


 CC-BY 4.0
CC-BY 4.0
1. Introduction
With the aim of providing energy-efficient and environmentally benign materials, a variety of sustainable energy conversion/storage devices for portable, mobile, and stationary power applications have attracted increasing research interest. Fuel cells are one of the most promising technologies to address future energy crises and environmental issues. Among the diverse fuel cell technologies, the direct liquid fuel cell is the most promising for portable devices. This is due to its easy and safe fuel storage/transport property [1]. Besides, methanol is considered the most electroactive alcohol fuel. Therefore, direct methanol fuel cells (DMFCs) play an important role in addressing the future energy crisis [2, 3]. The DMFC reaction involves the transfer of six electrons and produces CO2, H2O, and electricity. CO is the major reaction intermediate that tends to be adsorbed, blocking active sites of the anode (over the platinum catalyst) where methanol oxidation reactions (MORs) have taken place [3, 4, 5]. Significant effort has been devoted to address this poisoning phenomenon [4, 6, 7]. Pt-based bimetallic catalysts (often ruthenium, Ru) are considered the ideal alloy composition providing catalytic synergy to improve both power density and stability for methanol electro-oxidation [3, 4, 8, 9]. Adsorbed –OH species, generated on Ru at the platinum/ruthenium edge by water dissociation, promote CO oxidation and thus enhance tolerance against poisoning [1, 2, 10, 11]. The performance improvement obtained from multi-metallic catalysts can be attributed to distinct coordination effects, which are believed to accelerate the methanol adsorption/dissociation on the catalyst surface and further reduce the adsorption of carbonaceous intermediates [12]. Consequently, adding the second and even the third metal can adjust the binding energy, for example, the Pt 4f and/or outermost (5d9 6s1) electrons, through spin–orbit hybridization [12]. The result is a significant decrease in the adsorption energy of carbonaceous intermediates and enhancement of their oxidation. It has been reported that alloy catalysts containing tin (Sn) or tin oxide (SnO2) can cleave C–C bonds and increase the catalyst’s ability to resist deactivation [6, 13]. It was also revealed that an increase in the number of OH groups resulting from Sn doping facilitates the oxidation of poisoning CO intermediates adsorbed on Pt sites and significantly promotes the MOR according to the bifunctional mechanism [14]. For Pt–Ru–Sn trimetallic catalysts, the addition of Sn and Ru promoted the dissociation of adsorbed alcohol molecules and activated water molecules, respectively, leading to an enhanced oxidation rate of strongly adsorbed carbonaceous intermediates [15]. Zhou et al. determined that DMFCs with Pt–Sn alloys as anode catalysts achieved better performance than DMFCs with pure Pt because of the ligand effect exerted on Pt by Sn [16].
During the past decades, numerous studies attempted to disperse, confine, and stabilize metallic catalysts over various supporting materials [4, 8, 9, 17, 18, 19]. Instead of carbon nanotubes, graphene, and active carbon, carbon mesoporous materials (CMMs) enable a stable mesoporous structure with uniform porosity, desired pore size, and high specific surface area (SSA; m2/g). Thus CMMs are an ideal type material to host catalyst nanoparticles (NPs). Based on CMM electrodes, enormous improvements have been realized in various fields [20]. In general, mesoporous structures are beneficial for the dispersion of catalysts (i.e. Pt-based NPs). For example, NP catalysts can be clamped into the framework and their size confined to several nanometers. As a result, CMMs are excellent hosts for electrochemical applications [6, 21].
Here, CMK-3 type CMM was adopted as the host to prepare Pt/RuOu/SnOv/CMK-3 composite electrodes for DMFC applications. In this study, the effect of the Sn mass fraction on the catalyst chemical state, MOR mass activity, CO tolerance, and long-term stability was examined and discussed. The MOR activity of the P20R0S30/C3 catalyst was 23% and 118% higher than that of P20R10S0/C3 and P20R0S0/C3, respectively. Regarding the trimetallic catalyst, the MOR activity of P20R10S15/C3 was 60% and 154% higher than that of P20R10S0/C3 and P20R0S0/C3, respectively. Both P20R0S30/C3 and P20R10S15/C3 perform at the same level of CO tolerance compared to P20R10S0/C3. Besides, long-term stability and cyclic durability have long been considered as indexes for practical applications [8, 9, 22], and these have also been examined and compared with a commercial Johnson Matthey Pt/XC-72 catalyst in this report. In short, this study successfully demonstrated potential cost-effective catalyst compositions (i.e., P20R10S15 and P20R10S0) to replace the expensive Pt–Ru alloy catalysts for DMFC applications.
2. Material and methods
2.1. Preparation of porous silica templates
Mesoporous silica SBA-15 was prepared using a previously reported method [4], which was further adopted as the template for preparing mesoporous CMK-3. First, tetraethyl orthosilicate (TEOS; 2.3 g, 399.0%, Aldrich), the silica source, was added to an aqueous solution containing HCl (8.0 g, 37% HCl in 30.0 g H2O) and a triblock copolymer surfactant (1.0 g, P123; Acros) at 313 K. Second, the solution was aged at 373 K for 2 days after stirring for 2 h. Third, the suspension powder was filtered, washed, and dried. Finally, the product was calcined in air at 833 K for 6 h to remove the P123 soft template.
2.2. Preparation of mesoporous carbon CMK-3
The mesoporous CMK-3 adopted in this study was synthesized via a replication/carbonization process using SBA-15 as the hard template, referring to previously reported techniques [23]. First, the calcined SBA-15 was impregnated with a sucrose/H2SO4/H2O solution in a weight ratio of 1:1.25:0.14:5 (SBA-15/ sucrose/H2SO4/H2O). Second, the mixture was carbonized at 373 K (6 h) and 433 K (6 h). The above procedures were repeated prior to subjecting the sample to graphitization by pyrolysis at 1173 K for 1 h in an Ar environment. Finally, the mesoporous CMK-3 was obtained by removing the SBA-15 template by washing with a solution of 15% HF at 303 K.
2.3. Preparation of Pt–RuOu–SnOv/CMK-3 porous catalysts
The mesoporous catalysts, Pt–RuOu–SnOv/CMK-3 (where the subscripts u and v represent oxygen ratios in ruthenium and tin oxides, respectively), were prepared by referring to our previously reported reduction processes [4, 6, 7]. To obtain nominal metal loading, a mixture of metal chlorides including H2PtCl6⋅6H2O (99.9%, Acros), RuCl3 (99.9%, Acros), and SnCl4⋅5H2O (99.9%, Acros), in appropriate amounts depending on the desired catalyst composition, was dissolved in acetone and mixed with CMK-3. A uniform dispersion of the metal chlorides in CMK-3 was obtained by grinding the blend until the acetone evaporated, which yielded a uniform dispersion of the metal chlorides in CMK-3. Finally, the mesoporous catalysts were reduced in an H2 environment in an oven at 200 °C for 6 h. The resulting composites of Pt, RuOu, SnOv, and CMK-3 were designated PwRxSy/C3, where the subscript numbers w, x, and y represent the designated mass fractions of Pt, Ru, and Sn, respectively.
