


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Bio-oil from fast pyrolysis of biomass contains abundantly soluble and oxygenated organics such as carboxylic acids, guaiacols and aldehydes etc. [1, 2, 3, 4], which result in strong acidity, high viscosity, low calorific value and complex composition of bio-oil [5, 6, 7, 8]. For this reason, bio-oil needs to be upgraded (via esterification, aldol condensation, ketonization, cracking, and hydro-deoxygenation (HDO)) before it can be commercially applied [9]. Considering the difficulty in understanding routes of upgrading bio-oil, many of studies were concentrated on catalytic HDO of model molecules of bio-oil to obtain commercial biofuels and chemicals [10, 11, 12]. Recently, nickel phosphide supported on acid solids (Al2O3, HZSM-5, etc.) as catalysts for HDO attracted researcher’s attention since acid supports can promote synergic interactions between the metal active phase and their intrinsic acid sites [13, 14, 15]. Furthermore, the use of acid supports also favored sequential hydrogenation–dehydration–hydrogenation reactions in HDO of molecules [16, 17].
Noteworthy, HDO studies of carboxylic acids, guaiacols and aldehydes are mainly focused on nickel catalysts with acid solids, but fewer works use nickel phosphide catalysts with acidic supports. For instance, Chen and Falconer [18] studied the HDO of formic acid via temperature-programmed reaction (TPR) using a Ni/Al2O3 and found a total conversion of formic acid at 475 °C and high exposure (520 μmol/g catalyst). Results showed that CO was the main product followed by CO2 and a small amount of CH4. Similarly, Peng et al. [19] explored the HDO conversion of palmitic acid with a Ni/ZrO2 catalyst. The deoxygenation mechanism indicated hexadecanal and 1-hexadecanol were the initial products and then undergo decarbonylation to produce n-pentadecane and CO.
The HDO of another important family of bio-oil products, the guaiacols, has also been studied. Broglia et al. [20] performed experiments on guaiacol HDO using Ni/alumina-silica catalysts with varying Ni and silica amount. High conversions of guaiacol were achieved (up to 84%) in a very short time scale (1 h) and, generally, methylguaiacol and phenol were the primary products at 300 °C under H2 (50 bar). Zhang et al. [11] studied the catalytic HDO of guaiacol using a series of high-loading nickel phosphide catalysts supported on SiO2–TiO2. Cyclohexane, cyclohexanol, and 2-methoxycyclohexanol are the main products over all of these catalysts from 200 to 260 °C. In addition, Oyama et al. [21] investigated the guaiacol HDO using Ni2P/ASA, Ni2P/FCC, Ni2P/ZSM-5 catalysts. As a whole, the dominant products were cresol and phenol. Especially, the effect of contact time indicated that the main pathway on Ni2P/ASA was the conversion of guaiacol to the catechol as primary intermediate followed by dehydroxylation to phenol. Recently, Li et al. [22] studied the HDO of guaiacol on Ni/HZSM-5 catalysts and reported that a conversion rate of 74.8% and a selectivity of 87.1% for cyclohexane were achieved. Also, it was discovered that mesopores and strong metal-support interaction promoted the conversion of guaiacol and the hydrogenation of guaiacol. In another article, the HDO of guaiacol over Ni/γ-Al2O3 was carried out by Tran et al. [23]. Results showed that 96.88% guaiacol conversion and 58.98% of 1,2-dimethoxybenzene production were obtained over 10 wt% Ni/Al2O3 catalyst calcined at 450 °C. At the same time, it has been observed that guaiacols lead to easy coke formation [24], which results in obstacles in upgrading of bio-oil.
Besides, carboxylic acids and guaiacols, aldehydes (like furfural) are also one of the numerous families of pyrolysis oil components. Zhang et al. [25] investigated a one-pot hydrogenation/dehydration conversion of furfural using Ni/SiO2–Al2O3 bifunctional catalysts in a batch reactor and found a high selectivity to pentane and a conversion of 62.99% of furfural. In the study of HDO of n-hexane-extracted pyrolysis oil, Zhao and Lercher [26] found that furfural underwent HDO/hydrolysis to form n-pentane (64%) and tetrahydropyran (36%) over a Ni/HZSM-5 catalyst at 250 °C under 5 MPa H2 for 2 h. In addition, Wang et al. [27] studied the in situ HDO of furfural in aqueous solution over Ni/Al2O3 catalysts under 1 MPa N2. Results showed that high temperature and amount of Ni loading facilitated the conversion of furfural and a total yield above 85% for furan and 2-methylfuran was reached at 260 °C and at the methanol-to-water ratio of 2:1. More recently, Lan et al. [28] explored furfural HDO using SiO2-supported nickel phosphide catalysts. It was found that the improvement of P content on the catalysts weakens the furan-ring/catalyst interaction, which led to a decreasing activity of ring-opening and ring-hydrogenation reaction.
In this background, this work is dedicated to easily understanding the HDO mechanism of bio-oil by studying the HDO of various model molecules over Ni2P/HZSM-5 catalysts. Here, acetic acid, 4-ethylguaiacol, and furfural were selected as model molecules of bio-oil due to their abundance in pyrolysis oil [29]. The catalysts were already selected for their performance demonstrated in our previous study of acetone HDO [30], and characterized through the N2 adsorption/desorption (BET specific surface area and pore size calculation), Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES) elemental analysis, X-ray Powder Diffraction (XRD), pyridine adsorption by Fourier Transform Infrared Spectroscopy (FT-IR), and H2-TPR using Differential Scanning Calorimetry (DSC). Then, various parameters were examined, such as amount of supported phase, temperature and pressure. An in-depth characterization of liquid and gas products of model molecules HDO was performed. Probable HDO reaction routes for the conversion of the aforementioned molecules were proposed, which provide significant guidance for the selective preparation of liquid fuels and high-value chemicals from bio-oil.
2. Materials and methods
2.1. Materials
HZSM-5 (Si/Al ratio 38) was provided by ACS Material LLC in the form of pellets having a diameter of 3 mm and an average length of 10 mm. (NH4)2HPO4 (AR, ⩾99.0%) was bought from VWR Chemicals. Ni(NO3)2 ⋅ 6H2O (AR, 99.9985%) was purchased from Alfa Aesar. HNO3 (PrimarPlus, trace analysis grade, 70%), acetic acid (analytical reagent grade, ⩾99.7%), and isopropanol (IP) (GC analysis grade) were supplied by Fisher Scientific. Furfural (AR, 99%) and 4-ethylguaiacol (AR, ⩾98%) were acquired from Sigma-Aldrich. Nonane (AR, 99%) was obtained from Prolabo.
2.2. Preparation and characterization of the catalysts
The Ni2P/HZSM-5 catalysts were prepared by the incipient wetness impregnation method as a first step and temperature-programmed reduction (TPR) as the second step [30]. Here, the support HZSM-5 was first ground and sieved to 0.35–0.6 mm particle size before loading the active phase. The specific preparation process and the characterization results of Ni2P/HZSM-5 catalysts by N2 adsorption/desorption (BET surface), ICP-OES elemental analysis, and XRD can be found in our previous work [30]. In addition, the acidity of the catalysts was characterized by FT-IR (Spectrum BX, Perkin Elmer) at different pyridine desorption temperatures (100, 150, 275, and 400 °C) after pyridine adsorption and could be found in the Supplementary materials (Figure S1). The samples of the catalysts were saturated with a small amount of pyridine vapor. Prior to the pyridine adsorption, all the samples were dried at 200 °C for 2 h. The H2-TPR of the Ni2P/HZSM-5 catalysts’ precursors (not reduced) was performed using a DSC (SENSYS-EVO) instrument with a reduction temperature range from 100 to 600 °C and a total gas flow rate of 30 mL/min (5 vol% of H2 in N2).
2.3. Catalytic activity tests
2.3.1. Activity test in setup
The HDO reaction of acetic acid, 4-ethylguaiacol and furfural were performed in a continuous fixed-bed reactor. The experimental setup can be seen in Figure 1, and most of the operation conditions are the same as our previous study [30]. Here, the liquid reagent, acetic acid, 4-ethylguaiacol or furfural was vaporized in a preheater at a temperature of 150 °C, 260 °C or 200 °C, respectively. The total pressure increased to the desired value of 0.5 MPa or 2 MPa for acetic acid HDO and 0.5 MPa or 3 MPa for 4-ethylguaiacol or furfural HDO, and the temperature went to the designed value of 300–500 °C once the catalysts’ reduction process was completed at 450 °C and 0.1 MPa of H2.
