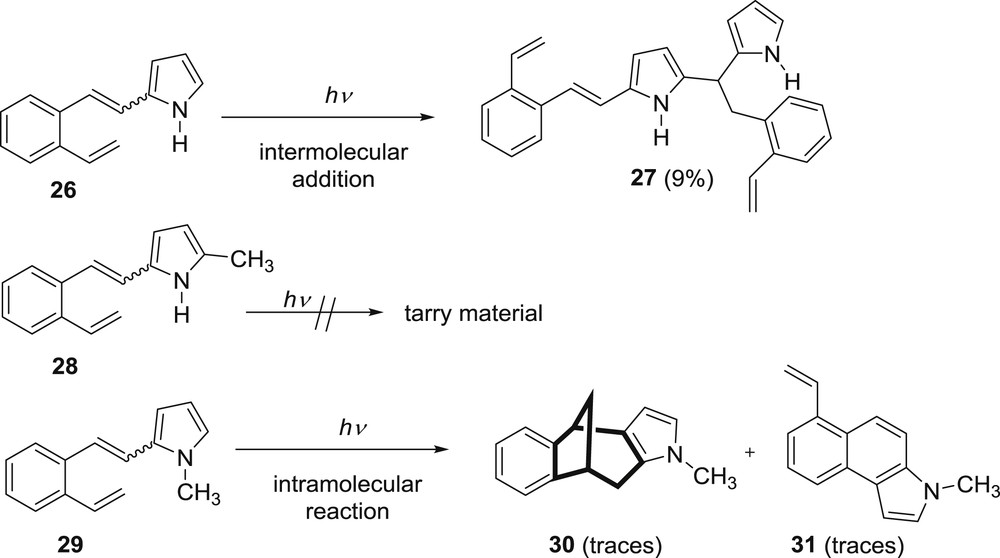
Photochemistry of β-pyrrolyl–substituted o-divinylbenzenes.
No intramolecular formation of the anticipated bicyclic product was observed. It was proposed that the formation of the dimeric product 27 occurs via the photoinduced electron transfer, followed by proton transfer and radical combinations (Scheme 9).
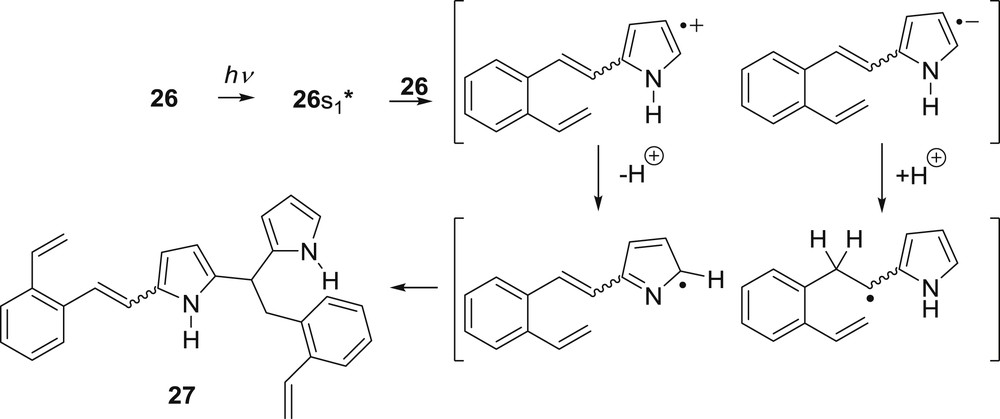
Proposed mechanism for the regiospecific intermolecular addition.
In an attempt to prevent further nucleophilic attacks, the position 5 on the pyrrole ring was blocked by methyl substituent (28) and in this case the only process that was observed was the cis–trans isomerization and decomposition to tarry material (Scheme 8) [29]. If the methyl group is introduced on the nitrogen atom of the pyrrole moiety the 5-methyl-2-(2-vinylstyryl)pyrrole (29) is formed. In the case of this derivative there is no intermolecular addition and the intramolecular cycloaddition gives the benzobicyclo[3.2.1]octadiene product only in trace amounts [28]. In an effort to find a suitable path for the formation of the desired benzobicyclo[3.2.1]octadiene, phenoxy- and ethoxy-carbonyl groups were introduced to the nitrogen atom (Scheme 10) [30–32].