2.4. Physicochemical characterization
X-ray diffraction (XRD) powder patterns for the synthesized electrocatalysts were obtained on a PANalytical X’Pert Pro X-ray diffractometer using a Cu Kα source (𝜆 = 0.1541 nm). The microstructure and surface morphology of specimens were observed using transmission electron microscopy (TEM; JEOL JEM 2100F; accelerating voltage of 200 kV) and scanning electron microscopy (SEM, JSM-7100F). The mass fraction of each composition in mesoporous PwRxSy/C3 catalysts was analyzed by an energy dispersive X-ray spectrometer. The textural properties (i.e., SSA and pore size dispersion (PSD)) of the samples were determined by Brunauer–Emmett–Teller (BET) N2 adsorption–desorption isotherms using a Quantachrome Autosorb-1 analyzer. The samples were degassed at 473 K for ⩾12 h under vacuum followed by N2 adsorption–desorption experiments conducted at 77 K. X-ray photoelectron spectroscopy (XPS) characterization was carried out with a Physical Electronics PHI 5600 multi-technique system using an Al monochromatic X-ray source at a power of 350 W.
2.5. Electrochemical characterization of PwRxSy/ C3 electrocatalysts
Cyclic voltammetry (CV) and chronoamperometry (CA) experiments were carried out to characterize MOR in a three-electrode glass cell with a CHI 6273E galvanostat/potentiostat. The counter electrode and reference electrode were Pt wire and Ag/AgCl, respectively. The glassy carbon thin-film working electrode was prepared by subjecting a mixture of the Pt-loaded electrocatalyst sample (10 mg) in deionized water (5 mL) to ultrasonication for 30 min. A 20 μL aliquot of the catalyst dispersion was drawn and injected into the glassy carbon electrode (diameter ∼5 mm). The electrode was then dried in air at 333 K for 1 h. Finally, a 1% Nafion solution (20 μL, DuPont) was added to cover the electrode. A mixed solution of 1 M CH3OH and 0.5 M H2SO4 was adopted as the electrolyte for the MOR experiments. Prior to each CV and CA measurement, N2 (99.99%) was purged for 30 min to deoxygenate all solutions. Cyclic voltammograms were recorded in 0.5 M H2SO4 between 0.2 and 1.0 V at a scan rate of 10 mV/s.
3. Results and discussion
3.1. Microstructure examination
Figure 1a depicts the low-angle XRD patterns of the as-prepared mesoporous silica SBA-15, CMK-3, P20R0S0/C3, P20R0S30/C3, P20R10S0/C3, and P20R10S15/C3 porous catalysts. Silica SBA-15 exhibits three small-angle peaks, which are characteristic of hexagonal ordered structures [23]. On the other hand, feature peaks at (1 0 0), (1 1 0), and (2 0 0) indicate a highly ordered periodic arrangement of mesoporous symmetry CMK-3, the replica of SBA-15 [24]. In addition, all P20RxSy/C3 catalysts exhibit (1 0 0) diffraction peaks, corresponding to ordered mesoporous CMK-3, indicating that the hexagonally ordered mesostructure was maintained in all the synthesized samples.
(a) Small-angle XRD patterns of SBA-15, CMK-3, and P20RxSy/C3; (b) wide-angle XRD patterns of the selected P20RxSy/C3.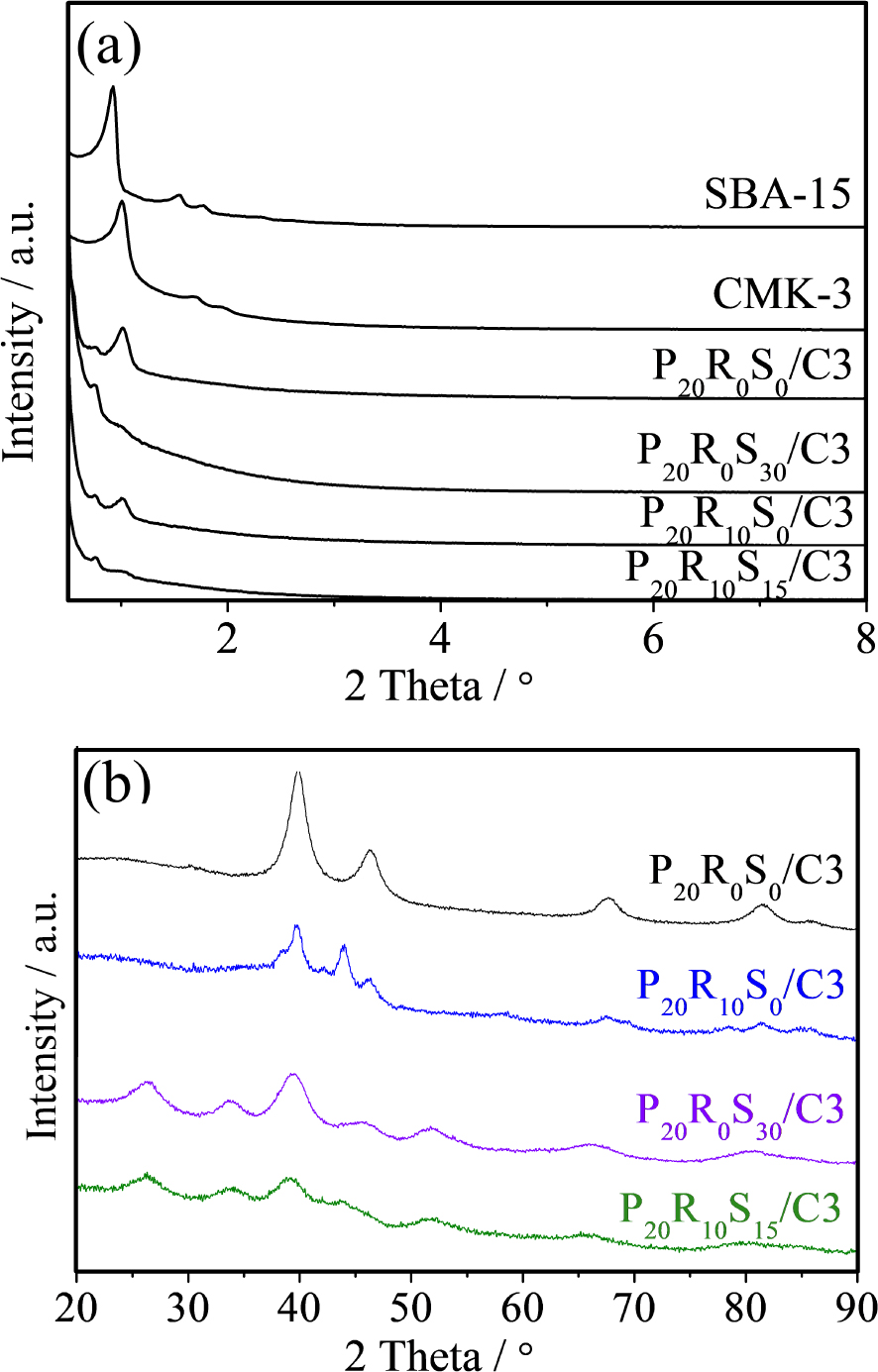
