

1 Introduction
The multifunctional therapeutic nanostructured systems are most important for numerous clinical applications and also well known for their essential adoptive effects and safe therapeutics. In the recent times, nanocomposites play a significant role in health care products; beside, they have wide range of applications in various biomedical fields like detection, diagnosis, and treatment of diseases such as different types of cancers, neurologic disorders, cardiovascular diseases, and so forth. Nanocomposites also play promising role as load-bearing composites for bone reconstruction through the interaction between the selected matrix and the filler. Bone is a mineralized hard tissue that exhibits highly complex and strongly hierarchical structure. In fact, natural bone tissue is composed of nanosized hydroxyapatite (n-HAp) crystals sporadically found in the collagen matrix. The apatite crystals present in human bone and enamel are all enormously small in size [1], which can be apprehended as nanosized materials. HAp [Ca10(PO4)6(OH)2] is the main mineral component of bone, which retains admirable osteoconductivity, bioactivity, biocompatibility, noninflammatory, and nontoxic properties and thus has been considered as the primary choice of material for orthopedic applications [2]. Owing to its high stability, HAp is used as a potent ceramic material. In addition, various utilities of HAp were found as lifesaving medicines, stabilizer, dentifrices, and nutrient supplements. Recently, HAp has gained a great attention toward the nanofiller of a polymer matrix nanocomposite. Basically HAp is a calcium phosphate compound, where the molar ratio of Ca/P is 1.67. HAp can also be used as a nanodrug carrier material in different biotechnological applications. Furthermore, HAp nanocomposite has been used to treat various types of major bone diseases including osteosarcoma [3], osteoporosis [4], osteomyelitis [5], osteoporosis [6], osteonecrosis [7], and so forth. In recent times, a keen interest has been raised toward the development of drug delivery systems and the first line chemotherapeutic for osteosarcoma. Osteosarcoma is a typical bone cancer most commonly found in young adults. A few research studies have been executed to treat different cancer cells by an anticancer drug doxorubicin (DOX)-coated HAp nanocomposite [8,9]. DOX is a common anticancer drug used in chemotherapy. DOX can be classified as anthracycline antibiotic. Basically DOX interacts with DNA and interferes with the nucleic acid synthesis, which produces a remarkable effect on DNA transcription [10]. Internal fixation by inhibiting tumor cells with the help of chemotherapy or radiation is not sufficient to manage bone metastases [11]. Increasing the structural strength and rapid release of high concentration anticancer drug to the targeted or infected site are also needed [12], which may be produced by DOX-encapsulated HAp polyvinyl alcohol (DOX-HAp-PVA) nanocomposite. The present research investigation consists of the development of a novel bioanalogue nanocomposite using PVA as the matrix and n-HAp particles as the filler encapsulate with anticancer drug DOX for osteosarcoma-affected bone tissue repair. The prior use of PVA as a matrix is because PVA is a synthetic water-soluble polymer and the presence of hydroxyl groups in PVA indicates the backbone chains of this polymer are highly interconnected by hydrogen bonds, which represent its outstanding chemical resistance character. Because of the hydrophilicity of PVA, it can make strong interaction with the hydroxyl groups present on the surface of the HAp, and this is evident that PVA can be ideal for scaffold formation [13]. Moreover, PVA is biocompatible [14] and it can improve the durability and the mechanical properties of HAp. High elastic modulus of PVA is the cause, which makes PVA as a polymer of choice in various biocomposite preparations [14,15]. Beside these, PVA is used as a potent biomaterial for the replacement of diseased or damaged articular cartilage and in a fixation method of arthritic applications for better adhesion [15]. After encapsulation, the synthesized materials have been characterized through Fourier transform infrared (FTIR) spectroscopy, X-ray diffraction (XRD), transmission electron microscopy (TEM), particle size distribution (PSD). Furthermore, the biocompatibility and the anticancer activity of the nanocomposite were measured by MTT (3-[4,5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide) assay study.
2 Materials and methods
2.1 Chemicals
All chemicals were of analytical grade and were purchased from commercial sources. Calcium nitrate tetrahydrate (98.0%) and diammonium hydrogen ortho-phosphate (98.5%) were procured from Rankem, RFCL, India. PVA (98%) of average molecular weight 85,000–124,000 was procured from Sigma–Aldrich, India, for nanocomposite formation. Ammonia solution (25%) and absolute ethanol (99.9%) were obtained from Changshu Yangyuan Chemical, China. DOX hydrochloride (95%) was purchased from TCI, Japan.
2.2 Preparation of HAp nanopowder
HAp was synthesized by using an in situ precipitation method. Stock solution (0.5 M) of each calcium nitrate tetrahydrate [Ca(NO3)2·4H2O] and diammonium hydrogen ortho-phosphate [(NH4)2HPO4] was prepared in distilled water. [Ca(NO3)2·4H2O] was taken in a beaker as a starting material and [(NH4)2HPO4] was added dropwise to it in a maintained molar ratio of Ca/P = 1.67 at pH above 10 environment and vigorously stirred in room temperature using a mechanical stirrer (2MLH, Remi) at 800–900 rpm. The pH of the reacting mixture was adjusted by gradually adding NH4OH solution (NH4OH/H2O = 1:2). The process was continued up to 5–6 h. Thus, a white gelatinous precipitate was obtained and filtered. The precipitate was thoroughly washed with ethanol–water solution [EtOH/H2O = 1:1] to remove the excess ammonia from the obtained white precipitate, and then freeze-dried for 15 h. Finally, a crystalline white powder was obtained.
2.3 Preparation of DOX-loaded HAp
DOX was coated onto the HAp particle surface using the absorption characteristics of HAp crystalline nanopowder with a molar ratio of DOX/HAp = 1:10. DOX was dissolved in 2 mL ethanol and aforementioned respective amount of HAp powder was taken in a sterile round-bottom flask. The solution of DOX was poured into the round-bottom flask and the mixture of DOX-HAp was then stirred at 500–650 rpm for 8 h and probe was sonicated with every 1-h interval at room temperature. Then the mixture was dried to powder using rota vapor and after that the powder was collected.