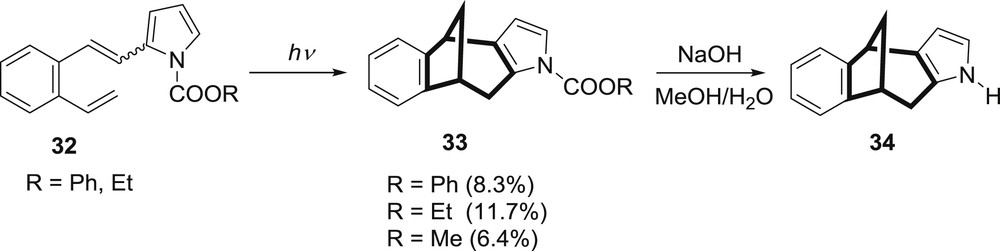
Photochemistry of N-protected pyrrole derivatives 32.
Upon irradiation of compounds 32 the benzobicyclo[3.2.1]octadiene products 33 were gained in relatively low yields and easily transformed by hydrolysis to desired polycyclic compound 34.
2.3 Oxazole and sydnone analogues of o-vinylstilbenes
In the continuing studies two more heterocycles, bearing nitrogen and oxygen, were introduced into the o-divinylbenzene moiety, sydnone and oxazole. Both of these moieties are five-membered heterocycles. Sydnone is mesoionic and can be represented as hybrids of a number of mesomeric ionic structures [32]. Upon photolysis of sydnonyl derivative 35 the formation of pyrazolonaphthalene was expected, by intramolecular trapping of photochemically formed nitrile imine with vinyl group. Upon irradiation of 4-aryl-3-(o-vinylstyryl)sydnones (35) only tarry material and no desired polycyclic products were isolated [33]. When the irradiations of 35 were conducted with the addition of acrolein (37) as dipolarophile, to trap the nitrile imine intermediate, styryl-pyrazoline products 38 were acquired in over 90% yield (Scheme 11). These pyrazoline products aromatized during purification and gave pyrazoles 39 and 40.
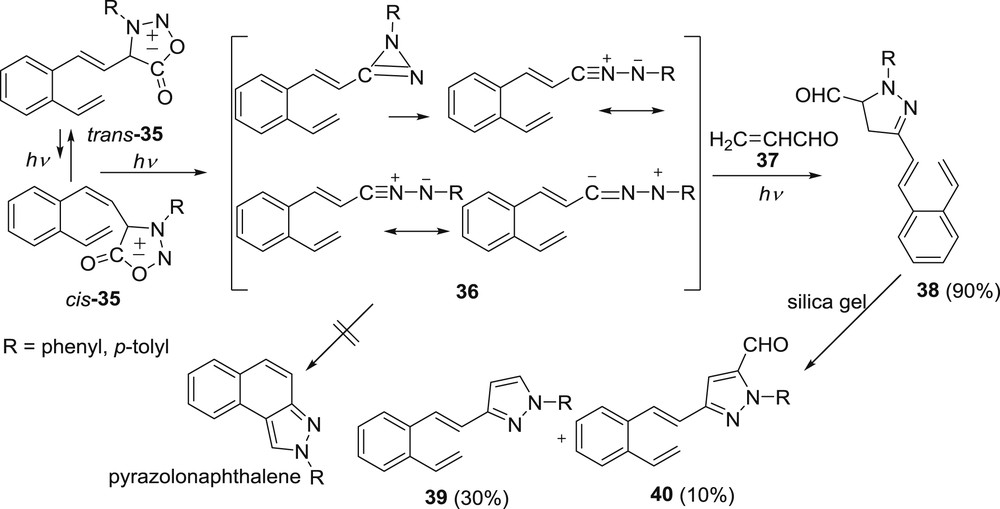
Irradiation of 35 in the presence of acrolein.
On the basis of the fact that intermolecular product 38 was obtained only in trans-configuration, it was concluded that sydnonyl-derivatives undergo competitive cis to trans isomerization and photolysis of the sydnone moiety, giving nitrile imine 36, which cannot react intramolecularly.
With the incorporation of the oxazole ring into the system it became plausible to attain polycyclic skeletons with two different heteroatoms incorporated into the system. Oxazole ring was placed into o-divinylbenzene moiety in all of the three available positions of the ring (2, 4, and 5) to study the influence of this placement on the excited state behavior. Several 4,5-subsituted 2-oxazolyl compounds 41a–d were synthesized, using again the one-pot Wittig strategy [34,35]. When irradiated, compounds 41a–d did not react by [2 + 2] intramolecular cycloaddition reaction to give 43 or 44 but, if appropriately substituted, they reacted by 10π,6π electrocyclization or formal [4 + 2] cycloaddition giving oxa-bridged quinoline derivatives 42b and c (Scheme 12).