Figure 1b shows the corresponding wide-angle XRD results of the following P20RxSy/C3 samples: P20R0S0/C3, P20R0S30/C3, P20R10S0/C3, and P20R10S15/C3. As can be clearly seen, all the samples exhibit the typical characteristics of a face centered cubic crystalline platinum structure. The samples exhibit diffraction peaks at 2θ = 39.9°, 46.3°, 67.7°, and 81.6°, corresponding to the (111), (200), (220), and (311) diffraction planes, respectively. Compared with the XRD pattern of the P20R0S0/C3 catalyst, the intensities of the (200), (220), and (311) planes of the P20R0Sy/C3 and P20R10Sy/C3 catalysts are indiscernible; the broadened peaks could be attributed to their nanosize. As seen in Figure 1b, as the Sn content increases, the intensities of the Pt20Ru0Sny and Pt20Ru10Sny composite planes become significantly reduced and the peaks in the XRD patterns for the Pt(111) planes are broadened accordingly. Indeed, the Pt crystalline features diminish compared to the diffraction peaks of the P20R0S0/C3 sample. The XRD peaks around 2θ = 25°, 34°, and 52° in Pt20R0S30/C3 and P20R10S15/C3 belong to the (110), (101), and (211) diffraction lines of tetragonal SnO2, respectively [6], indicating the presence of SnO2 in the catalysts. The average crystal size (ACS) (Table 1) in all the P20R0Sy/C3 (y = 0–40) and P20R10Sy (y = 0–20) catalysts was calculated by Scherrer’s equation tabulated in our previous report [25]. It is believed that the intrinsic stability of the SnO2 crystal helps to prevent Pt-based composites from agglomeration during sample preparation. Therefore, it is reasonable that the ACS decreased as the Sn content increased.
Designation of PwRxSy/C3 samples and corresponding If, Ib, and If∕Ib values
| Catalyst designationa | Designated metal amount/wt%b | SSAc (m2⋅g−1) | ACSd (nm) | Methanol | ||||
|---|---|---|---|---|---|---|---|---|
| Pt | Ru | Sn | If (mA⋅g−1) | Ib (mA⋅g−1) | If∕Ib | |||
| CMK-3 | N/A | N/A | N/A | 1150 | – | – | – | – |
| P20R0S0/C3 | 20 | N/A | N/A | 860 | 8 | 0.022 | 0.014 | 1.6 |
| P20R0S10/C3 | 20 | N/A | 10 | 683 | 7 | 0.035 | 0.015 | 2.3 |
| P20R0S15/C3 | 20 | N/A | 15 | 556 | 4 | 0.035 | 0.014 | 2.5 |
| P20R0S20/C3 | 20 | N/A | 20 | 500 | 3 | 0.037 | 0.014 | 2.6 |
| P20R0S30/C3 | 20 | N/A | 30 | 405 | – | 0.048 | 0.019 | 2.5 |
| P20R0S40/C3 | 20 | N/A | 40 | – | – | 0.039 | 0.019 | 2.1 |
| P20R10S0/C3 | 20 | 10 | N/A | 635 | 6 | 0.039 | 0.014 | 2.8 |
| P20R10S10/C3 | 20 | 10 | 10 | – | 5 | 0.044 | 0.023 | 1.9 |
| P20R10S15/C3 | 20 | 10 | 15 | 456 | 3 | 0.056 | 0.021 | 2.7 |
| P20R10S20/C3 | 20 | 10 | 20 | – | 3 | 0.054 | 0.031 | 1.7 |
a C3 stands for the mesoporous carbon supporter, CMK-3. b The deviation values of the exact metal amount for Pt, Ru, and Sn are 1.7, 1.4, and 1.4 wt%, respectively. c SSA: specific surface area. d ACS: average crystal size.
Figures 2a and 2c illustrate the N2 adsorption–desorption isotherms and the corresponding pore size distribution curves (Figures 2b and 2d) calculated from the isotherm adsorption branch by the Barrett–Joyner–Halenda (BJH) method for the P20R0Sy/C3 and P20R10Sy/C3 catalysts. The N2 adsorption–desorption isotherm of CMK-3 is close to type IV with an H1-type hysteresis loop, but the capillary condensation step is well defined. The ordered mesoporous carbon material CMK-3 has a much larger surface area than the Pt-loaded samples, where the decrease in surface area is mainly attributed to the average density increment caused by the heavy metals (i.e., Pt, Ru, and Sn). As shown in Table 1, the SSAs declined generally according to the percentage of metal. For example, P20R0S0/C3 contains 80% of CMK-3 and thus the SSAP20R0S0/C3 is approximately 80% of SSACMK-3. Figures 2b and 2d also show that the PSDs of all the catalysts are essentially identical. Overall, these results demonstrate clearly that our process maintains the CMK-3 mesostructure even at high Pt/Ru/Sn loadings.