Scheme of continuous fixed-bed reactor.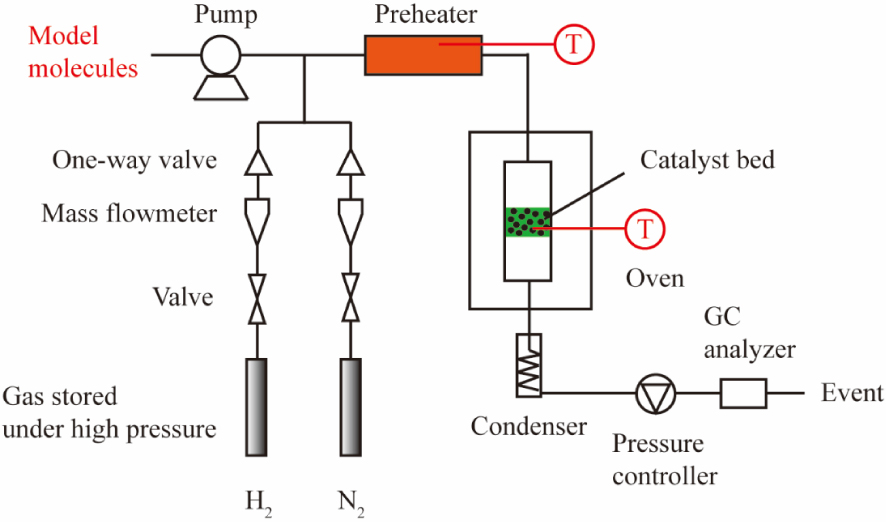
2.3.2. Products analysis
2.3.2.1. Liquid and gas compounds
The analysis of the liquid products was carried out using two different gas chromatographs equipped with the same medium polarity VF-1701-ms column (60 m × 0.25 mm × 0.25 μm film thickness) with two types of detector: mass spectrometer (GC-MS) (Clarus 580/SQ8S) system for identification and flame ionization detector (GC-FID) to quantify the amounts of compounds in liquid products. Isopropanol was used as the solvent to better analyze the liquid products in GC. Besides, the non-condensable products were gathered in a sampling bag and were then analyzed off-line using a Clarus 580 GC instrument from Perkin Elmer equipped with two detectors, a thermal conductivity (TCD) and a flame ionization detector (FID). The detailed analytical method of products was described in our previous work [30]. The level of experimental error was 2%, which was calculated by experiments repeated thrice.
In order to analyze the experimental results and to better understand the evolution of each model molecule, the conversion rate, selectivity, degree of deoxygenation (DOD), and mass yield were used. The expression of each term is given in the following:
The feedstock conversion X (%) is defined as:
| (1) |
nfeedout: the amount of feedstock in the liquid products, mol.
The selectivity [31] Seli (%) of the product chemicals was defined as:
| (2) |
ni: the amount of product i, mol.
nfeedconverted: the amount of feedstock converted, mol.
The DOD (%) was calculated according to the following equation:
| (3) |
mass of oxygen in : the total weight of oxygen in feedstock injected, g.
The yield Yi (%) of chemicals of the products (liquid, gas, and water) was determined using the formula:
| (4) |
3. Results and discussion
3.1. Catalysts’ characterization
3.1.1. N2 adsorption/desorption
The nitrogen adsorption–desorption isotherms for the catalyst samples are illustrated in Figure 2(A). It can be seen that both the 5% and 10% Ni2P/HZSM-5 catalysts basically show a combination of IUPAC types I and IV isotherms with a typical type H4 of hysteresis loop. Hence, it can be said that these samples exhibited the co-existence of microporous and mesoporous structures: the extensive adsorption observed below relative pressures (P∕P0 < 0.2) is characteristic of microporous structures, while intermediate relative pressures are attributed to mesoporous structures. Type H4 hysteresis is generally associated with narrow slit pores [32].
N2 adsorption/desorption of Ni2P/HZSM-5 catalysts: (A) isotherms; (B) pore size distributions.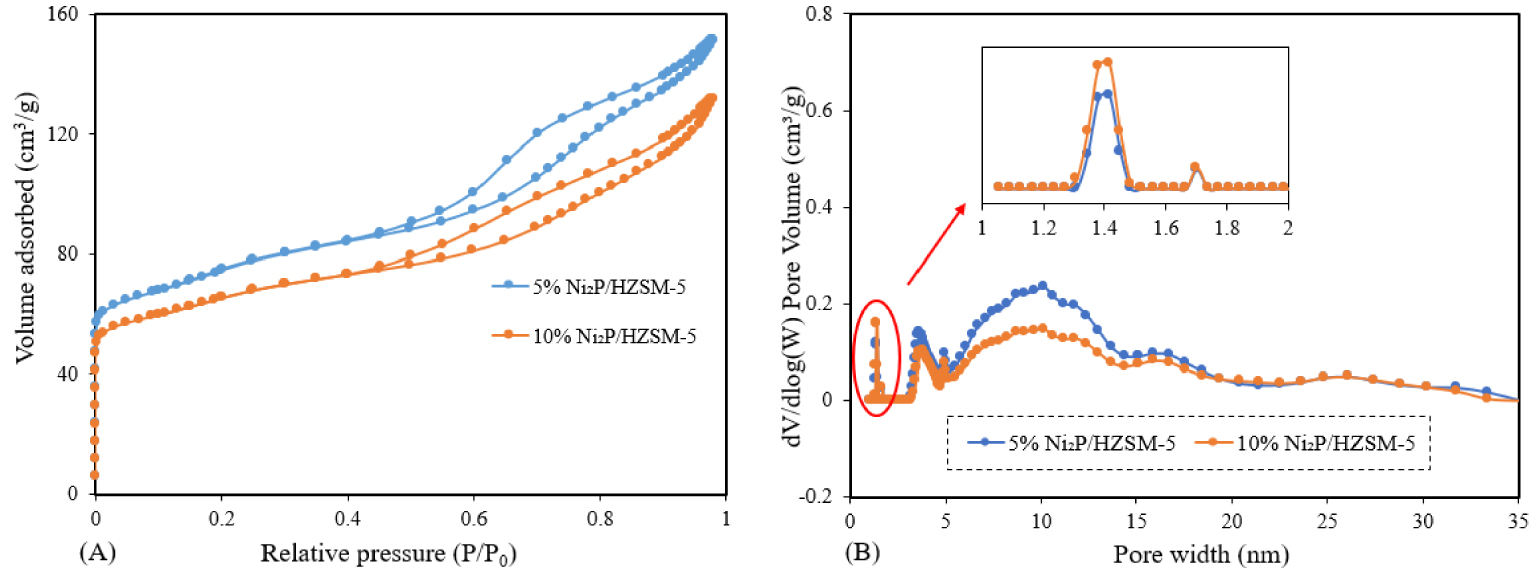
The pore size distribution of Ni2P/HZSM-5 catalysts was shown in Figure 2(B). It can be seen that Ni2P/HZSM-5 catalysts possess a majority of mesoporous distribution from 3 nm to 18 nm and a pinch of the microporous distribution below 2 nm. A higher mesoporous contribution for 5% Ni2P/HZSM-5 than the case of 10% Ni2P/HZSM-5 was observed. This can be probably attributed to the smaller Ni2P phase particles and lower Ni2P content on the 5% Ni2P/HZSM-5 compared to the 10% Ni2P/HZSM-5 catalyst.
3.1.2. H2-TPR characterization using differential scanning calorimetry (DSC)
The H2-TPR (DSC) of the Ni2P/HZSM-5 catalysts’ precursors with a reduction temperature range from 100 °C to 600 °C can be applied to study the formation process of the Ni2P active phase. As shown in Figure 3, a small peak was found between 250 °C–300 °C for 5% Ni2P/HZSM-5 precursor, indicating that some intermediates (for example, NixPyOz species) existed during this stage. No peak was observed during this stage in the case of 10% Ni2P/HZSM-5 precursor. This was probably because the higher Ni and P content formed bigger particles of active phase on the internal surface of pores and surface of 10% Ni2P/HZSM-5 than 5% Ni2P/HZSM-5 catalyst and thus prevented the internal NixPyOz intermediates from being transformed. The further transformation of intermediates to Ni2P via the reduction of NixOy and phosphorus species was associated with the presence of a broad peak at an onset temperature around 400 °C. This peak for 5% Ni2P/HZSM-5 was narrower than the case of 10% Ni2P/HZSM-5, which also indicated that the intermediates are easier to be reduced. In the literature, Oyama et al. also reported almost the same reduction peak of NiO species in the H2-TPR result of Ni2P/ASA precursor [21], and Chen et al. reported a higher onset reduction temperature around 500 °C of oxidized nickel species and phosphorus species (such as PO, P2O, and (PO)n) to form Ni2P phase [33].
H2-TPR (DSC) profiles of catalysts’ precursors.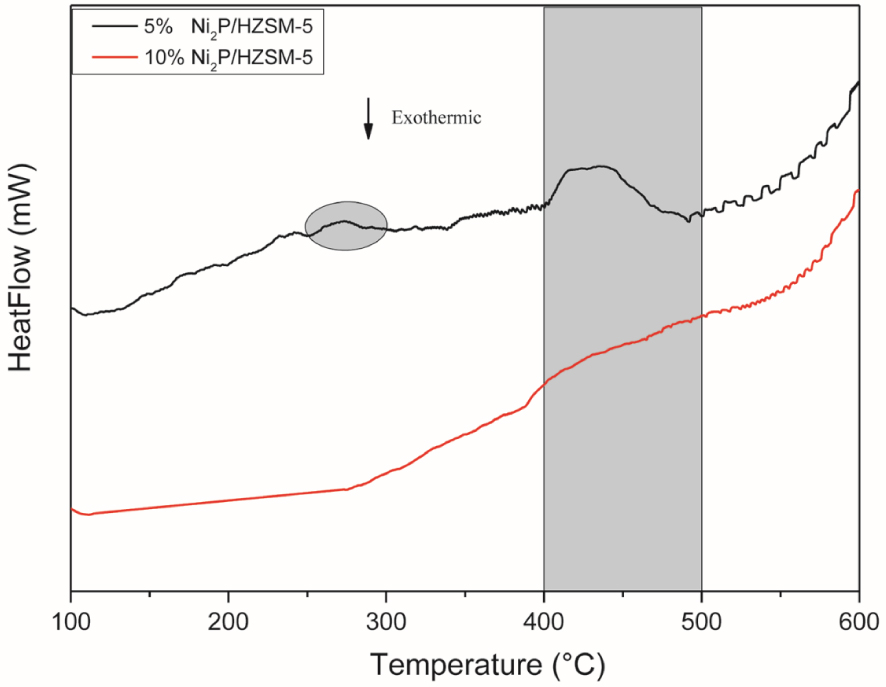
3.2. HDO of acetic acid
To get a liquid product with a low oxygen content, high yield of liquid phase is important and interesting. First of all, HDO of acetic acid using both the 5% and 10% Ni2P/HZSM-5 catalysts was carried out. The specific results were presented in Supplementary materials (Table S1 and Figure S2). In summary, both the catalysts exhibited an excellent catalytic activity for HDO reaction, and their similar behavior was observed. Subsequently, the 5% Ni2P/HZSM-5 catalyst was selected for further study.