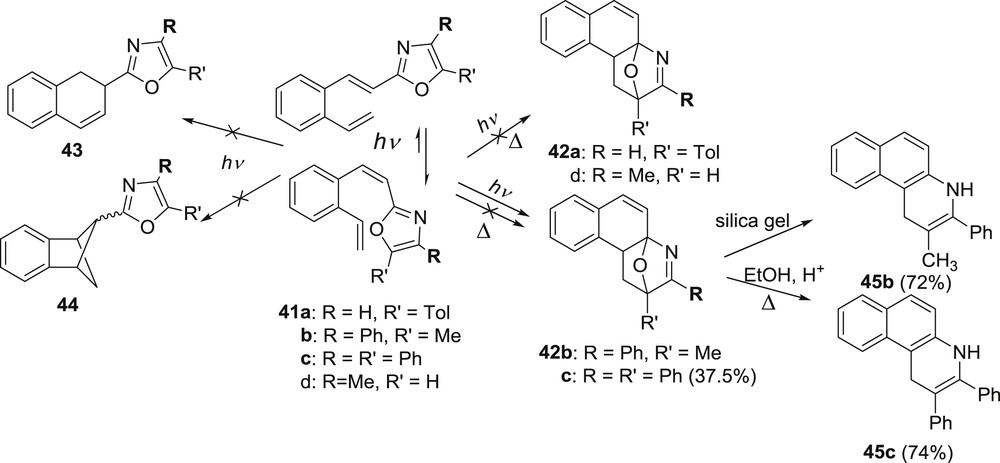
Photochemical reaction of 2-(2-vinylstyryl)oxazoles 41a–d.
We have seen the occurrence of [4 + 2] cycloaddition process with some furan derivatives and formation of phenanthrenes as minor products (Scheme 5) but this process is preferred in the case of 2-oxazole derivatives. Time-dependent density functional theory (TD-DFT) calculations were performed [34] and revealed that the intramolecular photocyclization of 2-(2-vinylstyryl)oxazoles to form oxa-bridged benzo[f]quinoline derivatives proceeds on the S1 potential energy surface (PES) via a stepwise pathway. It was also obviously from the experiments and confirmed by calculations that the reactivity of the photocyclization step depends on the substitution pattern at the positions 4 and 5 on the oxazole ring, where the aryl group in the position 5 definitely deactivates the reaction. The oxa-bridged derivatives 42 were spontaneously transformed or easily converted to benzo[f]quinolines 45, which are compounds with significant pharmacological properties [36–41] (Scheme 12).
To further investigate the impact of the placement of the heteroatoms (N, O) in the oxazolyl o-divinylbenzene system, 4- and 5-(o-vinylstyryl)oxazoles were synthesized [35,42]. For the synthesis of the unsubstituted 46, 5-(o-vinylstyryl)oxazole, a new synthetic route had to be developed [42] because of the volatility problem with the needful oxazole-5-carbaldehyde for the one-pot Wittig strategy. Photoexcitation of 4- or 5-oxazolyl o-divinylbenzenes afforded by photochemical intramolecular cycloaddition reaction benzobicyclo[3.2.1]octadiene products with the oxazoline ring incorporated into the skeleton (48, 50, Scheme 13). The substituted 4-(2-methyl)oxazolyl derivative gave analogue results. The 1,3-H shift, which happened in the previously mentioned photochemistry of furan and thiophene analogues of o-vinylstilbenes, did not happen here.
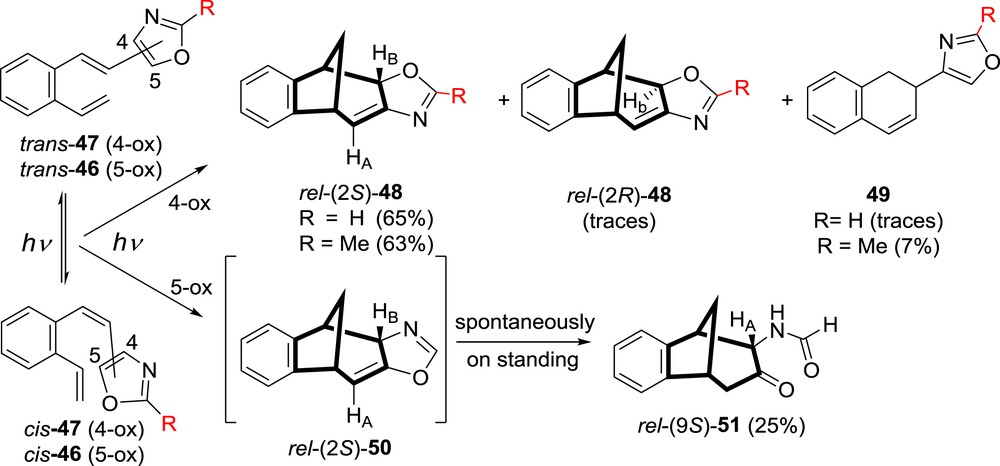
Irradiation of 4- (47) and 5-(2-vinylstyryl)oxazoles (46).