77 K nitrogen adsorption/desorption isotherms for the catalysts: (a) CMK-3 and P20R0Sy/C3, and (c) P20RxSy/C3; (b) and (d) are the corresponding pore size distributions, respectively.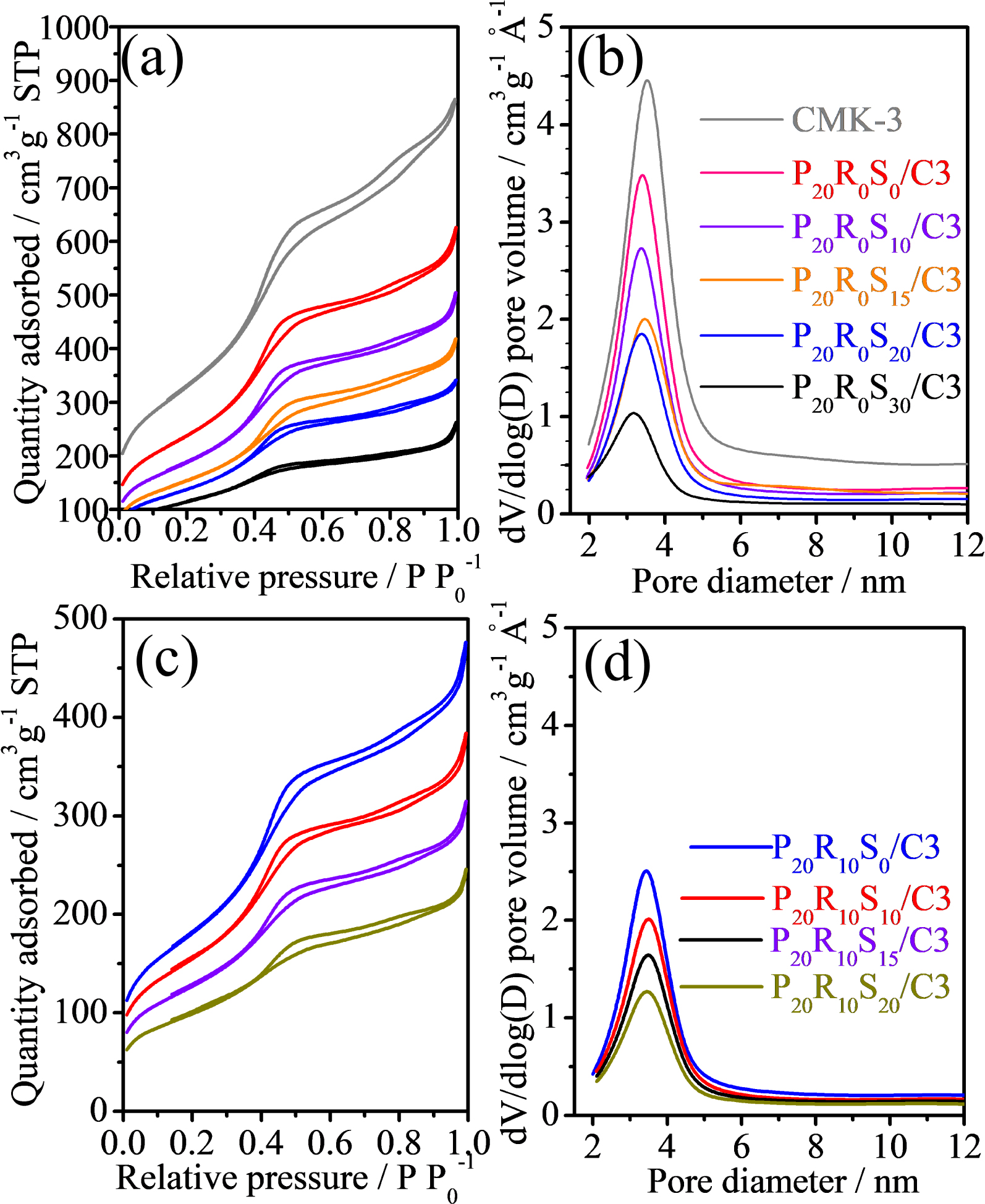
Figure 3 shows typical high-resolution TEM (HRTEM) images of the CMK-3, P20R0S30/C3, and P20R10S15/C3. TEM images confirm that CMK-3 is a highly ordered mesostructure that exhibits an inverse replica structure of the SBA-15 template (Figure 3(a) side view and (inset) cross-section view). The images also clearly show that Pt–SnO2 NPs are well dispersed on CMK-3 with discernible internal pore structures (Figure 3(b)). Further, after the dispersion of Pt–RuOu–SnO2 NPs on the carbon material support, all the NPs are uniformly distributed inside the pores of CMK-3 without noticeable aggregation (Figure 3(c)). Based on these observations, it can be determined that the average particle size of ∼3–5 nm of the metal alloy NPs is consistent with that calculated from Scherrer’s equation (Table 1). The low- and high-magnification SEM images of the Pt-loaded CMK-3 catalysts are shown in Figure 4. All the samples exhibit a similar configuration and pore size with CMK-3, indicating that the morphology was maintained during the carbonization step and the subsequent removal of the silica SBA-15 template without any significant modification or alteration, even at high metallic loadings. Nanoparticle dispersion can be clearly observed from the high-magnification image of the P20R10S15/C3 sample (Figure 3c). The good dispersion of the metal NPs is conducive to avoiding particle agglomeration and maintaining a high active surface area of the composite [26]. The presence of metal elements on the porous CMK-3 framework was revealed by EDS elemental mapping (Figure 5), which indicates the desired amount of metals. Besides, they also confirmed that metallic components such as Pt, Sn, and Ru were dispersed uniformly on the mesoporous carbon CMK-3 for the P20R0Sy/C3 and P20RxSy/C3 catalysts.
TEM images of the (a) CMK-3, (b) P20R0S30/C3, and (c) P20R10S15/C3 catalysts.
SEM images of (a, b) P20R0S0/C3, (c, d) P20R0S30/C3, (e, f) P20R10S0/C3, and (g, h) P20R10S15/C3.
EDS mapping images of (a) P20R0S0/C3, (b) P20R0S30/C3, (c) P20R10S0/C3, and (d) P20R10S15/C3.
Thermogravimetric weight loss analyses of the P20R0Sy/C3 and P20RxSy/C3 catalysts recorded by TGA experiments under an air atmosphere are shown in Supplementary Figures S1a and S1b, respectively. The maximum weight loss of pristine CMK-3 occurred at approximately 600 °C, whereas the maximum weight loss of the P20R0Sy/C3 and P20RxSy/C3 catalysts occurred between 350 °C and 450 °C. These results suggest that the Pt–Ru–SnO2 NPs are active and can potentially accelerate the oxidation reaction. As shown in the insets of Supplementary Figures S1a and S1b, the residue after the decomposition of carbon (CMK-3) was found to be in the range of 19–48 wt% for P20R0Sy/C3 and 28–51 wt% for P20R10Sy/C3 as the Sn content increased, respectively. The residual weight percentage approximately represents the total weight of Pt, Ru, and Sn, which is consistent with the EDS mapping results.
3.2. Effect of Sn ratio on catalyst chemical states
XPS was used to examine the chemical states of the elements, their contents, and valence states. Figure 6 shows the XPS Pt 4f peaks of the Pt20/C3 catalyst. Deconvolution of the Pt 4f XPS doublet (4f7/2 and 4f5/2) gives rise to two pairs of peaks, corresponding to dominant Pt0 atoms and a smaller number of surface Pt(II) ions, respectively. The principal peaks with lower binding energies at 71.4 eV (4f7/2) and 75.1 eV (4f5/2) can be attributed to zero-valence Pt (Pt0), and the deconvoluted peaks at 72.2 and 76.0 eV and at 74.0 and 77.5 eV can be assigned to Pt in the 2+ and 4+ oxidation states, respectively [27]. The Pt peak found in its zero-valence state and that found in its ionic form are most commonly assigned as Pt(OH)2 and PtO2 and/or Pt–O–Sn phases [28, 29, 30]. Figure 7 shows the XPS Pt 4f peaks of the P20R0Sy/C3 (Sn wt% = 0, 10, 20, 30) and P20R10Sy/C3 (Sn wt% = 0, 10, 15, 20) catalysts. It is clear that the overall Pt peak shifts slightly toward lower binding energy following the inclusion of Sn NPs, that is, as the “y” values increase in P20R0Sy/C3 and P20R10Sy/C3. This shift also suggests that alloying Pt with Sn can alter the electronic structure of Pt due to lattice strain and charge transfer [31]. As seen in Figure 7, increasing the Sn content tends to reduce Pt from an oxidized state to a metallic state in both P20R0Sy/C3 and P20R10Sy/C3. This result is consistent with Zhou’s suggestion, by which the interaction between Pt and the SnO2 NPs may prevent Pt surface oxidation, which leads to Pt–O bonds. Moreover, the result suggests that Pt can be more efficiently utilized by increasing the Sn content in both P20R0Sy/C3 and P20R10Sy/C3. In short, the XPS results suggest higher catalytic activity may be achieved with a higher Sn content in the catalyst.