Effect of temperature and pressure on acetic acid HDO
| Item | Conditionsa | |||
|---|---|---|---|---|
| 0.5 MPa | 0.5 MPa | 0.5 MPa | 2.0 MPa | |
| 350 °C | 400 °C | 450 °C | 350 °C | |
| Conversion rate & DOD (wt%) | ||||
| XAcetic acid | 61 | 80 | 97 | 69 |
| DOD | 59 | 77 | 96 | 68 |
| Yield of products (wt%) | ||||
| YLip. (free water) | 43 | 33 | 28 | 41 |
| YGas | 40 | 41 | 50 | 45 |
| YH 2O | 17 | 26 | 22 | 14 |
a Fixed conditions: 0.43 g 5% Ni2P/HZSM-5 catalyst, 0.05 mL/min of acetic acid, H2: 40 mL/min, N2: 10 mL/min, 90 min.
3.2.1. Effect of temperature and pressure
3.2.1.1. Conversion rate, DOD, and yield
Table 1 showed the effect of temperature and pressure on the conversion rate, DOD, and yield (liquid, gas, and H2O) of acetic acid HDO. The results exhibited a significantly higher conversion rate (97%) and DOD (96%) at 450 °C than at 350 °C, suggesting that temperature had a relevant effect on the catalytic activity of Ni2P/HZSM-5. The maximum yield of 50% for gas products was obtained at 450 °C accompanied by the minimum yield of liquid products (28%, H2O free). This evolution can be attributed to a notable increase in the hydrogenolysis of C–C bonds for acetic acid followed by dehydration and hydrogenation reactions to give more gas products, especially, CO and CO2. In a previous study, Badari et al. [34] found similar results during acetic acid HDO using a Ni/SiO2 catalyst, namely, that this catalyst favored the C–C bond hydrogenolysis to form lighter gaseous products. Concerning the water content, it first increased, and then decreased when the temperature increased from 350 °C to 450 °C, suggesting that the main reaction of acetic acid HDO at a medium temperature is dehydration.
The influence of temperature and pressure on acetic acid HDO (0.43 g 5% Ni2P/HZSM-5, 0.05 mL/min acetic acid, 40 mL/min H2, 10 mL/min N2, reaction time: 90 min): (A) Selectivity of major liquid products; (B) Selectivity of gas products.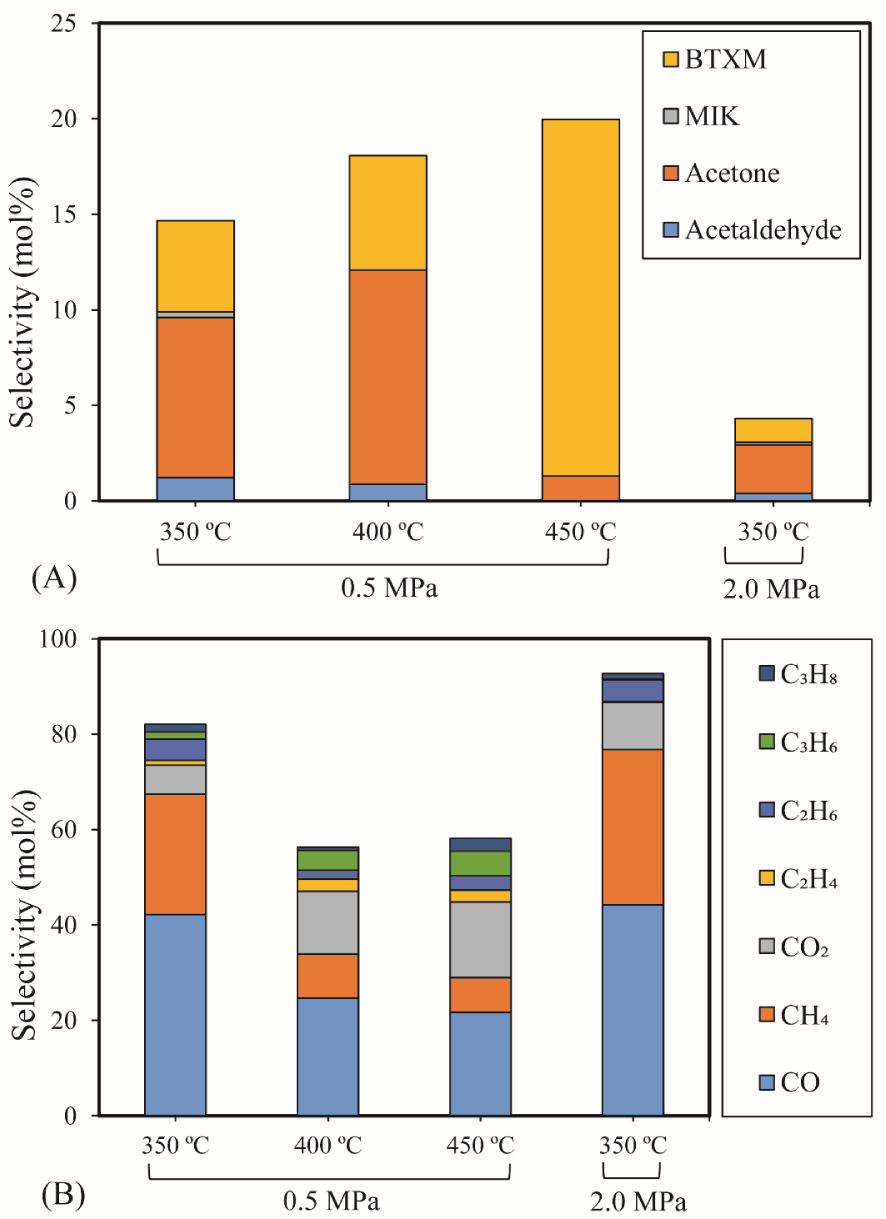
3.2.1.2. Selectivity in the liquid phase
The selectivity of major products for acetic acid HDO by varying the temperature and pressure is shown in Figure 4(A). Details of the chemicals identified in the liquid phase can be seen in Supplementary materials (Table S2). The results exhibited a significantly increasing trend in the selectivity of BTXM (benzene, toluene, xylene, and mesitylene) from around 5% to 19% when the temperature increased from 350 °C to 450 °C. In another work, Bej and Thompson [35] reported that mesitylene was formed via aldol condensation between mesityl oxide (MO) and acetone during the investigation of acetone condensation over molybdenum nitride and carbide catalysts. Gamman et al. [36] also proposed the same reaction route during the synthesis of methyl isobutyl ketone (MIK) over Pd/MgO/SiO2. On the other hand, MO was not detected in our work, which can be explained by the fact that the reaction from MO to MIK is spontaneous (enthalpy: 𝛥H400 K (kcal/mol) = −14.5; Gibbs free energy: 𝛥G400 K (kcal/mol) = −12.1) [36, 37]. Here, it can thus be speculated that the formation of BTXM can be caused by a further aldol condensation between mesityl oxide (MO) and acetone, followed by dehydration, hydrogenation, and isomerization.
Acetone was identified as the main product below 400 °C due to the self-ketonization reaction of acetic acid. This reaction was also observed in the catalytic upgrading of carboxylic acids by Huo et al. [38]. The linear MIK was probably obtained at 350 °C through an aldol condensation reaction of acetone [39]. These results indicate that high temperature promotes the condensation reaction to form dimers and trimers. Above 400 °C, the selectivity of acetone decreased sharply from 11% to 1%, which can be attributed to the conversion of acetone to aromatic hydrocarbons [40]. In addition, acetaldehyde was observed at low temperatures (350 °C and 400 °C) owing to the reduction of acetic acid.
3.2.1.3. Selectivity in the gas phase
Figure 4(B) shows the selectivity for gas products over the 5% Ni2P/HZSM-5 catalyst at different temperatures and pressures. Gases obtained were CO, CH4, CO2, C2H4, C2H6, C3H6 and C3H8. At 350 °C, similar values were observed under the two different pressures, and the highest selectivities for CO (44%) and CH4 (33%) were obtained under 2.0 MPa. The lower values of the selectivity of CO, CH4 and C2H6 were attained at 400 °C and 450 °C compared to those obtained at 350 °C. At 400 °C and 450 °C, the results also demonstrated a higher selectivity of gaseous olefins (C2H4 and C3H6) compared to 350 °C. C2H4 and C3H6 are derived from the direct deoxygenation and hydrogenation of acetaldehyde and acetone over the acid sites of the HZSM-5 support, and are favored by high temperature. Significant amounts of CO and CO2 were released because of the cracking of either carbonyl (RR′CO) or carboxyl (RCO2H) functional groups [41]. The further hydrogenation of C2H4 and C3H6 can lead to C2H6 and C3H8.