2.4 Preparation of DOX-HAp-PVA nanocomposite
DOX-HAp-PVA composites with different HAp contents were prepared by a solution-based chemical method (Scheme 1). The most probable structure of DOX-HAp-PVA nanocomposite has been depicted in Fig. 1. At first, 1% PVA solution was prepared by dissolving 500 mg PVA powder into 50 mL of hot distilled water. From this PVA solution, 1 mL was taken and diluted with 5 mL ethanol, then 1 mL of the prepared PVA–ethanol mixture was taken into a 25-mL round-bottom flask and DOX-modified HAp nanoparticles were added slowly with vigorous stirring using a magnetic stirrer (500–650 rpm) at room temperature, and the stirring was allowed for 8 h with intervallic sonication. The resulting mixture was kept in a vacuum desiccator to remove the bubbles and rotavapored followed by high vacuum pumping (FD 12, Crompton Greaves Ltd., India) for the drying purpose.
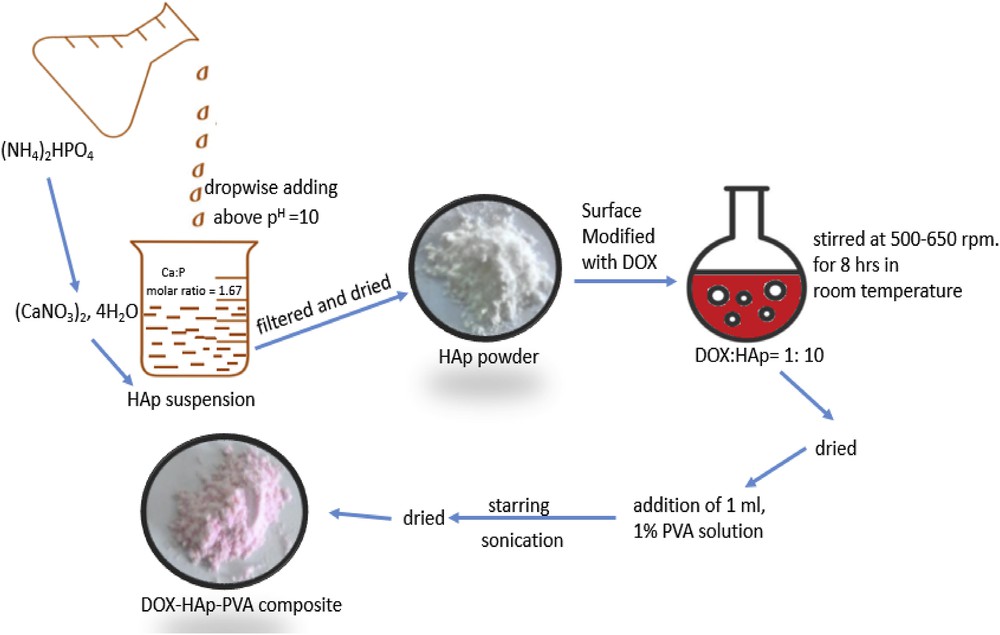
Synthesis of DOX-HAp-PVA nanocomposite.
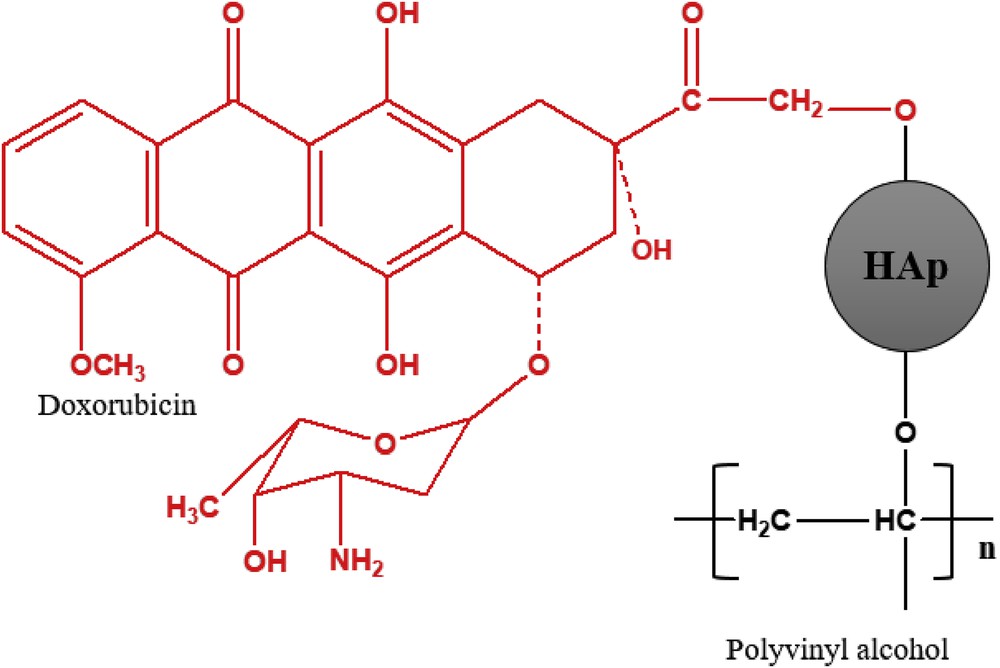
Most probable chemical structure of DOX-HAp-PVA nanocomposite.
2.5 Characterization
The solid raw materials and the prepared nanocomposites were weighed using analytical balance (Model ME204, Mettler Toledo, India). For drying purpose rota vapor (Model R-100, BUCHI) and high vacuum pump (FD 12, Crompton Greaves Ltd.) were used. For the sonication purposes, probe sonicator (Model Processor Sonopros PR-250 MP, Oscar Ultrasonics Pvt Ltd., India) was used.
The functional groups present in the HAp powder, PVA, and nanocomposite samples were identified and the nature of bonding between the particles and the polymer matrix was analyzed by FTIR spectrometry analysis (Model NEXUS870, FTIR, Thermo Nicolet, Madison, WI) within the scanning range of 4000–400 cm−1. The pellet was prepared by mixing 2 mg of the sample with 200 mg of oven-dried spectroscopy grade potassium bromide (Sigma–Aldrich, Germany).