When there was a methyl substituent in the position 4 of the oxazole ring, 5-(4-methyl)oxazolyl derivative 52, only benzobicyclo[2.1.1]hexene product was obtained. With the placement of phenyl substituent on the position 2 of the ring, 5-(2-phenyl)oxazolyl compound 53, no bicyclic products were formed (Scheme 14).
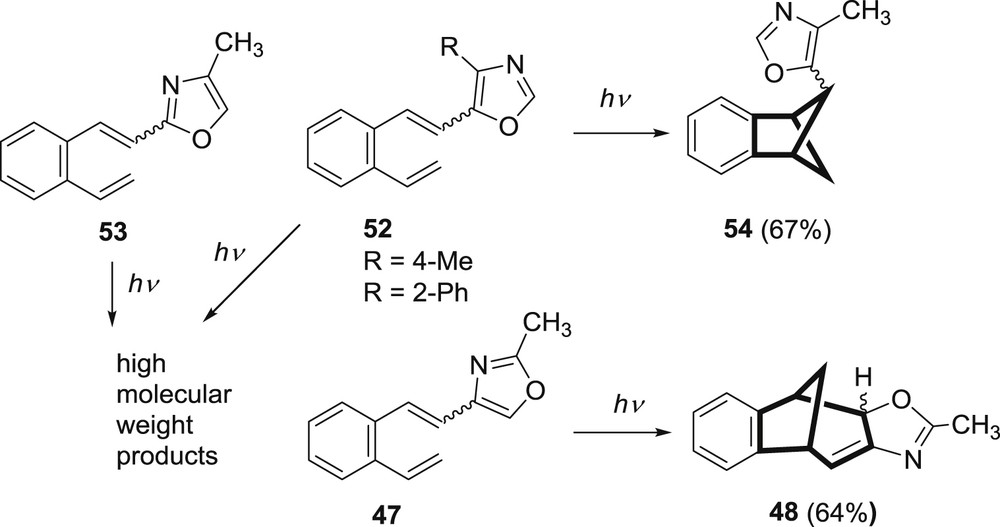
Photoreactivity of substituted 2/4/5-oxazolyl-o-divinylbenzenes.
Oxazoline compound 50, obtained upon irradiation of 5-oxazolyl o-divinylbenzene 46, was extremely unstable and was seen only in NMR tube. The oxazoline ring spontaneously opened and the tricyclic formamido derivative 51 was isolated (Scheme 13). For the oxazoline compounds 48, which were formed by irradiation of 4-oxazolyl derivatives 47, a number of reactions were performed and they were easily transformed further to various functionalized derivatives 55–60 (Scheme 15) [35,42].
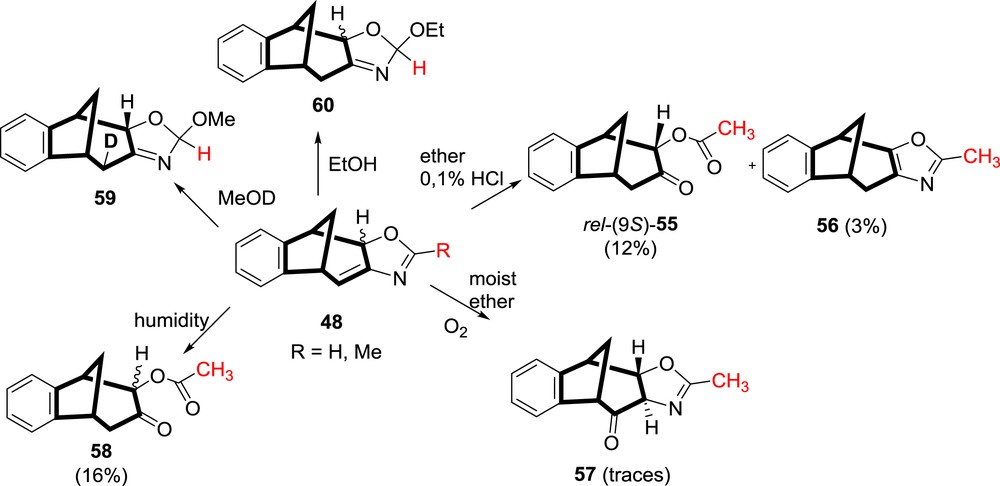
Reactions of photochemical oxazoline product 48.