3.2.2. Proposed reaction pathways for acetic acid HDO
Based on the literature [38, 39, 42] and products formed, a set of reaction pathways for the HDO of acetic acid is proposed in Table 2. Reaction (1) is an acetic acid direct self-ketonization and decarboxylation reaction on a coordinatively unsaturated metal site of catalyst to a ketene (R2C=C=O) intermediate, and then to form acetone, CO2 and H2O [43]. Acetone is subsequently hydrogenated and dehydrated to C3H6 over the acidic function HZSM-5 in reaction (2) [44]. Reaction (3) corresponds to the reduction of acetic acid to acetaldehyde, together with the formation of H2O. Reaction (4) is a hydrogenation/dehydration reaction of acetaldehyde produced by the reaction (3), forming C2H4 and H2O. Reaction (5) is an aldol condensation reaction of acetone molecule to produce diacetone alcohol and MO intermediates followed by dehydration to form MIK [35, 39]. Mesitylene can be formed by further aldol condensation between MO and acetone molecules followed by dehydration in reaction (6). Mesitylene can be further converted to other aromatic hydrocarbons via demethylation and/or alkyl substitution, such as BTX (benzene, toluene and xylene) and ethylbenzene. Reaction (7) is the main hydrogenated reaction to form propane.
3.3. HDO of 4-ethylguaiacol
3.3.1. Effect of temperature and pressure
3.3.1.1. Conversion rate, DOD, and yield
Table 3 illustrates the influence of temperature and pressure on the conversion rate of 4-ethylguaiacol, DOD, and yield of products for HDO of 4-ethylguaiacol. The evolution of the conversion rate of 4-ethylguaiacol and DOD showed a small improvement when the temperature increased from 300 °C to 500 °C. At 400 °C, a significant increase of the conversion rate (84%) and DOD (51%) under 3.0 MPa was obtained compared to 0.5 MPa, demonstrating that pressure had a greater effect on the conversion rate and DOD than temperature.
The yield of gas products and the amount of H2O exhibited an increasing trend as the temperature increased. This can be attributed to an increase of the direct deethylation of 4-ethylguaiacol to form C2H6, C–O bond cleavage of the methoxy group for producing CH4, and further dehydroxylation of phenol intermediates to give H2O. The highest yield of 32% for gas products and of 14% for H2O was accompanied by the lowest yield of liquid products (50%, H2O free) at 500 °C.
Proposed reaction pathways of HDO of acetic acid
| Main products | Main reaction pathways | |
|---|---|---|
| Acetone | 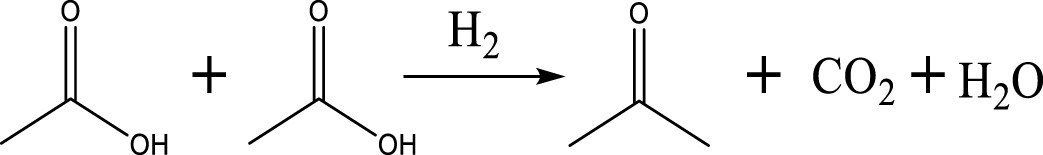 | (1) |
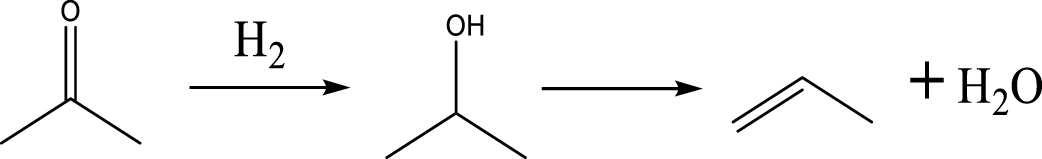 | (2) | |
| Acetaldehyde | 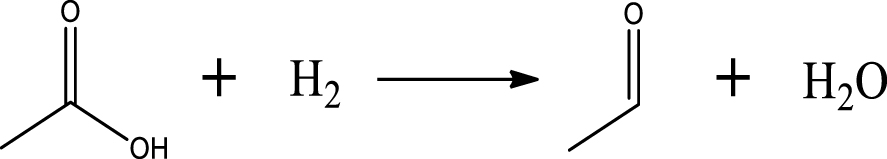 | (3) |
| Ethylene | 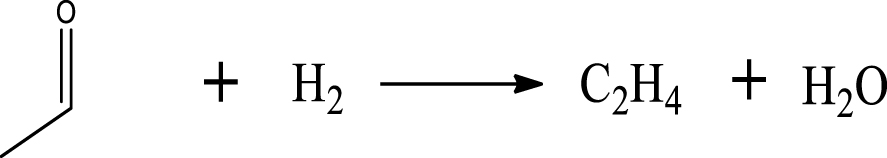 | (4) |
| Methyl isobutyl ketone | 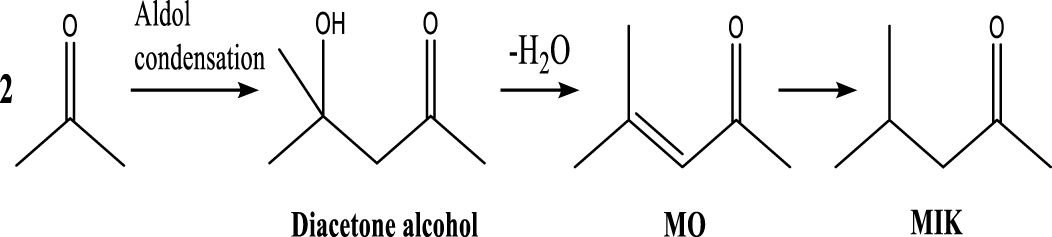 | (5) |
| Mesitylene | 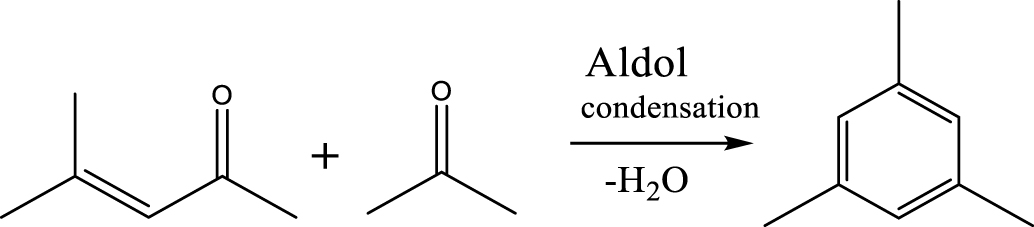 | (6) |
| Propane |  | (7) |
Effect of temperature and pressure on 4-ethylguaiacol HDO
| Item | Conditionsa | |||
|---|---|---|---|---|
| 3 MPa | 3 MPa | 3 MPa | 0.5 MPa | |
| 300 °C | 400 °C | 500 °C | 400 °C | |
| Conversion rate & DOD (wt%) | ||||
| X4-Ethylguaiacol | 75 | 84 | 83 | 44 |
| DOD | 43 | 51 | 53 | 25 |
| Yield of products (wt%) | ||||
| YLiq. (free water) | 79 | 71 | 50 | 87 |
| YGas | 12 | 20 | 32 | 8 |
| YH 2O | 9 | 9 | 14 | 5 |
| YCoke | - | - | 4 | - |
a Fixed conditions: 0.43 g 5% Ni2P/HZSM-5 catalyst, 0.05 mL/min of 4-ethylguaiacol, H2: 40 mL/min, N2: 10 mL/min, 90 min.
In addition, a notable amount of coke (4%) was formed at 500 °C, although no coke formation was observed at lower temperatures (300 °C and 400 °C). It is clear that coke formation is favored at a high temperature, probably via the condensation of 4-ethylguaiacol during the demethoxylation reaction. In another study, Gayubo et al. [45] found a notable coke formation from the upgrading of 2-methoxyphenol over ZSM-5 at high temperature.