Pt 4f peak deconvolution of P20R0S0/C3.
XPS Pt 4f peaks of (a) Pt20R0Sny/C3 and (b) P20R10Sy/C3 catalysts.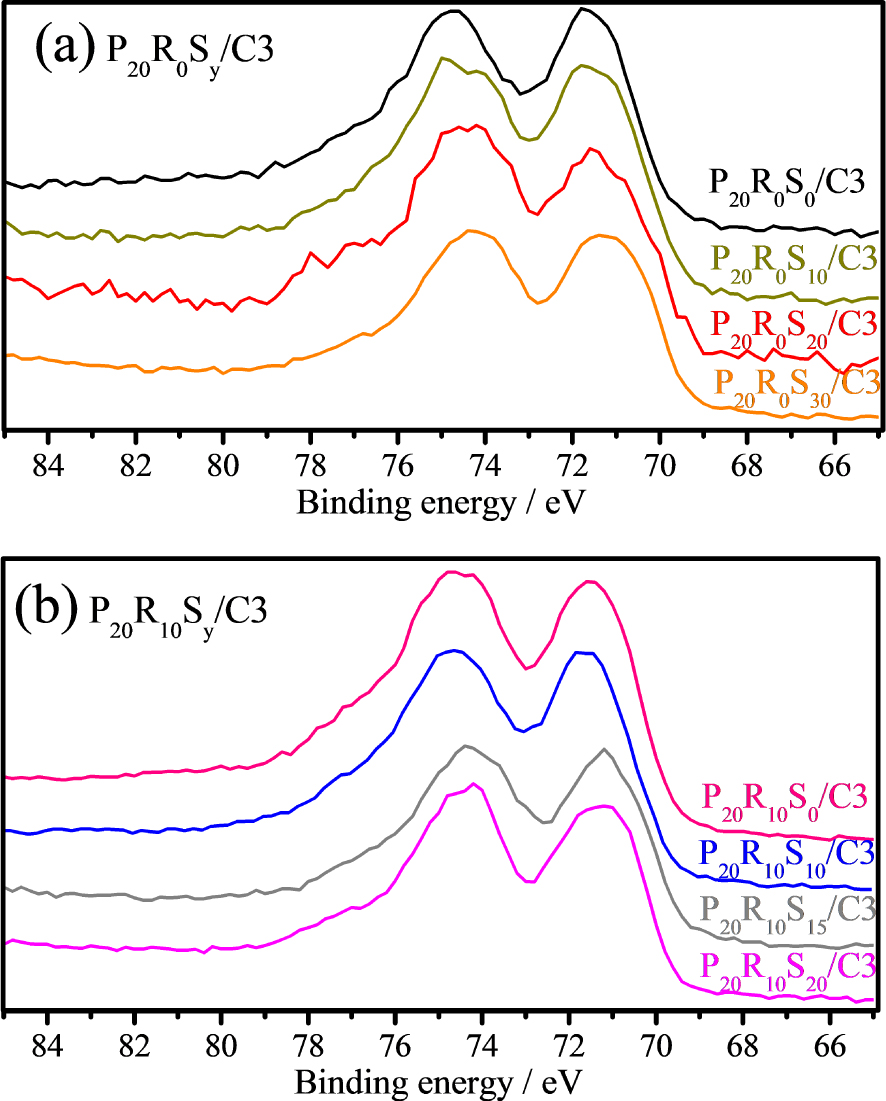
The chemical states of Sn in the catalyst were also examined by XPS, as shown in Figures 8 and 9. Figure 8 shows the XPS Sn 3d peak deconvolution peaks of the P20R0Sy/C3 (Sn wt% = 10, 15, 20, 30) catalysts. The Sn 3d spectra clearly exhibit intense doublets attributable to 3d3/2 (494.7 eV) and 3d5/2 (486.4 eV), indicating the presence of Sn4+ in SnO2 [32, 33, 34]. It is clear that the metal state of tin (Sn0) only appears in P20R0S10/C3. A further increase in the Sn content would lead to a greater amount of SnO2, which is consistent with our expectation on account of previously reported XRD patterns [6]. Further, the XPS studies on the P20R10S10/C3 catalysts also agree with this finding.