2.4 Pyridine analogues of o-vinylstilbenes
The next step in the research of o-vinyl-heterostilbenes was incorporation of pyridine as a six-membered heterocyclic ring with nitrogen. Nitrogen has been proven in the previous sections as a heteroatom that changes the overall excited state charge distribution and thus the reactivity. The 2/3/4-(2-vinylstyryl)pyridines 61–63 were synthesized and irradiated. The 2- and 3-pyridyl-isomers afforded diverse polycyclic products by photochemical intramolecular cycloaddition and cyclization reactions. The 4-pyridyl-isomer 63 did not react and upon prolonged irradiation times gave only high-molecular-weight products. Both the 2- (61) and 3-pyridyl derivatives (62) undergoing electrocyclization reaction gave benzoquinoline derivatives 65 and 68. Photocycloaddition products benzobicyclo[2.1.1]hexene 64 and 66 as well as benzobicyclo[3.2.1]octene 67 were obtained in small or trace quantities (Schemes 16 and 17).
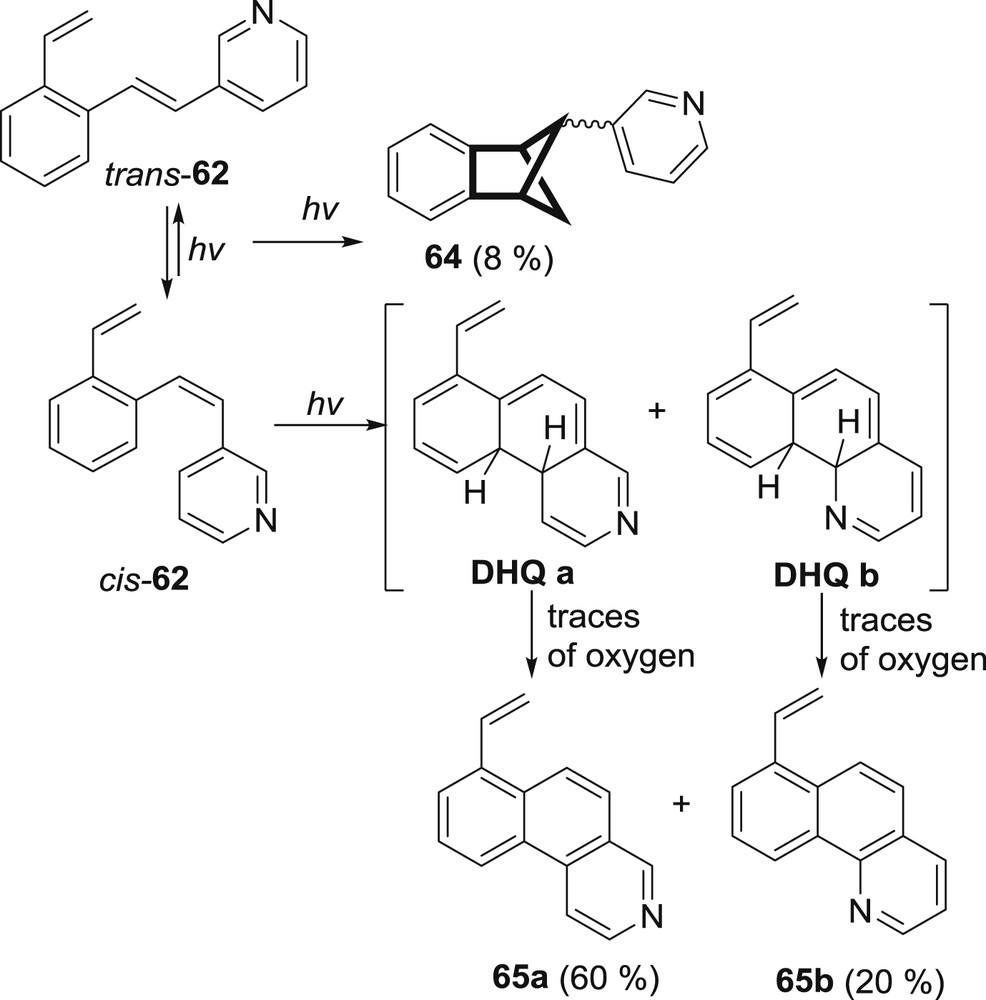
Photochemistry of 3-2[(2-vinylphenyl)ethenyl]pyridine (62).