The influence of temperature and pressure on 4-ethylguaiacol HDO (0.43 g 5% Ni2P/HZSM-5, 0.05 mL/min 4-ethylguaiacol, 40 mL/min H2, 10 mL/min N2, reaction time: 90 min): (A) Selectivity of major liquid products; (B) Selectivity of gas products.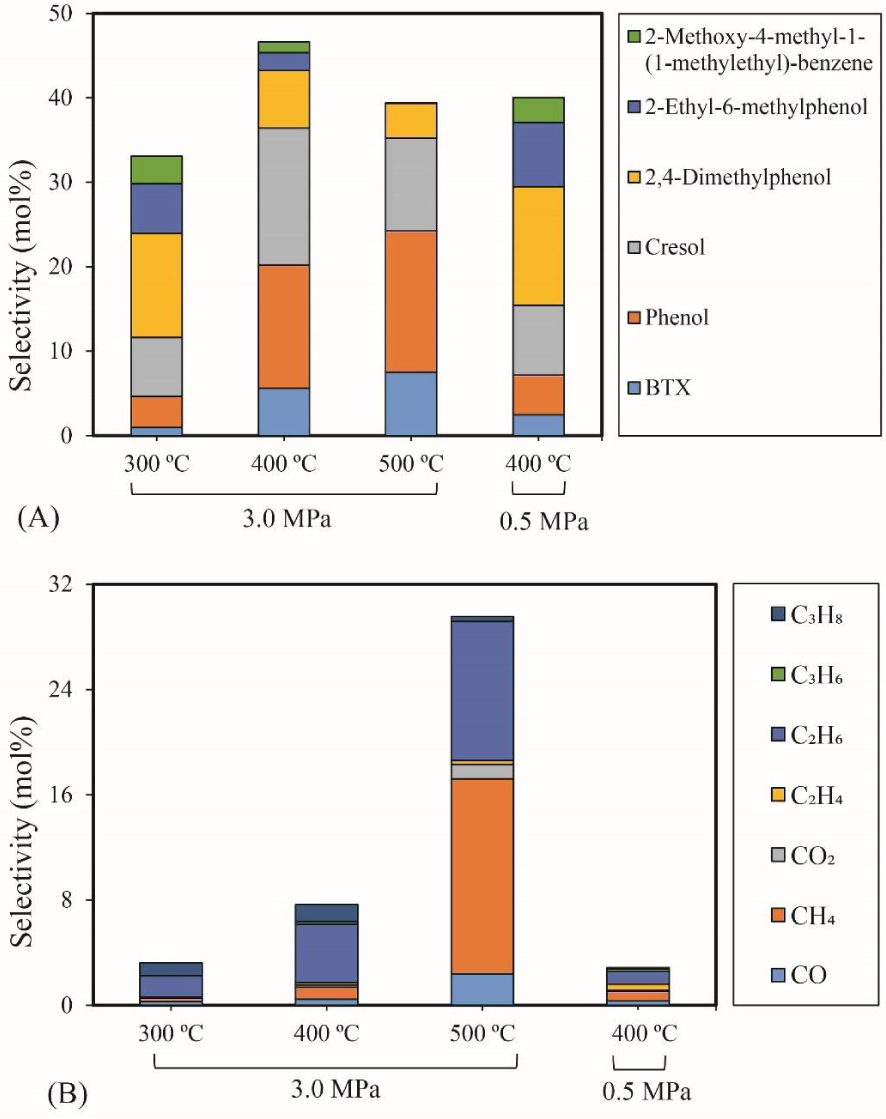
3.3.1.2. Selectivity in the liquid phase
BTX selectivity. Figure 5(A) shows the selectivity values of major products of 4-ethylguaiacol HDO as a function of temperature and pressure. The detail of the chemicals in the liquid products can be seen in the Supplementary materials (Table S3). The results revealed that the Ni2P catalyzed the HDO of 4-ethylguaiacol to produce oxygen-free aromatic hydrocarbons (benzene, toluene, xylene, etc.), mono-oxygenated aromatics (phenol, cresol (o- & p-), 2,4-dimethylphenol (DMP), etc.). At 300 °C, selectivities of 12% for DMP and 3% for 2-methoxy-4-methyl-1-(1-methylethyl)-benzene (MMMEB) were obtained under 3 MPa. The results under 3 MPa showed that the selectivity of MMMEB and DMP decreased when the temperature increased from 300 °C to 500 °C. This suggested that 4-ethylguaiacol was first converted to DMP via deethylation and methoxy removal, followed by alkylation/transalkylation, and the MMMEB was formed by dehydroxylation, alkylation/transalkylation and/or isomerization of 4-ethylguaiacol. These reaction pathways can be explained by the presence of a Lewis basic benzene ring of guaiacol molecules, which should be chemisorbed on Lewis acidic Ni sites of Ni2P phase at the outset of the reaction, and then be transformed into derivatives [46]. In particular, the outset reactions needed active H species to cleave Caromatic-OCH3 bonds since the PO-H groups on Ni2P phase allowed spillover of the H species to the chemisorbed guaiacol on the Ni sites of Ni2P [31], and then further transalkylation occurred on Brønsted acid sites [47].
Cresol selectivity. In particular, the selectivity of cresol first increased and then decreased as the temperature increased. In addition, the selectivity of phenol and BTX increased as the temperature rose. These results can be explained by the fact that the initial increase of cresol is due to the demethylation of DMP, and subsequent decrease to further demethylation and/or dehydroxylation to form phenol and aromatic hydrocarbons. High temperature favored C–C and C–O bond cleavage between the aromatic ring and its branched chains. The highest selectivity of cresol (16%) was obtained at 400 °C and 3 MPa.
MMMEB selectivity. At 400 °C and under 0.5 MPa, it was observed that the selectivity of MMMEB (3%) and DMP (14%) was higher than that obtained under 3 MPa, while the selectivity of cresol (8%), phenol (5%) and BTX (2%) decreased significantly. It is therefore clear that high pressure favored the further reaction of MMMEB and DMP to form cresol, phenol and BTX. Subsequently, cresol and phenol underwent dehydroxylation and/or demethylation to produce BTX. Other phenols identified in the liquid phase, such as 3-methyl-4-isopropylphenol and 2-ethyl-6-methylphenol, are probably produced via the isomerization of reaction intermediates. Notably, the selectivity of 2-ethyl-6-methylphenol first decreased due to a further dealkylation and then increased due to coke formation. Probably, the coke affected the pore size of the catalysts, which suppressed further dealkylation.
3.3.1.3. Selectivity in the gas phase
Gas products selectivity. Figure 5(B) shows the evolution of the selectivity of gas products for 4-ethylguaiacol HDO over the 5% Ni2P/HZSM-5 catalyst at different temperatures and pressures. As can be seen, the selectivity of gas products increased with the increase of temperature. The main gas product was C2H6 at 300 °C and 400 °C, followed by C3H8 or CH4. At 500 °C, CH4 and C2H6 were the major products in gas phase. C2H6 can be produced via deethylation of 4-ethylguaiacol followed by hydrogenation. CH4 can be formed via various reaction routes, mainly via methoxy removal from 4-ethylguaiacol followed by further C–O bond to give some CO and, to a lesser extent, via direct demethylation of cresol intermediate.
The increase in selectivity of CH4 and C2H6 revealed that high temperature promotes C–O bond cracking of the methoxy group and multiple C–C hydrogenolysis of 4-ethylguaiacol and other intermediates. Chen et al. [48] found that methane was yielded as a main gas product in the hydrogenation of m-cresol via C–C bond cleavage on silica supported Ni catalyst. The highest selectivity of C2H6 (11%) and CH4 (15%) was obtained at 500 °C. Under 3 MPa, a significant increase in the selectivity of total gas products was observed and particularly C2H6 and C3H8. This behavior proves that higher pressure improved the HDO of 4-ethylguaiacol to form gas products and especially favored C2H6 and C3H8. This last was possibly formed by an addition reaction between CH4 and C2H4.
The proposed HDO pathways of 4-ethylguaiacol
| Main products | Main reaction pathways | |
|---|---|---|
| 4-Ethylphenol & 2-methoxyphenol | 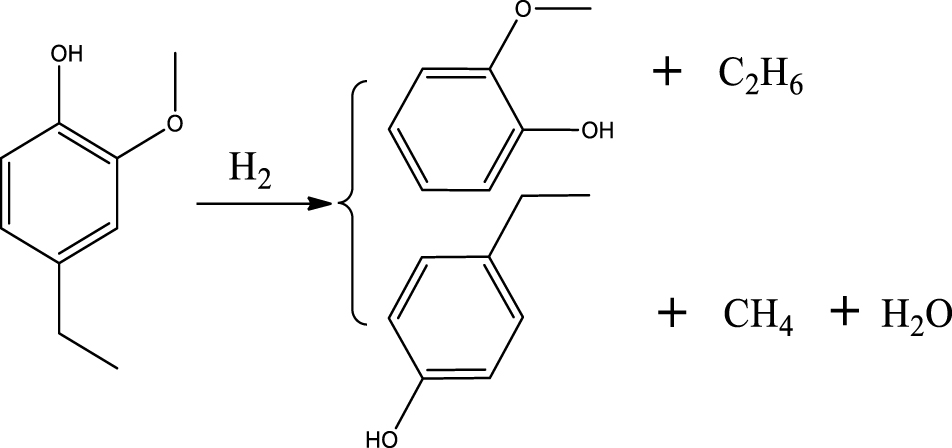 | (1) |
| DMP | 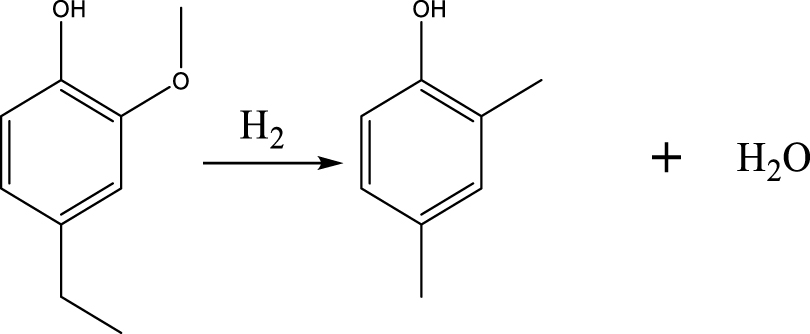 | (2) |
| MMMEB | 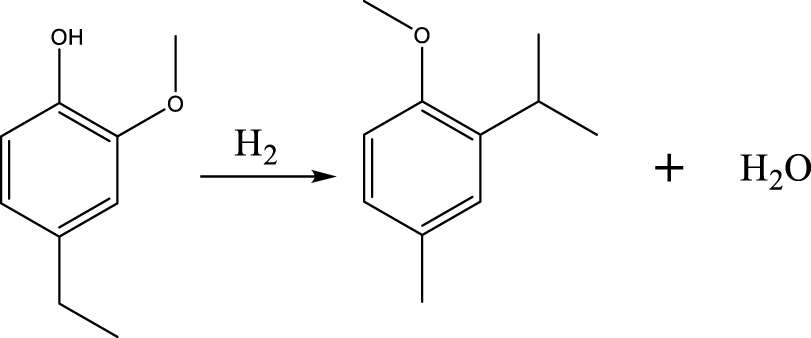 | (3) |
| Cresol (o- & p-) & Phenol | 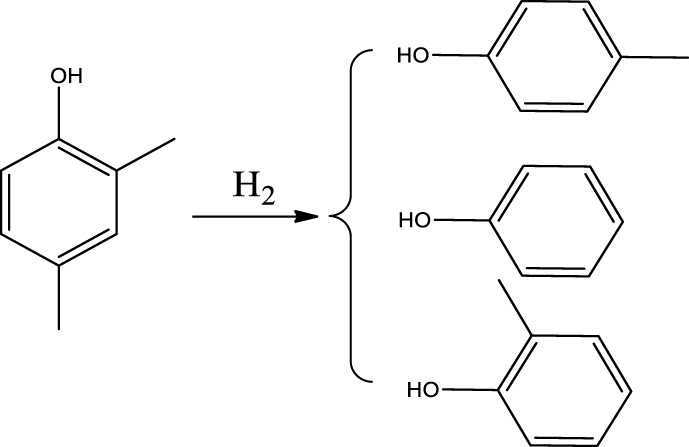 | (4) |