XPS Sn 3d peak deconvolutions of (a) P20R0S10/C3, (b) P20R0S15/C3, (c) P20R0S20/C3, and (d) Pt20R0S30/C3.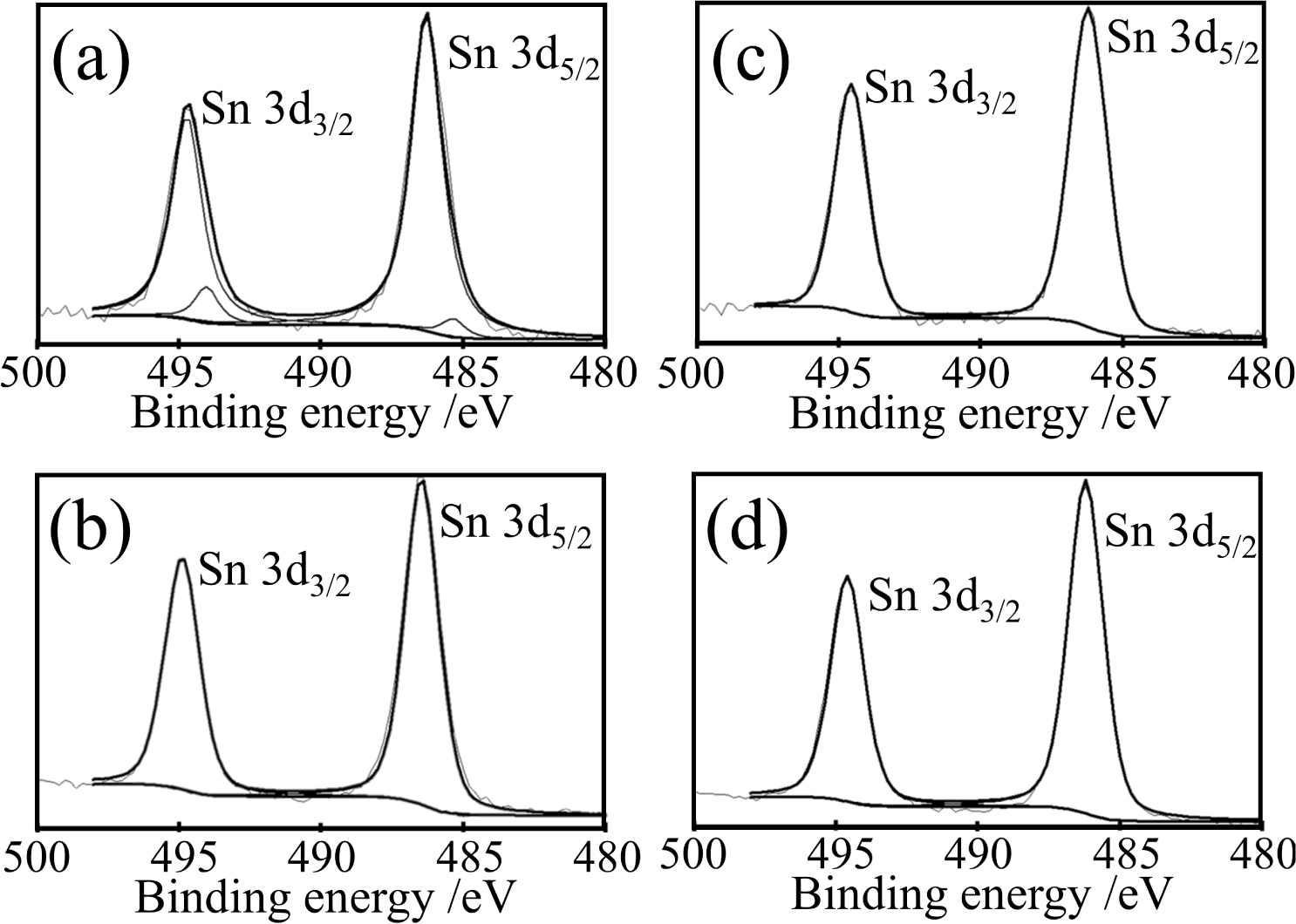
XPS peaks of (a) P20R10S10/C3, (b) P20R10S15/C3, and (c) P20R10S20/C3.
Figure 9 shows the XPS wide spectra with binding energies between 480 and 500 eV, where the Ru 3p1/2 spectrum overlaps with Sn 3d3/2 and Sn 3d5/2 spectra. Since the binding energies of Sn0, Ru0, RuO2, and RuO2⋅zH2O (z represents the molar ratio of hydrate) overlap within this range, it is exceedingly challenging to distinguish them accurately. For an easy comparison, however, the pair of sharp peaks at 486 and 495 eV can be attributed to SnO2, and a pair of broad peaks is designated to represent the mixture of Sn0, Ru0, RuO2, and RuO2⋅zH2O, as shown in Figure 9a. Except for the major SnO2 phase, mixed phases are also present in the P20R10S10/C3 catalyst. Additionally, the aforementioned mixed phases decrease as the Sn content increases from 10 to 20, as shown in Figures 9a–c. In other words, the decreasing tendency for the presence of Sn0 in P20R10Sy/C3 coincides with that in P20R0Sy/C3.
The effect of the Sn content may also influence the chemical state of Ru. To this end, Ru 3p3/2 XPS analyses were also performed for the P20R10Sy/C3 catalysts (Figure 10). It is clear that RuO2 is the major phase in P20R10S0/C3, and Ru0 and RuO2⋅zH2O phases are equivalent. The addition of 10 wt% Sn decreases both the RuO2⋅zH2O and RuO2 phases while Ru0 becomes the major phase. This is reasonable given that the standard reduction potential of Ru is greater than that of Sn. Further, the addition of 15 wt% and 20 wt% Sn did not show obvious effects on the Ru0/RuO2⋅zH2O/RuO2 ratio. Ruthenium oxide and hydrated ruthenium oxide are considered to be the most effective choice for generating OH groups, which lead to best CO tolerance [35, 36]. Therefore, the addition of Sn may negate the positive effect of Ru on CO tolerance (vide infra).
XPS Ru 3p3/2 peaks of P20R10Sy/C3 catalysts.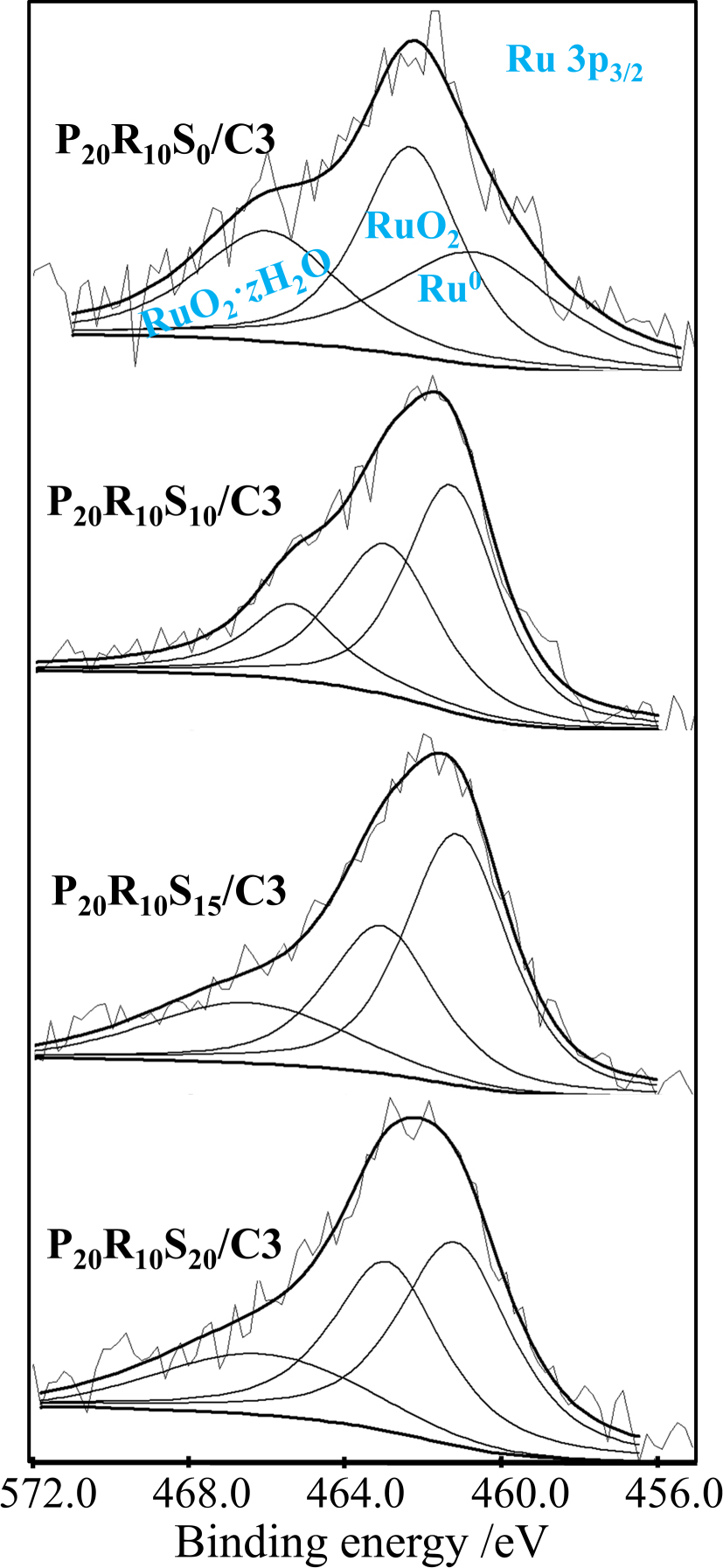
3.3. Effect of Sn ratio on MOR mass activity and CO tolerance
Figure 11a shows the CV curves of the P20R0Sy/C3, and Figure 11b shows the CV curves of the P20RxSy/C3 and commercial 30 wt% Pt/XC-72 samples in an acidic methanol medium (0.5 M H2SO4 and 1 M CH3OH). Here, the effects of the Sn content on the catalytic activities of both P20R0Sy/C3 (y = 0–40 wt%) and P20R10Sy/C3 (y = 0–20 wt%) were investigated for the MOR. All the CV curves exhibit two representative oxidation peaks, denoting the forward oxidation current (If) ∼ 0.6 V and the backward oxidation current (Ib) ∼ 0.4 V. The If arises from the oxidation of methanol on the catalyst NP surface, during which a large number of oxygen-containing species such as CO2 and CO are produced. The Ib value can be attributed to the further oxidation and removal of incompletely oxidized carbonaceous species (e.g., CO) [7]. These current values (If and Ib) and the If∕Ib ratio are significantly influenced by the Sn wt%, as depicted in Table 1. Overall, the If increased following the addition of Sn to both the P20R0Sy/C3 and P20R10Sy/C3 systems. These results suggest that Pt–SnO2 NPs provide more MOR active sites than pure Pt particles. The P20R0S30/C3 and P20R10Sn15/C3 catalysts with 30 wt% and 15 wt% Sn are the optimized conditions for the P20R0Sy/C3 and P20R10Sy/C3 systems, respectively. Increasing the Sn wt% further leads to a slight decline in the If value for both systems. These trends match the variation in the ACS calculated by Scherrer’s equation, suggesting that the MOR activities of P20R0Sy/C3 and P20RxSy/C3 depend greatly on the SSA of the alloy NPs. Besides, the optimized P20R10Sy/C3 demonstrates higher MOR activity than commercial 30 wt% Pt/XC-72, indicating that P20R10Sy/C3 is a competitive electrocatalyst.