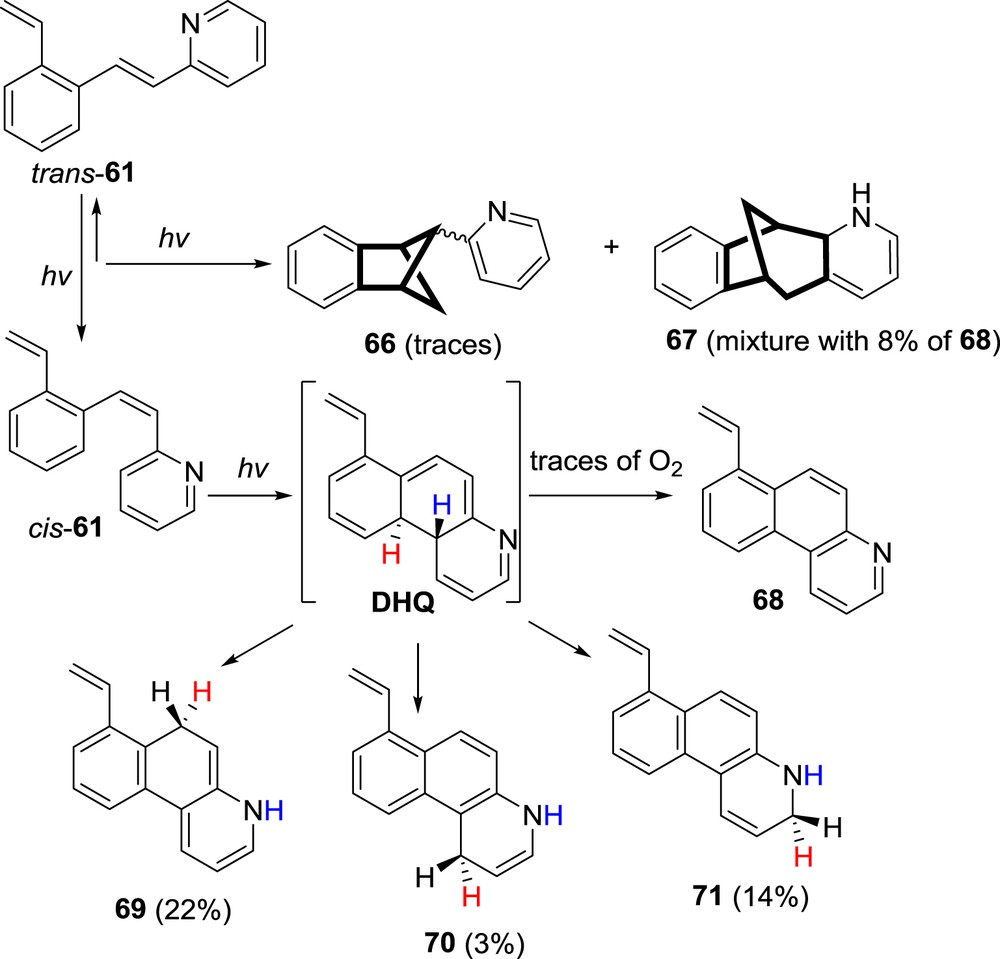
Photochemistry of 3-2[(2-vinylphenyl)ethenyl]pyridine (61).
Interesting part of this research was the isolation and characterization of stable isomerized dihydro-benzo[f]quinolines 69–71. It was argued that the stability of the 69–71 is probably connected to the fact that one of the hydrogen shifts to the nitrogen atom and such species is unable to neither revert back to the starting o-vinyl-styrylpyridine nor aromatize to the benzoquinoline product (Scheme 17).
3 Photochemistry of pyrrole, furan, and thiophene diheterostilbenes
In continuation of the work that was done on the o-vinyl-heterostilbenes, diverse symmetrically substituted diheterostilbenes were synthesized with pyrrole, thiophene, and furan as the heterocycle incorporated into the skeleton.
Irradiation of the dipyrrole derivative 72 gave a mixture of dimeric stereoisomers 74 in a yield of 40% and traces of intramolecular product 73 (Scheme 18) [43,44].