| Phenol | 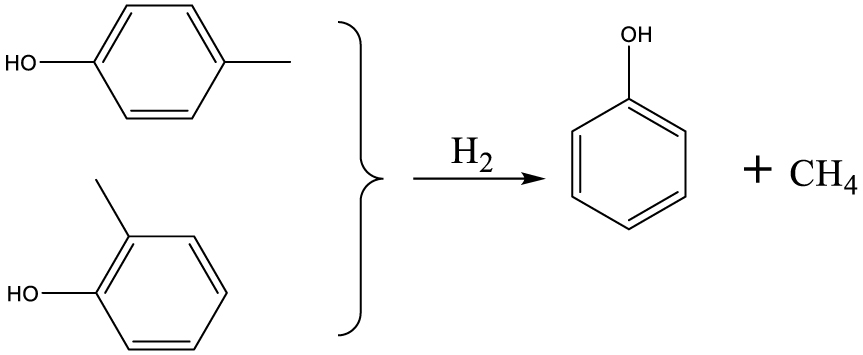 | (5) |
| Benzene | 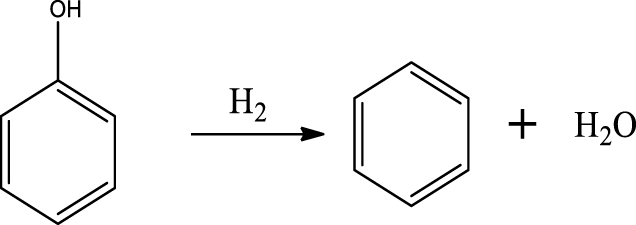 | (6) |
| Toluene & Xylene (p- & m-) | 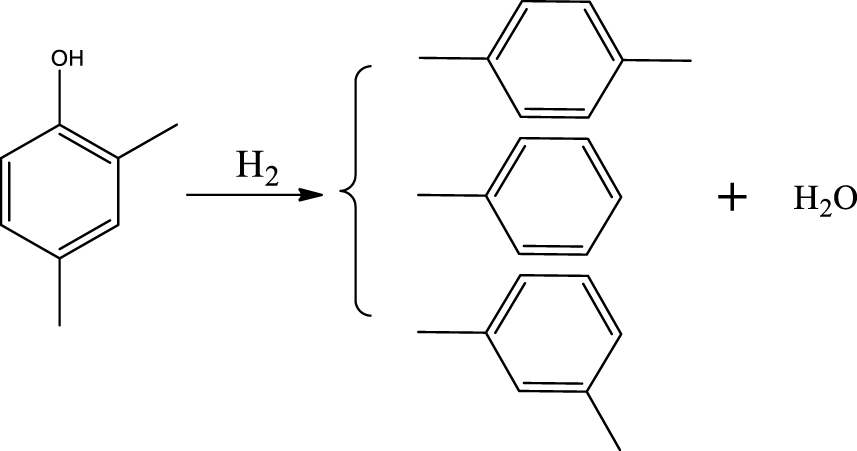 | (7) |
3.3.2. Proposed reaction pathways of 4-ethylguaiacol HDO
Based on the products formed, and the discussion in Section 3.2.1, a set of reaction pathways for the HDO of 4-ethylguaiacol is proposed and reported in Table 4. Reaction (1) corresponds to the dealkylation of 4-ethylguaiacol to 2-methoxyphenol and demethoxylation to 4-ethylphenol on Lewis acidic Ni sites, with the formation of C2H4, CH4 and H2O. The C2H4 was then hydrogenated to C2H6. Reaction (2) is a direct demethoxylation and alkylation reaction of 4-ethylguaiacol forming DMP below 400 °C. This is a key reaction because DMP is the most primary product at 300 °C. Reaction (3) is a dihydroxylation and alkylation reaction of 4-ethylguaiacol to form MMMEB at 300 °C and 400 °C. Instead of the alkylation reaction favored by relatively low temperatures, the dealkylation reaction, namely, the C–C bond breakage between benzene ring and its side alkyl group, was promoted by high temperatures. The DMP underwent subsequent demethylation to form cresol (p- & o-) and phenol in reaction (4). Reaction (5) is a dealkylation reaction of cresol to produce phenol and CH4. Similarly, phenol and CH4 were also identified as important products of guaiacol HDO [20, 49]. Aromatic hydrocarbons can be formed by the further dehydration reaction of alkylphenols, as shown in reactions (6) and (7). Other alkylphenols, (3-methyl-4-isopropylphenol, for example), were probably produced via the isomerization of certain intermediates.
3.4. HDO of furfural
3.4.1. Effect of temperature and pressure
3.4.1.1. Conversion rate, DOD, and yield
Table 5 presents the effect of temperature and pressure on the conversion rate, DOD, and yield of products for furfural HDO. Results under 3 MPa pressure exhibited a significant increase in conversion rate (from 54% to 100%) and DOD (from 39% to 92%) when the temperature increased from 300 °C to 500 °C. The highest yields of gas products (51%) and H2O (10%) were obtained at 500 °C and 3 MPa and were accompanied by the lowest yield of liquid products (39%, H2O free). At 400 °C and 3 MPa, the results showed a slight increase of the conversion rate and a decrease of liquid products and H2O compared to the results under 0.5 MPa. These results indicated that high pressure promoted the C–C hydrogenolysis between the furan ring and its branched chains and reduced the direct dehydration reaction to some extent.
Effect of temperature and pressure on furfural HDO
| Item | Conditionsa | |||
|---|---|---|---|---|
| 3 MPa | 3 MPa | 3 MPa | 0.5 MPa | |
| 300 °C | 400 °C | 500 °C | 400 °C | |
| Conversion rate & DOD (wt%) | ||||
| XFurfural | 54 | 66 | 100 | 57 |
| DOD | 39 | 51 | 92 | 35 |
| Yield of products (wt%) | ||||
| YLiq. (free water) | 75 | 64 | 39 | 65 |
| YGas | 19 | 28 | 51 | 26 |
| YH 2O | 6 | 8 | 10 | 9 |
a Fixed conditions: 0.43 g 5% Ni2P/HZSM-5 catalyst, 0.05 mL/min of furfural, H2: 40 mL/min, N2: 10 mL/min, 90 min.
3.4.1.2. Selectivity in the liquid phase
The selectivity of major products of furfural HDO is illustrated in Figure 6(A). The detailed chemical composition of products can be found in the Supplementary (Table S4). Generally, high selectivities of furan, 2-methylfuran (2-MF), butanal, BTXM, and 1-methylindan were observed in these conditions.
3.4.1.3. Furan selectivity
Furan was observed as the most primary product of furfural HDO, with the selectivity of 17% (300 °C and 3 MPa), 22% (400 °C and 3 MPa) and 17% (400 °C and 0.5 MPa). It can be deduced that direct aldehyde removal was the main reaction of furfural HDO in these conditions, and this removal was promoted by the pressure increase. Furthermore, deoxygenation of the aldehyde group of furfural under 3 MPa was also a significant reaction, since high selectivities of 2-MF (17% and 10%) were achieved at 300 °C and 400 °C. Accordingly, these results proved that the high pressure favored the aldehyde removal and deoxygenation of furfural but less furan ring-opening on the acid sites of 5% Ni2P/HZSM-5 catalyst. Similarly, Cai et al. [50] reported that HDO reactions of furfural mainly happened on its aldehyde group, but little on furan ring using Cu/SiO2 catalyst. At 500 °C, a very low selectivity of furan was obtained demonstrating that temperature promoted the cracking of furan.
3.4.1.4. BTXM and 1-methylindan selectivities
A greater amount of BTXM was obtained at higher temperatures of 400 °C and 500 °C than at 300 °C. In this case, the formation of aromatic hydrocarbons can be attributed to the oligomerization of mono-olefins (C2H4, C3H6 and C4H8) and/or dienes followed by cyclization at high temperatures. Under 3 MPa, the selectivity of BTXM (6%) at 500 °C decreased slightly compared to the value of 10% at 400 °C, probably due to the further oligomerization reaction of BTXM and olefins to 1-methylindan with a high selectivity of 1-methylindan (21%). The various olefins can be produced by the ring-opening reaction of furan, 2-methylfuran and other intermediate reactions. In the literature, Cai et al. also mentioned the role of oligomerization and cyclization of olefins to produce aromatic hydrocarbons [50].