On increasing the Sn content, clear increases in the If∕Ib ratio were observed for both types of catalysts. For the P20R0Sy/C3 catalysts, the If∕Ib ratio increased dramatically from 1.6 to 2.3 following the addition of 10 wt% Sn. Increasing the amount of Sn further to 15, 20, and 30 wt% led to only a slight increase in the ratio (to 2.5, 2.6, and 2.5, respectively). Notably, these observations are consistent with a bifunctional mechanism [37]. It has also been reported that the electronegativity difference induced by the Sn content may contribute to weaker absorption of carbonaceous intermediates on the Pt alloy surface [12]. Zhou et al. suggested that SnO2 may generate more OH groups, leading to more effective oxidation of chemisorbed CO at the Pt–SnO2 interface [38]. For the P20RxSy/C3 catalysts, the P20R10S0/C3 catalyst exhibits the largest If∕Ib ratio achieved in our study. The addition of 10 wt% Sn decreased the If∕Ib ratio from 2.8 to 1.9. Further increasing the amount of Sn to 15 wt% (i.e., P20R10S15/C3) increased the If∕Ib ratio to 2.7, which is almost identical to the analogous value for P20R10S0/C3. As a result, both P20R0Sy/C3 and P20R10Sy/C3 catalysts demonstrate outstanding performance for both the MOR activity and CO tolerance. In short, these results suggest that Sn can potentially be used to replace Ru in Pt-based catalysts in order to improve the MOR activity.
Although the If∕Ib ratio clearly increased following the addition of Sn (e.g., P10R0Sy/C3), the Ib values also increased slightly, suggesting that more CO species were adsorbed and that CO tolerance improved. However, it was previously reported that the co-addition of Ru can further augment the performance of Pt alloys [39]. Thus, three-metal Pt–RuO2–SnO2 composite catalysts have also been examined as P20R10Sy/C3 catalysts in this study. The results show an obvious increase in Ib and If∕Ib as the Sn content increases. The If∕Ib ratio of P20R10S15/C3 (∼2.7) is second only to that of P20R10S0/C3 (∼2.8), and it is a great improvement over that of P20R0S0/C3 (∼1.6). In short, the MOR activities of both the two-metal P20R0Sy/C3 and three-metal P20RxSy/C3 alloys, as evidenced by their If values, have been improved by the addition of an optimal amount of Sn. In addition, their CO tolerance, as evidenced by the If∕Ib ratio, is almost identical to that of P20R10S0/C3. As predicted, however, the addition of Sn did eliminate the positive effect of Ru on CO tolerance (vide supra); thus, Pt20Ru10/C3 still shows the highest If∕Ib ratio observed in this study.
3.4. Effect of Sn ratio on long-term stability and cyclic durability
The long-term stability of our proposed mesoporous catalysts Pt20RuxSny and commercial 30 wt% Pt/XC-72 was examined by chronoamperometric measurements at a forward peak potential of 0.7 V for 2000 s in 0.5 M H2SO4 and 1 M CH3OH. Figure 12 shows the specific current density versus time (I–t) curves. The current densities of the P20R0S0/C3, P20R0S30/C3, P20R10/C3, and P20R10S15/C3 nanocatalysts at 2000 s are 1 mA/g, 4 mA/g, 5 mA/g, and 9 mA/g, respectively. The long-term stability of 20R0S30/C3, P20R10/C3, and P20R10S15/C3 is obviously better than that of commercial 30 wt% Pt/XC-72 (Figure 12). Although P20R0S0/C3 achieved steady state immediately after the reaction began, the current densities of the three other catalysts declined slightly to various extents before stabilizing. This initial fast decay can be attributed to the adsorption of MOR intermediates such as COads, CH3OHads, and CHOads during the electrochemical MOR [40, 41]. The current decayed gradually for an extended period of time before the pseudo-steady-state was achieved; this decay can be attributed to adsorbed anions on the catalyst’s surface, which can restrict the MOR [40]. The lower stability can also be partly caused by larger and/or agglomerated metal NPs on the carbon support. Under anodic oxidation conditions in the acidic medium, degradation of catalytic performance generally stems from the tendency of noble metal NP aggregation due to Ostwald ripening [42, 43].