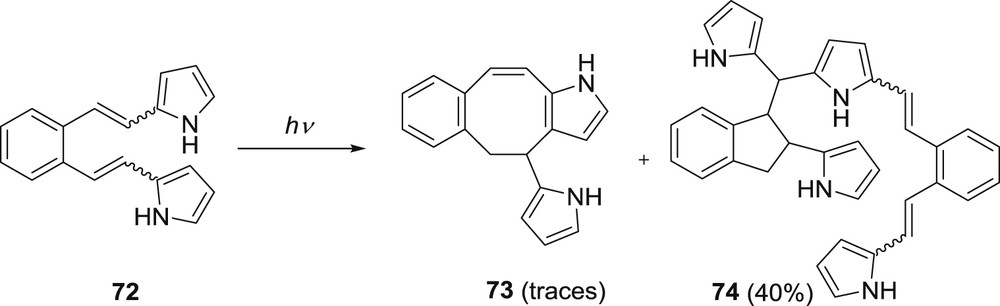
Photochemistry of 2,2′-(o-phenylenevinylene)dipyrrole (72).
Formation of these products can be explained by electron transfer followed by hydrogen transfer and ring closure to the indane derivative or to the fused octatriene product 73. The formed indane derivative reacts further, with the starting compound 72, to give the dimeric product 74. To prevent a nucleophilic attack of the starting compound on the indane derivative, a methyl-substituted starting compound 75 was prepared where the position 5 on the ring was blocked. Irradiation of the methyl-substituted derivative gave a mixture of two products 76 and 77 (Scheme 19) [44].
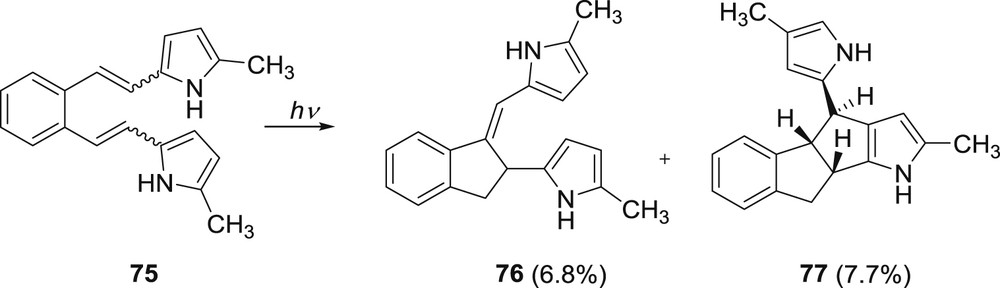
Photochemistry of 5-methyl-dipyrrole derivative 75.
In the case of derivative 75 there is electron transfer followed by the hydrogen transfer and a regioselective ring closure forming two intermediates. Both of them undergo a 1,7-H shift to a more stable indanylidene product 76. One of the intermediates can undergo a second intramolecular addition of the pyrrole moiety at the position 3 thus giving the benzopentaleno-pyrrole compound 77.
In further studies the photochemical behavior of β,β′-di(2-furyl)–substituted o-divinylbenzenes 78 (Scheme 20) was described [45]. In o-vinyl-heterostilbenes, increase in annelation increases the intermolecular complexation resulting in formation of dimeric products [24–26].
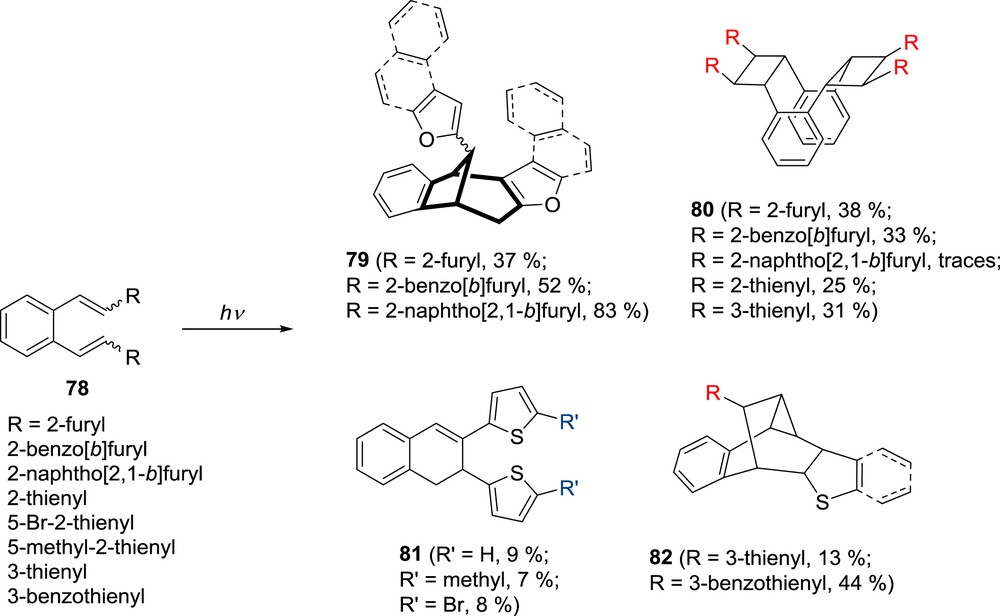
Photoproducts of β,β′-di(furyl/thienyl)–substituted o-divinylbenzenes 78.