3.4.1.5. 2-vinylfuran selectivity
A small amount of 2-vinylfuran was detected, which might be linked to a route of direct substitution reaction between furan and C2H4. Although, 2-vinylfuran was observed as a secondary product in the case of hydrotreatment of the furfural–acetone condensation reaction by Ulfa et al. [51] and the catalytic reduction of furfural–methanol by Grazia et al. [52], a few studies reported that 2-vinylfuran was a product of furfural hydrotreatment without other co-reagents. The 2-vinylfuran can potentially be obtained from the methylenation reaction of furfural involving several consolidating approaches. The most frequently used methods for furfural conversion reaction are the Wittig [53] and Horner–Wadsworth–Emmons reactions [54], which have the common feature of employing the co-reagent of phosphorus-substituted carbanions. One can then suggest that the active Ni2P phase of the Ni2P/HZSM-5 catalyst favored the olefin substitution reaction of furan via the interaction between Ni2P and ethylene.
The influence of temperature and pressure on furfural HDO (0.43 g 5% Ni2P/HZSM-5, 0.05 mL/min furfural, 40 mL/min H2, 10 mL/min N2, reaction time: 90 min): (A) Selectivity of major liquid products; (B) Selectivity of gas products.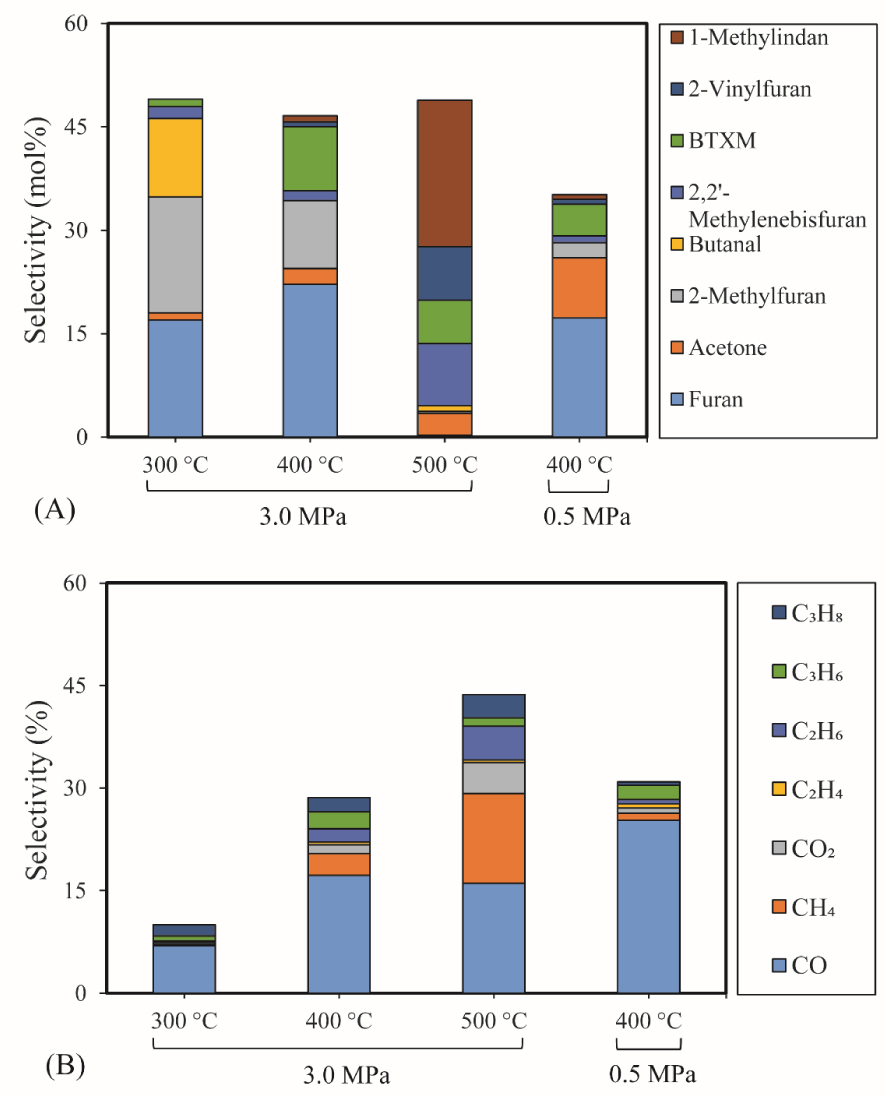
The proposed HDO pathways of furfural
| Main products | Main reaction pathways | |
|---|---|---|
| 2-Methylfuran | 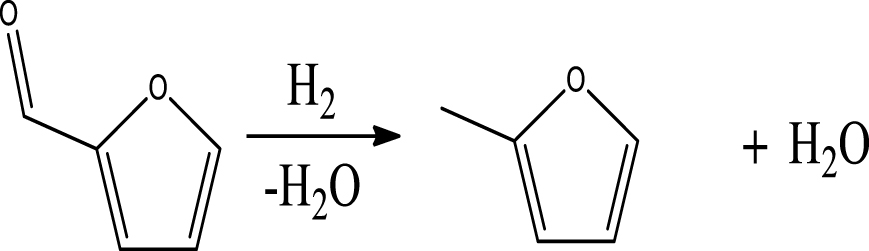 | (1) |
| Furan & CO | 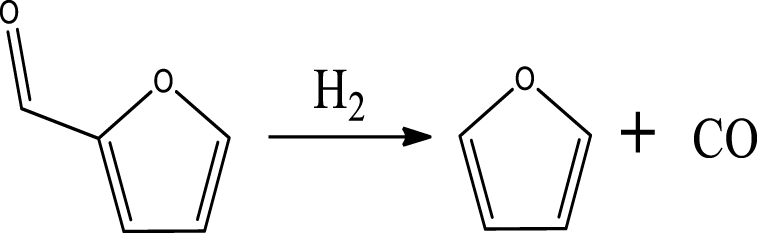 | (2) |
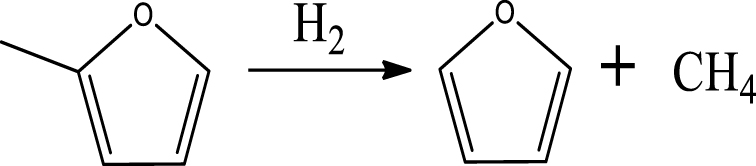 | (3) | |
| Acetone | 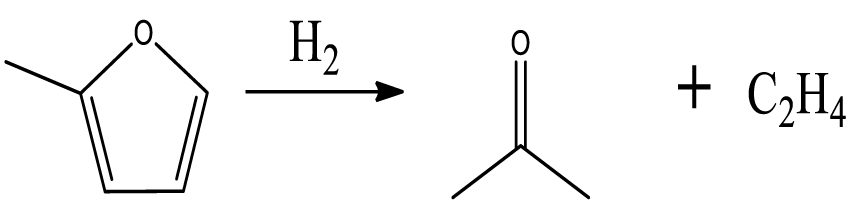 | (4) |
| Butanal & C4H8 | 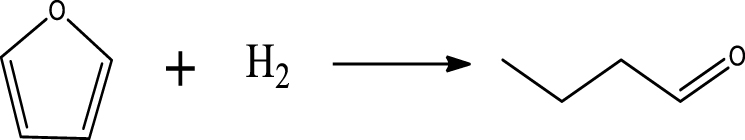 | (5) |
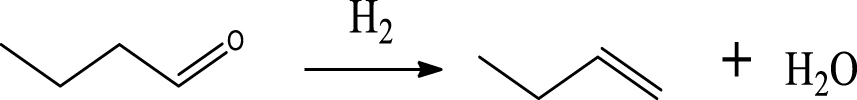 | (6) | |
| C3H6 & C3H8 | 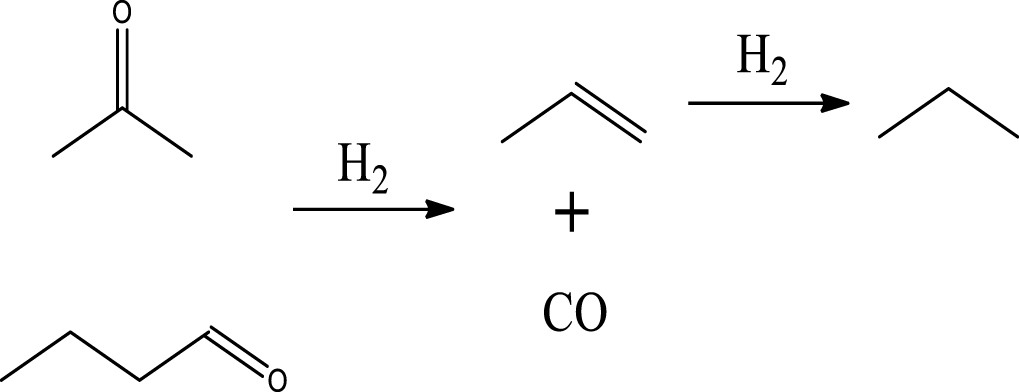 | (7) |
| 2-Vinylfuran | 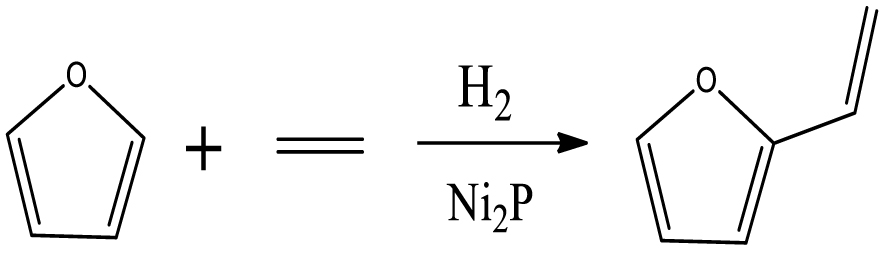 | (8) |
| 2,2-Methylenebisfuran | 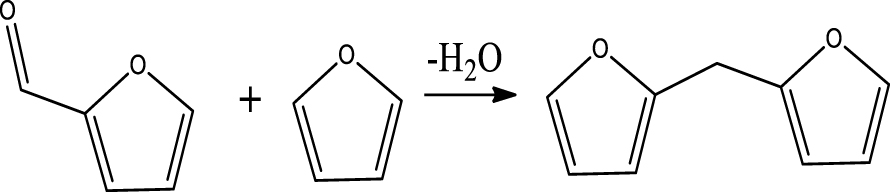 | (9) |
| Aromatic hydrocarbons | 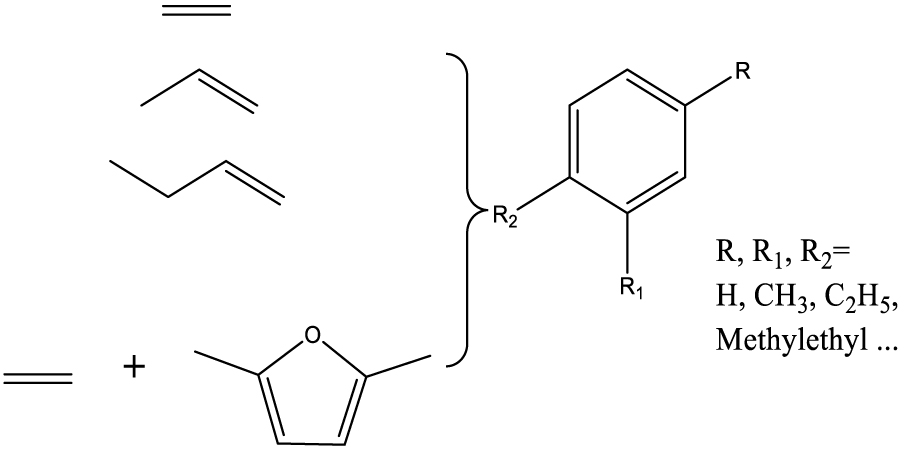 | (10) |
3.4.1.6. Other compounds
At low temperature, the formation of butanal can be attributed to the ring-opening reaction and further hydrogenation of furan. The acetone was possibly produced via the ring-opening reaction of 2-MF followed by the further hydrogenolysis reaction of C–C bond together with the release of C2H4.
3.4.1.7. Selectivity in the gas phase
Figure 6(B) shows the analysis of various gas products during the furfural HDO reaction. The results showed a very low gas production at 300 °C due to the low conversion rate of furfural at low temperatures. The main gas product was CO with a selectivity of 17%. High selectivity of other gases (CH4, CO2, C2H6, C3H6 and C3H8) at 400 °C and 3 MPa was also observed. CO was mainly formed by the direct decarbonylation of furfural and aldehyde intermediates (like butanal). CO2 was possibly produced by the 2-MF ring-opening reaction followed by C–C bond cracking at high temperatures.
Among these gaseous products, light alkanes were formed by the hydrogenation reaction of intermediates of furfural HDO since the ring-opening reaction produced olefins. Notably, the selectivity of CH4 (13%) at 500 °C is significantly higher than at 400 °C, but the selectivity of CO was lower compared to 400 °C. CH4 can be formed mainly by the demethylation of 2-MF and probably by hydrogenation of CO and CO2, as reported in the literature in the case of metal Ni under high pressure of H2 [55]. These results suggested that the selectivity of alkanes depends on the relative rates of the C–C bond cleavage, dehydration and hydrogenation reactions of furfural and its intermediates. Similarly, Davda et al. also stated that the selectivity of various alkanes is strongly linked to the relative rates of the C–C bond cleavage, dehydration and hydrogenation reactions during the aqueous-phase processing of sorbitol [55]. At 400 °C, the gas product selectivity under 0.5 MPa showed a relatively narrow distribution compared to 3 MPa, and CO was the most important product, with 25% of selectivity. Thus, it can be deduced that pressure had a significant effect on the distribution of gas products of furfural HDO and that high pressure facilitated the ring-opening reactions.
3.4.2. Proposed reaction pathways of furfural HDO