XRD (Model PW 1729, Philips, Holland) was done for the phase analysis of prepared pure HAp powder and the composite samples using 35 mA and 40 kV current, with a monochromatic Cu Kα (target) radiation (λ = 1.5405 Å) with a step size of 0.04° 2θ, a scan rate of 0.02° 2θ/s, and a scan range from 2θ = 20°–60°. The crystallite size and crystallinity of the HAp crystals in the nanocomposite samples was determined using XRD technique.
TEM of HAp powder, DOX-HAp, and DOX-HAp-PVA nanocomposites was done through a Phillips CM 200 to observe the morphology, particle size, and dispersion of the particles in the polymer matrix with an acceleration voltage of 200 kV. A small amount of pure HAp powder and DOX-HAp-PVA nanocomposite samples was dispersed in ethanol and bath sonicated for total 8 h at room temperature. Then the TEM samples were prepared by depositing a few drops of the HAp powders and DOX-HAp-PVA nanocomposite solution on two different copper grids.
Stoichiometry of HAp was observed through the energy-dispersive X-ray (EDX) spectroscopy.
The particle size of DOX-HAp-PVA nanocomposite was also analyzed through dynamic light scattering, Zetasizer nanoparticle analyzer (Model Merlvern). For that purpose, 0.5 mg of DOX-HAp-PVA nanocomposite powder was dissolved in 5 mL of deionized water and stirred well by sonication for 2 h to reduce the particle agglomerations. Then the solution was diluted with 5 mL water and again stirred and sonicated for 30 min. Next, 0.5 μL double diluted DOX-HAp-PVA nanocomposite solution was taken and added in 1 mL distilled water and the particle size of processed solution was analyzed.
2.6 Drug loading content
Considering the UV absorption interference, the drug loading capacity was indirectly calculated as follows:
To determine the DOX loading efficiency, we used a UV–vis spectrometer (JASCO 720, Japan) at a wavelength of 480 nm.
2.7 Analysis of drug release from DOX-HAp-PVA nanocomposite
At first, four clean autoclaved glass vials were taken. Then 1 mg/vial conjugates were taken and 2 mL of phosphate-buffered saline (PBS) (pH 5.5) was added to each vial. The release was performed in a constant-temperature shaker at 37 °C. After 24 h, first vial was centrifuged at 10,000 rpm to separate the free drugs from the nanocomposite system and the collected supernatants were passed through m-columns to ensure that there were no residual nanoparticles. Likewise all of the supernatants were collected after every 24 h of interval. For the determination of released drug present in the supernatants UV spectrophotometer was used. All experiments were performed in triplicate to get perfection.
2.8 Cell viability and cell cytotoxicity assay
Cell viability was investigated using an MTT assay. First, osteosarcoma bone cancer cells (MG 63) were cultured in a 96-well culture plate at a density of 3 × 103 cells/well and allowed to attach for overnight. Then the cell-attached plate was washed three times with PBS solution and immersed in serum-free Dulbecco's Modified Eagle Medium (DMEM) medium (0.5 mL/well) containing different concentrations (10, 50, and 100) of 20 μL of HAp sample and DOX-HAp-PVA nanocomposite suspension. After incubation for different periods, the nanoparticle-immersed plate was washed several times with PBS solution to remove the nanoparticle residue. The MTT stock solution (5 mg/mL) was then diluted 10 times with serum-free DMEM medium and added to each well (0.5 mL/well). These cells were further incubated for 4 h to allow the yellow dye to transform into blue formazan crystals. The unreacted dye was then removed by aspiration, and dimethylsulfoxide (DMSO) (400 μL) was added to each well to dissolve the blue formazan crystals. Finally, the dissolved DMSO solution was transferred to a 96-well culture plate (100 μL/well), and its optical density was measured with an ELISA reader (Thermo Molecular Devices Co., Union City) at a wavelength of 570 nm. Tissue culture plate was taken as control. The experiment was performed in triplicate.
3 Results and discussion
The particle size of HAp powder was evaluated from TEM micrograph, which shows diameter approximately 10–12 nm and length of 25–35 nm with paddy seeds–like acicular crystals. Ca/P molar ratio of HAp powder is 1.67 with the specific surface area of 98.21 m2/g. The use of synthesized HAp powder with DOX provides an active step to produce drug–ceramic nanocomposites. Furthermore, the DOX-HAp system was modified with PVA, which is used as a matrix in the DOX-HAp-PVA nanocomposite. PVA helps to decrease the rapid release of DOX from the nanocomposite, for that the biodegradable polymers are used to prepare DOX loaded bioceramic scaffolds. Sustained release of drugs from porous bioceramic materials and good mechanical strength are expected from those nanocomposites.
The broad peaks were observed at 3600–3100 cm−1 in the FTIR spectrum of HAp (Fig. 2). The presence of structural OH (3569.36 cm−1), H2O (3416.74 and 1638.46 cm−1), and PO43− (1090.87, 1030.11, 964, 871.68, 604.31, and 564.37 cm−1) were found in the FTIR spectrum of HAp. The bands at 1090.87 and 1030.11 cm−1 are assigned to the PO stretching vibrational mode (ν3) of PO43−, whereas the bands at 964, 871.68, 604.31, and 564.37 cm−1 are due to PO bending vibrational modes of PO43− in the HAp crystals [16,17]. Thus, from the analysis of characteristic peaks found in the FTIR spectrum of HAp, it can be stated that the synthesized white powder is of stoichiometric HAp. Also some distinct peaks at wave numbers 669.53, 1032.01, 1099.40, and 3443.11 cm−1 were found from the FTIR spectrum of DOX-HAp. These peaks correspond to alkyne CH, aromatic CH, aromatic CH, and H-bonded OH stretch (Table 1) [18]. The band at 1622 cm−1 of DOX loaded HAp nanocomposite may be due to the overlapping bands of pure DOX at 1637.12 cm−1 and HAp at 1630.72 cm−1 indicating association of DOX with HAp particles [19]. Beside these, a band at 2043.54 cm−1 was found in the DOX-HAp FTIR spectrum, which may be due to overlap of 2077.63 cm−1 of HAp and 2085.67 cm−1 of DOX. The CH2 asymmetric stretching band of PVA at 2947.23 and 831.31 cm−1 was found from the DOX-HAp-PVA FTIR spectrum. The asymmetric bending vibration of the CH2 group of PVA molecule at 1384 cm−1 and structural OH at 1618 cm−1 were found to be shifted, which correspond to 1381.03 and 1613.68 cm−1 in the DOX-HAp-PVA spectrum. The structural OH at 3569.36 cm−1 was found almost at the same position for all three FTIR spectra. The stretching vibration of CO is traced at 1095.57 cm−1 [20]. The PO vibrational mode (ν3) of PO43− at 1030.11 cm−1 in HAp crystals was found shifted at 1032.01 cm−1 in both FTIR spectra of DOX-HAp and DOX-HAp-PVA. This observation indicates that the prepared material is composed of HAp, DOX, and PVA particles.