Cyclic voltammogram curves measured for (a) P20R0Sy/C3 and (b) P20RxSy/C3 and Pt/XC-72 using 0.5 M H2SO4/1 M methanol electrolyte solutions.
Chronoamperometric curves of P20R0Sn0/C3 (black), P20R10S0/C3 (red), P20R0S30/C3 (green), P20R10S15/C3 (blue), and Pt/XC-72 (pink).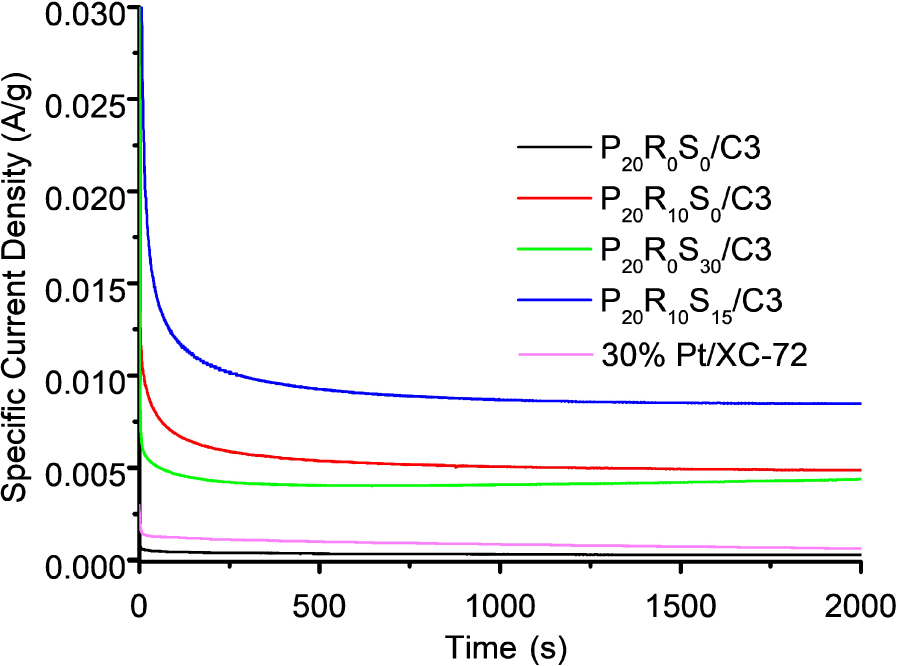
Among all these catalysts, only P20R0S0/C3 shows poor durability. Due to CO poisoning, P20R0S0/C3 demonstrates a steep decline in its current density to 1 mA⋅g−1. The addition of 30 wt% Sn (i.e., P20R0S30/C3) effectively enhances the catalyst’s durability, where the current at 2000 s reaches 4 mA/g. Notably, this density is almost identical to that achieved by the well-known Pt20Ru10 catalyst composition (i.e., P20R10S0/C3; 5 mA⋅g−1 at 2000 s). Although P20R0S30/C3 exhibits 80% performance of the P20R10S0/C3 catalyst, the price of Sn is only a fraction (1/114) of the cost of the precious metal Ru (Ru: 66 USD/ounce; Sn: 0.58 USD/ounce). Given the current price of Pt (980 USD/ounce), our proposed composition could reduce the anode catalyst costs by up to 6%. Clearly, the results suggest that it is economically prudent to replace Ru by Sn in DMFC anode catalysts.
Furthermore, the P20R10S15/C3 catalyst shows the most moderate decline among these catalysts (Figure 8). This result indicates that the P20R10S15/C3 catalysts have the highest durability for the MOR relative to the other catalysts, which may be due to the synergistic effect involving the Pt, Ru, and Sn atoms. Due to the activation of interfacial water preferentially adsorbed at the SnO2 sites, the P20R10S15/C3 catalyst demonstrates superior resistance to CO poisoning and exhibits a higher current density [12]. In addition, an abundance of metal oxides (i.e., SnO2 in this study) can protect Pt NPs from aggregation. Thus, the P20R10S15/C3 catalysts with an optimal Sn content demonstrated the best stability during the MOR.
Finally, the cyclic durability of our proposed electrocatalyst and commercial 30 wt% Pt/XC-72 was also examined, as shown in Supplementary Figure S2, which indicates that the MOR activity improved by 15% after 500 cycles. However, the MOR activity of the commercial 30 wt% Pt/XC-72l reduced to 40% of its original value. It can be suggested that our proposed catalyst composition provides a potential route to improve the cyclic durability of DMFC.
4. Conclusions
The present work has successfully demonstrated the potential of P20S30/C3 and P20R10S15/C3 for application as DMFC anodes. Both the BET and small-angle XRD results indicate that the preparation process applied herein is moderate and can effectively disperse alloy NPs on the CMK-3 framework without modifying its mesostructure. The XPS results show that reduced Pt tends to form in the presence of Sn in both the P20Sy/C3 and P20R10Sy/C3 catalysts. The MOR If-P20R0S30/C3 current of 0.048 mA/g is two-fold higher than If-P20R0S0/C3 (0.022 mA/g) and even higher than If-P20R10S0/C3 (0.039 mA/g). For the P20R10Sy/C3 catalyst, the MOR If-P20R10S15/C3 reached 0.056 mA/g, which is higher than the analogous values for the two-metal catalysts P20R0S30/C3 and P20R10S0/C3. Regarding CO tolerance, P20R10S0/C3 exhibits the highest If∕Ib ratio (2.8) of the catalysts examined in this study, though P20R0S30/C3 (2.5) and P20R10S15/C3 (2.7) also show high If∕Ib ratios. Finally, the long-term stability tests suggest MOR activity in the following order: P20R10S15/C3 > P20R10S0/C3 > P20R0S30/C3 > P20R0S0/C3. In short, this study has demonstrated a potential cost-effective catalyst composition (i.e., P20R10S15 and P20R10S0) to replace the expensive Pt–Ru alloy catalysts for DMFC applications. These proposed catalysts can potentially reduce the cost of DMFC anode catalysts by up to 6%.
Acknowledgments
This work was supported by the Ministry of Science and Technology, Taiwan (grant numbers MOST-108-2221-E-167-009-MY3 and MOST-107-2221-E-167-019-). The authors thank the BET, XRD, and SEM/EDS measurement services provided by the Green Energy and Engineering Materials Research Center of National Chin-Yi University of Technology (NCUT), Taiwan.