Table 6 summarizes a series of reaction pathways for the HDO of furfural according to the main products obtained. Reaction (1) corresponds to the hydro-dehydration of furfural to 2-MF, together with the formation of H2O. Reaction (2) is a direct decarbonylation reaction of furfural forming furan and CO [56]. This reaction is the primary route of furan formation due to the highest selectivity of CO among other gas products (Figure 6B). Reaction (3) can be considered a minor reaction for furan production, associated with the formation of CH4. Acetone was produced by the ring-opening reaction of 2-methylfuran accompanying the release of C2H4 in reaction (4). C3H6 and C3H8 can be produced mainly from the dehydration–hydrogenation of acetone in reaction (7), and C2H4 can be easily converted to C2H6 by hydrogenation.
Furan can be converted by hydro-opening-ring and hydrogenation to form butanal in reaction (5). Subsequently, butanal can be hydrogenated, and then dehydrated forming C4H8 in reaction (6) and/or directly decarbonylated to form CO and C3H6. C3H8 is then formed by hydrogenation of C3H6 in reaction (7). Reaction (8) is the probable pathway allowing the formation of 2-vinylfuran from furan and C2H4. Reaction (9) is a condensation reaction between furfural and furan molecules to synthesize the 2,2-methylenebisfuran. Reaction (10) is a probable route of aromatic hydrocarbons via the addition reaction of olefins on Ni2P and subsequent cyclization on HZSM-5 acidic sites. As stated in the literature, aromatic hydrocarbons can be formed by cyclization–dehydration of dienes, which comes from the oligomerization of mono-olefins (C2H4, C3H6 and C4H8) [50] or the Diels–Alder cycloaddition of 2,5-dimethylfuran with ethylene and subsequent dehydration to form p-xylene, followed by alkylation to other aromatics [57, 58]. Thus, it can be deduced that 1-methylindan was also formed by these routes.
4. Conclusion
The HDO of acetic acid, 4-ethylguaiacol, and furfural as model molecules of bio-oil with the use of prepared Ni2P/HZSM-5 catalysts was studied, which could provide significant guidance for the upgrading of crude bio-oil. The results showed that the reaction temperature has a pronounced effect on the conversion rate and DOD of acetic acid and furfural using 5% Ni2P/HZSM-5 catalyst. However, pressure had a greater effect on the conversion rate and DOD of 4-ethylguaiacol HDO than temperature.
For acetic acid HDO, the temperature and pressure mainly affected the decarboxylation, hydrogenation, and further decarbonylation associated with the release of CO, CH4, and CO2. Aromatic hydrocarbons were obtained via a further aldol condensation between MIK and acetone molecules followed by dehydration and hydrogenation.
The results of 4-ethylguaiacol HDO illustrated that 2,4-dimethylphenol, cresol, and 2-ethyl-6-methylphenol were the most important reaction intermediates. The final products, such as phenol and BTX (benzene, toluene, and xylene), can be produced from those intermediates via dealkylation, dehydroxylation and isomerization. Notably, the coke formation had a slight effect on the conversion rate of 4-ethylguaiacol, but significantly affected the further dealkylation of the 2-ethyl-6-methylphenol intermediate.
Moreover, it was proved that the principal reaction of furfural HDO was the direct decarbonylation with furan and CO formation. Higher temperatures and pressures promoted the ring-opening reaction and C–C hydrogenolysis of furfural HDO. Aromatic hydrocarbons from furfural HDO were probably formed by the addition reaction and subsequent cyclization–dehydration of dienes, which come from the oligomerization of mono-olefins during the ring-opening and C–C bond cleavage process.
Abbreviations
| HDO | Hydro-deoxygenation |
| DMP | 2,4-Dimethylphenol |
| MO | Mesityl oxide |
| MIK | Methyl isobutyl ketone |
| BTX | Benzene, Toluene and Xylene |
| BTXM | Benzene, Toluene, Xylene and Mesitylene |
| 2-MF | 2-Methylfuran |
| DOD | Degree of deoxygenation |
| BET | Brunauer–Emmett–Teller |
| XRD | X-ray Powder Diffraction |
| TPR | Temperature-programmed reduction |
| ICP-OES | Inductively Coupled Plasma-Optical Emission Spectrometry |
| FT-IR | Fourier Transform Infrared Spectroscopy |
| DSC | Differential Scanning Calorimeter |
| X (%) | Conversion rate |
| Seli (%) | The selectivity of product chemicals |
| Yi (%) | The yield of products |
| W (%) | Water content |
Conflicts of interest
There are no conflicts of interests to declare.
Acknowledgments
This work was supported by China Scholarship Council (CSC) and UT-INSA program between China and France. The authors give great thanks to the GenComm Interreg project that offered the opportunity for the internship in this work.
Supplementary data
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.122 or from the author.