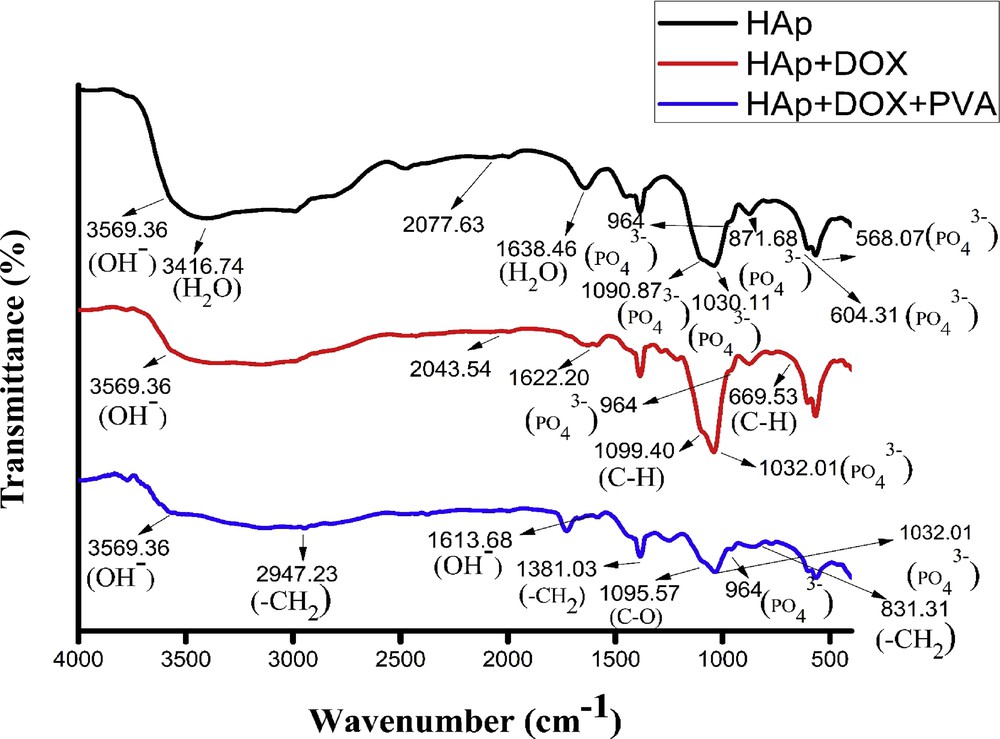
FTIR spectra of HAp, HAp-DOX, and HAp-DOX-PVA nanocomposites.
FTIR peak positions of various functional groups of HAp, DOX-HAp, DOX-HAp-PVA nanocomposite before and after composite formation.
| Sl. No. | Assignments | HAp (cm−1) | HAp-DOX (cm−1) | HAp-DOX-PVA (cm−1) |
| 1 | Structural OH | 3569.36 | 3569.36 | 1613.68, 3569.36 |
| H2O | 3416.74, 1638.46 | – | – | |
| 2 | PO vibrational mode (ν3) of PO43− | 1090.87, 1030.11 | 1032.01 | 1032.01 |
| 3 | PO bending vibrational mode of PO43− | 964, 871.68, 604.31, 564.37 | 964 | 964 |
| 4 | Alkyne CH | – | 669.53 | – |
| 5 | Aromatic CH | – | 1032.01, 1099.40 | – |
| 6 | H-bonded OH stretch | – | 3443.11 | – |
| 7 | CH2 asymmetric stretching | – | – | 2947.23, 831.31 |
| 8 | CH2 asymmetric bending | – | – | 1381.03 |
| 9 | CO | – | – | 1095.57 |
The XRD pattern of the synthesized apatite powder has been presented in Fig. 3. The d-values correspond to that of calcium HAp [Ca10(PO4)6(OH)2]. The broadening of XRD peaks indicates the synthesized HAp powder is nanocrystalline in nature. Scherrer's equation (
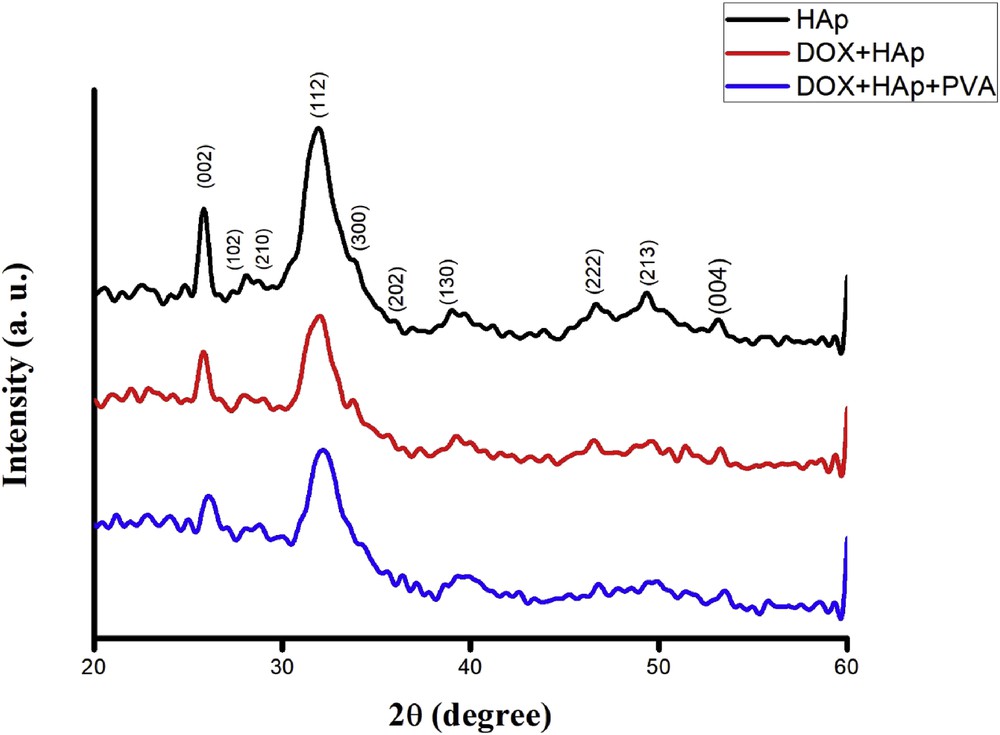
XRD patterns of HAp, DOX-HAp, and DOX-HAp-PVA nanocomposites.