It was expected that introduction of the second annelated furan moiety to the o-divinylbenzene (Scheme 20) could result in an intramolecular complexation, which would give interesting annelated bicyclo[3.2.1]octadiene structures as was afforded (Scheme 20) [45]. In concentrated solutions β,β′-difuryl–substituted o-divinylbenzenes 78 produced regio- and stereo-specifically cyclophane structures 80. These structures have potential as molecular tweezers, having hydrophobic cavity and an electron rich recognition site of the heteroatom. Among the examined annelated β,β′-difuryl–substituted o-divinylbenzenes, the most selective is the β,β′-di-naphtho[2,1-b]furyl derivative, which due to the π–π intra- or intermolecular complexation gives, at low concentration, only the exo-bicyclo[3.2.1]octadiene derivative 79 and at high concentration the cyclophane derivative 80. On irradiation of 2,2′-(o-phenylenedivinylene)dithiophenes, 3,3′-(o-phenylenedivinylene)dithiophene, and 3,3′-(o-phenylenedivinylene)dibenzothiophene 78 in dilute solution [46] (Scheme 20), gave products of intramolecular cycloaddition. The electrocyclization products, 1,2-dihydro-2,3-dithienylnaphthalenes 81, were isolated from the 2-thiophene derivatives while the 3-thiophene derivatives gave the polycyclic structures 82. The cyclophane derivatives 80 are formed as the result of intermolecular double [2 + 2] photocycloaddition in all of the cases studied. Intramolecular photochemical reactions of the same thiophene analogues 78 were also studied in acidic media at low concentrations [47] and a 1,6- and 1,5-ring closures of the hexatriene system leading to dihydronaphthalene or indene derivatives, respectively, were observed.
The incorporation of further heterocyclic nuclei into the o-divinylbenzene and investigation of their photochemical behavior is expected as a future continuation of studies because the benzobicyclo[3.2.1]octane skeleton comes up as a basic framework for a vast number of important natural compounds that exhibit significant biological activity [48,49]. Properly functionalized derivatives with the bicyclo[3.2.1]octane skeleton have proved to be powerful building blocks in organic synthesis [49–51]. This is a driving force behind the study and development of new methodologies and new synthetic approaches for the preparation of functionalized derivatives with the integrated benzobicyclo[3.2.1]octane skeleton [50,51] or the unsaturated benzobicyclo[3.2.1]octene/octadiene skeleton. The unsaturated structures with the heterocyclic nuclei incorporated into it can easily be further modified and transformed [22,42,52] to even more biologically active forms or they can serve as convenient building blocks for further synthesis.
4 Conclusions
In this article an overview is given on the research done on the photochemistry of heterostilbenes where furan, pyrrole, sydnone, thiophene, oxazole, and pyridine nuclei are incorporated into the basic stilbene-type skeleton. Throughout this long-lasting study of excited state reactivity of various o-vinyl-heterostilbenes and diheterostilbenes it has become evident that the heterocycle that is incorporated into the system changes its reactivity. This comes as a consequence of the heteroatom(s) that comes into play in such moieties and contributes to the excited state charge distribution. The photochemistry of these compounds is of great importance as it gives a pathway to new polycyclic products that are impossible to obtain by ground state organic chemistry approaches. It was mentioned that the star representatives of this group of polycyclic products are the benzobicyclo[3.2.1]octadiene photoproducts, which might show biological activity. All of the other products that were obtained, either by photocycloaddition or photocyclization reactions, come as valuable building blocks that can further be modified and functionalized.