XRD patterns of the DOX-HAp and DOX-HAp-PVA nanocomposites are presented in Fig. 3. Scherrer's equation is also used to calculate the crystallite size of both nanocomposites. The crystallite size of DOX-HAp was found to be 22 nm using Scherrer's equation, whereas the crystallite size was decreased to 17 nm for DOX-HAp-PVA nanocomposite. The XRD peaks of HAp (210) shifted to higher and (102) shifted to lower 2θ in DOX-HAp XRD micrographs as compared with HAp, that is, from 28.78° to 29.08° and from 28.08° to 27.98°, respectively. XRD peak (210) of DOX-HAp further shifted to 29.08°–28.82° in case of DOX-HAp-PVA. It is possibly because of compression from the contracting polymeric matrix through interfacial bonding. A peak of HAp at 56.76° is totally vanished from DOX-HAp XRD graph and also appeared at 55.80° in DOX-HAp-PVA XRD graph. The XRD peak at (213) (2θ = 49.36) of HAp has been vanished in DOX-HAp and DOX-HAp XRD graph. Simultaneously, a shift in the crystalline peak of drug ceramic composite has been observed at 2θ = 37.34. For DOX-HAp-PVA nanocomposite, it is shifted to 2θ = 37.16. Also a new peak has been found at 2θ = 36.42 in the polymer matrix drug ceramic XRD graph. The peak positions before and after composite formation are given in Table 2. The shift and decrease/increase in crystallinity of each peak of the HAp in DOX after composite formation clearly indicate the presence of bonding/bond formation between HAp particles with drug and polymer matrix [15].
XRD peak positions of (210), (102), and (213) planes of HAp and crystalline peaks of polymer nanocomposite before and after composite formation.
| Peaks | XRD peak positions (2Ө values in degree) | ||
| HAp | HAp-DOX | HAp-DOX-PVA | |
| (210) Peak of HAp | 28.78 | 29.08 | 28.82 |
| (102) Peak of HAp | 28.08 | 27.98 | – |
| – | 56.76 | – | 55.80 |
| (213) Of HAp | 49.36 | – | – |
| – | – | 37.34 | 37.16 |
| – | – | – | 36.42 |
The morphology and the particle sizes of synthesized n-HAp powders are shown in the TEM micrographs. Fig. 4 shows the TEM micrographs of the synthesized HAp. The crystals are paddy seeds–like in shape for all cases. In case of HAp, the particles are more or less uniform in sizes, and micrograph depicts the acicular paddy seeds–like crystals in the nanometer range having approximately 10–12 nm diameter by 25–35 nm length (Fig. 4a). A little bit uniformity of particles was found in the TEM micrograph of DOX-HAP nanocomposite, with acicular paddy seeds–like crystals in the nanometer range having approximately 10–15 nm diameter by 45–50 nm length (Fig. 4b). The presence of agglomerations between the particles was seen in the micrographs of HAp and DOX-HAP nanocomposite. In a comparison of HAp and DOX-HAp less agglomerations were observed in the TEM micrograph of prepared DOX-HAp-PVA nanocomposite. In case of DOX-HAp-PVA nanocomposite, the particles are of approximately 10–15 nm in diameter with 65–85 nm in length (Fig. 4c). Polycrystallinity of HAp powder was found in all of the TEM micrographs. Moreover, less agglomeration is observed in case of DOX-HAp-PVA TEM micrographs for the use of PVA as a polymer matrix, which may be due to lowering of the surface energy of HAp nanoparticles.

TEM micrographs of (a) HAp powders, (b) DOX-HAp nanocomposite (c) DOX-HAp-PVA nanocomposite. Selected area electron diffraction (SAED) pattern of (d) HAp, (e) DOX-HAp nanocomposite, and (f) DOX-HAp-PVA nanocomposite.
The TEM micrograph (Fig. 4) of HAp depicts the acicular paddy seeds–like crystals in the nanometer range having approximately 10–12 nm diameter by 25–35 nm length with agglomerations. The diameter and the length of HAp particles found in TEM micrographs are almost same as found by the other researchers elsewhere [21,22]. According to the nucleation–aggregation–agglomeration growth mechanism theory HAp particles can undergo through the following steps: (a) nucleation and growth to form HA nanocrystallites; (b) aggregation of elemental nanocrystals by molecular attractions of different nanometric/colloidal scale forces, which cause surface free energy minimization; and (c) further crystal growth, at a constant residual supersaturation, acting as a cementing agent inside the aggregates to form agglomerate [23]. Nanoparticles have a very high surface area. So if we think about the surface of any nanoparticulates, the cohesive forces between molecules work on the neighboring atoms. However, the unsatisfied atoms exist on the surface of the material, which do not have neighboring atoms above them. So a stronger attractive force between the nearest neighbors on the surface is exhibited by surface atoms, that is, surface tension. This may be the cause of nanoparticle agglomerations and the size (94 nm) of pure small HAp particles represented through PSD (described in Fig. 5).
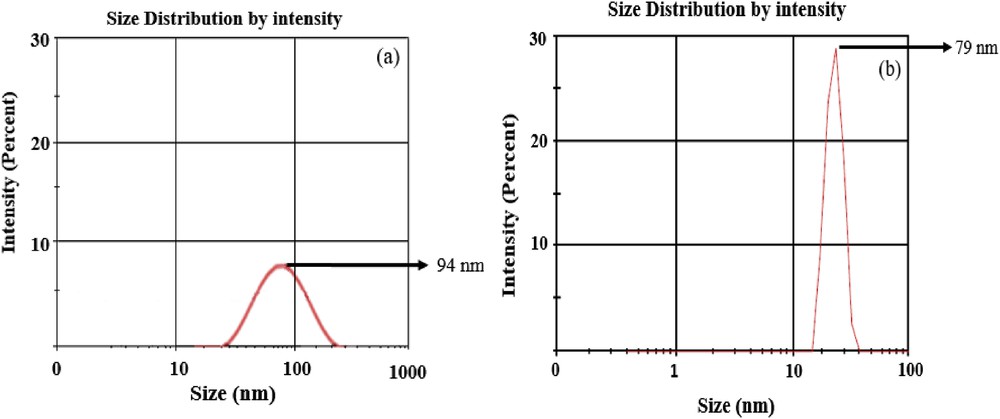
PSD of (a) pure HAp and (b) DOX-HAp-PVA nanocomposite.
Nanosized crystallites are found in many powders but most are agglomerated when found in the dry state and redispersion is quite difficult. Generally PSD (described briefly in Fig. 5) is a manner to measure the degree of agglomerations, range of distribution of agglomerates, and also is an important aspect to define the suitability of powders for various applications [24], whereas TEM microscopy may depict the singular particular dimensions of nanoparticles with a few or less agglomerations [24].
In the given EDX, Fig. 6 shows the presence of Ca and P in HAp and DOX-HAp-PVA composite. The given HAp and DOX-HAp-PVA composite gives the stoichiometric ratio of Ca/P = 1.674 and 1.651, respectively, in the elemental analysis (Table 3). The observed Ca2+/PO43− number ratio (1.674 and 1.651) against a calculated value (1.67) indicates that the synthesized HAp and the HAp used in the preparation of DOX-HAp-PVA nanocomposite are stoichiometric in nature.
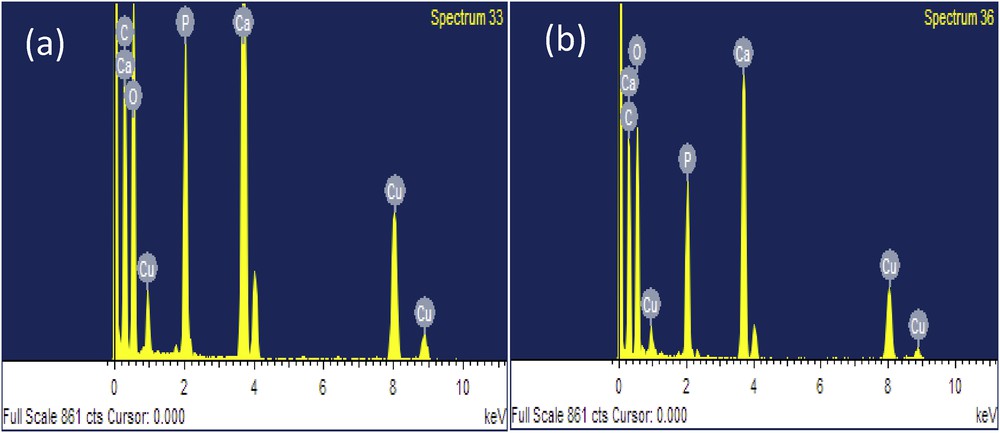
EDX of (a) HAp and (b) DOX-HAp-PVA nanocomposite.
EDX data of HAp powders and DOX-HAp-PVA nanocomposite.
| Element | Synthesized HAp (atom %) | HAp from drug polymer composite (atom %) |
| C K | 38.23 | 45.05 |
| O K | 28.31 | 26.54 |
| P K | 11.10 | 9.92 |
| Ca K | 18.59 | 16.38 |
| Cu K | 3.78 | 3.11 |
| Total | 100 | 100 |
| Observed Ca+/PO43− number ratio | 1.67 | 1.65 |
| Calculated Ca+/PO43− number ratio | 1.67 | 1.67 |
The PSD of HAp particles and the PVA-coated DOX-HAp nanocomposite sample were done by using dynamic light scattering technique (Fig. 5). The hydrodynamic size of HAp nanoparticles was received as approx. 94 nm (Fig. 5a), whereas the hydrodynamic size of DOX-HAp-PVA nanocomposite was found to be approx. 79 nm (Fig. 5b). Thus the result clearly supports the TEM study of HAp and DOX-HAp-PVA nanocomposites, which depicts that agglomeration is decreased for DOX-HAp-PVA nanocomposite rather than only particular solution. This may be due to the use of DOX, which acts as a surface anchoring agent to prevent particle agglomerations; for that it may control the hydrodynamic size of particles and uniformity in dispersion of particles present in the polymer matrix, which may also be responsible for the improvement of physicochemical and biological properties in DOX-HAp-PVA nanocomposite.
In the present study, the burst release of DOX was observed at 37 °C and at low pH. A time-dependent DOX release study was done by Akbarzadeh et al. [25] and also concluded that the cumulative release rate of DOX increases in low pH with increased temperature. Four clean autoclaved glass vials were taken. Then 1 mg/vial conjugates were taken and 2 mL of PBS (pH 5.5) was added to each vial. The release of DOX was performed in a constant-temperature shaker at 40 °C. After 24 h, first vial was centrifuged at 10,000 rpm to separate the free drugs from the nanocomposite system and the collected supernatants were passed through m-columns to ensure that there were no residual nanoparticles. Likewise all of the supernatants were collected, respectively, after 24, 48, 72, and 96 h of interval (Fig. 7). The determination of drug release was performed by UV–vis spectrophotometer. Five different concentrations of DOX were prepared and the absorbance of the each sample was taken by UV spectrophotometer at 480 nm, and the standard curve of DOX was prepared using the equation, y = mx + c. Also the release curve was obtained by plotting the UV data of supernatants with respect to the DOX standard curve. All experiments were performed in triplicate.

Supernatants of DOX-HAp-PVA nanocomposite at different time durations.
Sustain release of DOX from synthetic biodegradable polymers has some advantages that the loaded drugs can release from the scaffolds over a period of days to several weeks [26], and the treatment of cancer by chemotherapy over a certain time is the primary needs to inhibit cancer proliferation. On this concern, it is also possible to monitor the different stages and physiological conditions of the tumor cells after giving medications, which may open another window for further research. Basically interconnected polymer fibers create pores in resulting scaffolds, which inhibit the illusion of drug from scaffolds [27], and the fine pores of fibrous polymer may be responsible for the release of DOX in a controlled manner from composite. In addition, drug (DOX) diffusion through layers of polymer may cause for the more controlled and prolonged release rate of DOX from DOX-HAp-PVA composite [27]. From this release curve (Fig. 8), it can be stated that the burst release of DOX was increased in first 72 h and after that the release was same or stopped.
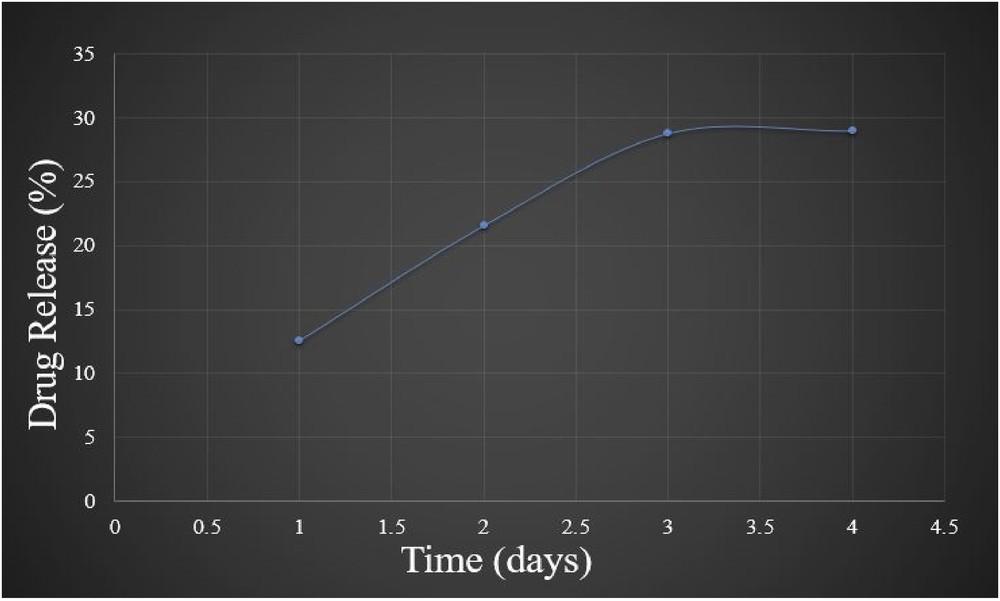
In vitro release pattern of DOX from DOX-HAp-PVA nanocomposite into PBS (pH = 5.5) at 37 °C.
The MTT assay is a colorimetric assay for assessing cell metabolic activity. The yellow tetrazolium MTT reagent is reduced by metabolically active cells and resulting intracellular purple formazan can be solubilized and quantified by spectrophotometric means. In the reaction of MTT, the yellow color tetrazolium salt losses its color and turned into purple. The color reduction is directly proportional to the number of living cells. The measured optical density is directly proportional to the number of viable cells present in the culture medium. Therefore, MTT assay can be used as a quantitative assay to determine the cytotoxicity of materials and the viability/proliferation of the cells. The effect of pure HAp nanocrystals on osteosarcoma bone cancer cells (MG 63) was examined to evaluate its cytocompatibility. Two different concentrations of pure HAp were used and the obtained data are presented in Fig. 9 through graphical presentation. MTT is a water-soluble tetrazolium salt, which reacts with the mitochondria (mitochondrial dehydrogenase) of metabolically active cells and converts the soluble yellow MTT tetrazolium salt into an insoluble purple formazan. Formazan is an impermeable product accumulated in cell membranes with loss of integrity, and using this method, living cells can be estimated. The analyzed MTT result exposes a good cytocompatibility of HAp nanocrystals and no apparent toxicity of HAp toward MG 63 cells, even at higher concentrations of 10 and 50 μg/mL. Also the viability was obtained over 90%. This study based on cytocompatibility of HAp nanoparticle narrates that it is a potential bioactive material for biomedical applications, especially as a drug carrier in nanodrug delivery systems.
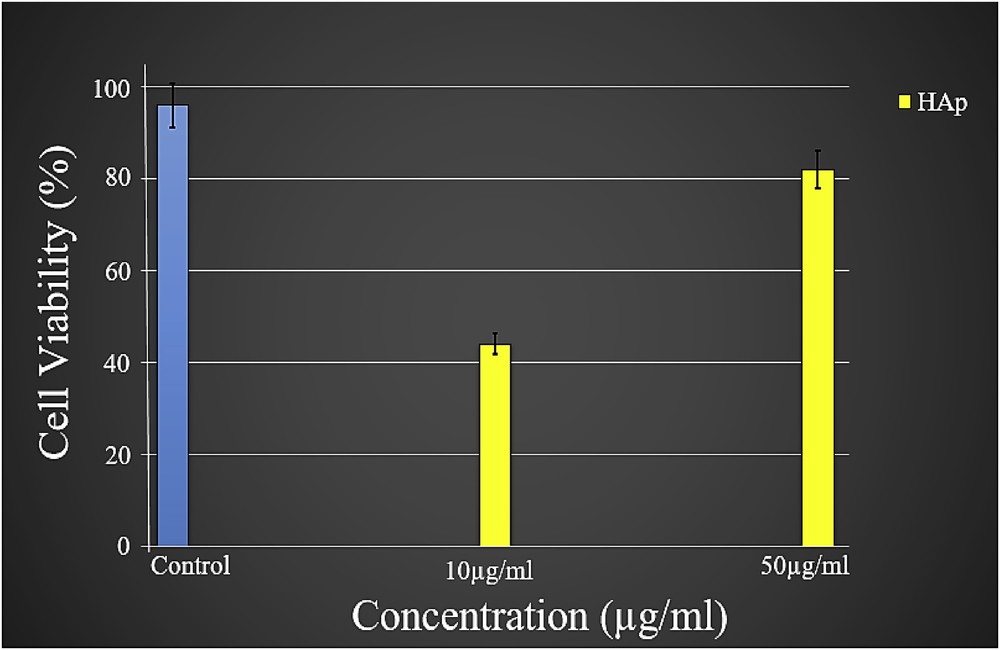
The MTT assay of cells (MG 63) cultured on HAp samples with different concentrations. Error bars represent the mean ± SD of three samples (n = 3).
Cytotoxicity studies of DOX-HAp and DOX-HAp-PVA nanocomposites were assessed on the MG 63 osteosarcoma cell line using an MTT assay (Fig. 10). MG 63 cells were treated with different concentrations of DOX-HAp and DOX-HAp-PVA nanocrystals (Fig. 11) and allowed to incubate for the periods of 24 and 48 h, respectively, at 37 °C in a CO2 5% humidified incubator. It was observed that the cell inhibition rate was increased when the concentration of the samples increased during the incubation periods. Fig. 10 describes that MG 63 was more sensitive to DOX-HAp nanocomposite as compared with DOX-HAp-PVA nanocomposite for 24 h. But it was observed that DOX-HAp-PVA nanocomposite was found to be more sensitive than DOX-HAp nanocomposite at the next 48 h. The differences in cytotoxicity of the two nanocomposites explained by many researchers that the mechanism of cellular uptake of the drug occurs through a passive diffusion mechanism [28]. In the case of polymer-coated drug ceramic nanocomposite, the drug has to be released in a time-dependent manner from the polymer matrix. Therefore, the concentration of the released drug from the DOX-HAp-PVA nanocomposite was increased with proportional to time and inhibited the MG 63 cell growth in a time-dependent manner.
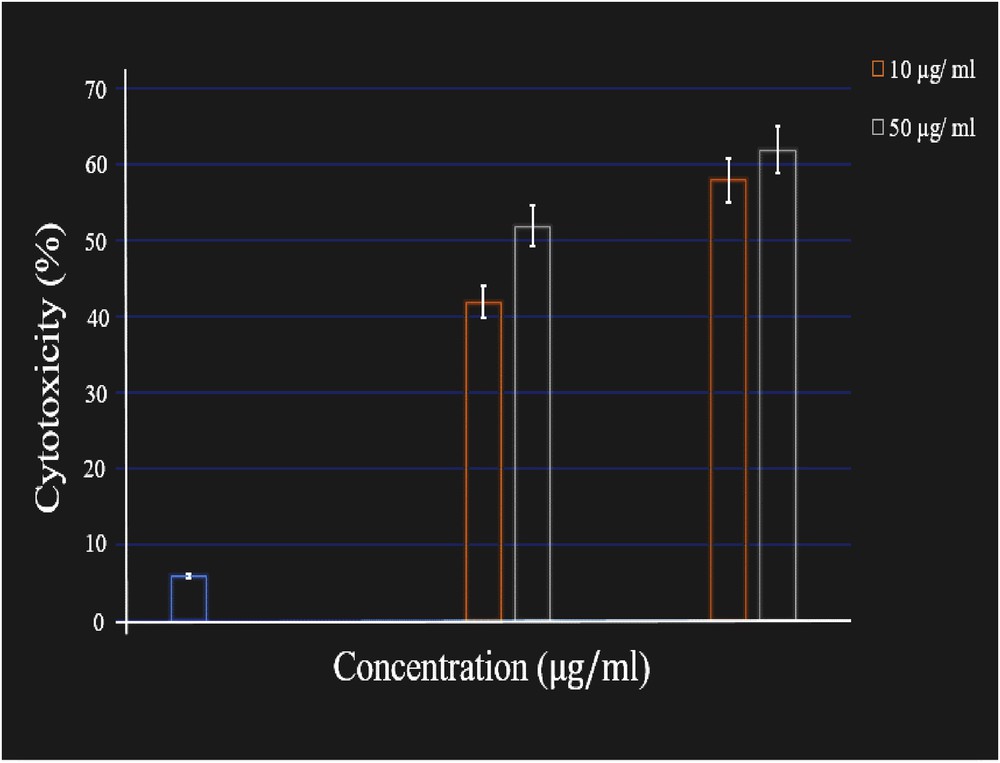
The MTT assay of MG 63 cells cultured on nanocomposite samples with different concentrations. Error bars represent the mean ± SD of three samples (n = 3).

Optical micrograph of (a) control, (b) DOX-HAp-treated MG 63 after 24 h, (c) DOX-HAp–treated MG 63 after 48 h, (d) DOX-HAp-PVA–treated MG 63 after 24 h, and (e) DOX-HAp-PVA–treated MG 63 after 48 h.
Morphological observation through microscopic investigation (Fig. 11) is one of the most consistent tests for determining and quantifying cell viability and cell death. MG 63 cells were treated with various concentrations of DOX-HAp-PVA nanocomposite for 24, 48, and 72 h. After 72 h, the cells were observed under microscope. The treated cells were found in round shape and cell shrinkage was also observed, which depicts the apoptotic changes. Also the apoptotic bodies were found bounded with the membrane.
4 Summary and conclusions
A novel bioanalogue DOX-HAp-PVA nanocomposite with uniform dispersion of nanoparticles has been synthesized successfully using a simple solution-based chemical method with a significant improvement in physicochemical and biomedical properties. The use of DOX has provided an effective method toward the improvement in physicochemical and biological properties of nanocomposites. FTIR analysis clearly confirms the presence of interfacial bonding interaction between the filler and polymer matrix of the prepared nanocomposites. Changes/shifts in XRD peaks of DOX-HAp and DOX-HAp-PVA nanocomposites after composite formation have indicated the bond formation between HAp particles with drug and the polymer matrix. The use of PVA as a polymer matrix has produced a remarkable effect on the dispersion of nanoparticles, which is further proved by TEM analysis. The particulate dimensions measured from XRD, PSD, and TEM studies are found to be comparable and in the nanoscale range. The cytocompatibility of prepared HAp was found from MTT assay, and it can be stated that the prepared HAp may supply the nutrient to bone for regeneration. A remarkable cytotoxicity toward osteosarcoma cells (MG 63) was observed when HAp is conjugated with DOX in the PVA matrix (DOX-HAp-PVA nanocomposite) with improved physicochemical properties followed by improved biological properties.
Acknowledgments
Authors are grateful to the Indian Council of Medical Research (ICMR), New Delhi, India, for providing financial support (Project Grant No. 5/7/1263/2015-CH).
Vous devez vous connecter pour continuer.
S'authentifier