


 CC-BY 4.0
CC-BY 4.0
1. Introduction
1.1. Ethylene production
Ethylene is a critical building block for the chemical industry, with a total consumption exceeding 150 million tons in 2017 [1], currently produced mainly by steam cracking of naphtha and ethane at temperatures above 750 °C. The process is highly carbon and energy demanding, with emissions of up to two tons of CO2 for every ton of ethylene [2] and shows a significant amount of work loss [3], with little room for improvements without major process intensification [4]. Recent developments of alternative approaches, such as electrochemical, membrane or chemical looping catalytic systems developed for feedstocks from methane to naphtha, are more and more appealing due to increased ethylene production. Although these technologies have not yet been implemented, some oxidative approaches may reduce the CO2 emissions and energy consumption up to an order-of-magnitude compared to steam cracking [4].
Ethylene can be produced (Table 1) either by oxidative routes, such as ethane oxydehydrogenation (ODH), oxidative dehydrogenation with carbon dioxide (CO2-ODH), oxy-cracking of naphtha and oxidative conversion of methane (OCM), or a non-oxidative route, such as non-oxidative ethane dehydrogenation (NDH) and non-oxidative conversion of methane (NCM).
Desired reactions and secondary products obtained in non-oxidative and oxidative ethylene generation processes [4]
| Ethylene production process | Desired reactions | By-products |
|---|---|---|
| OCMa and electrochemical OCM | 2CH4 + O2→C2H4 + 2H2O | C3+, CO, CO2, coke |
| NCMb and electrochemical NCM | 2CH4→C2H4 + 2H2 | H2, C2H2, C6H6, C6H5–CH3, coke |
| C2H6→C2H4 + H2 | ||
| ODHc, CO2-ODHd and electrochemical ODH | C2H6 + 1∕2O2→C2H4 + H2O | C3+, CO, CO2, coke |
| C2H6 + CO2→C2H4 + H2O + CO | ||
| NDHe and electrochemical NDH | C2H6→C2H4 + H2 | H2, C2H2, C6H6, C6H5–CH3, coke |
| Naphtha oxy-cracking | CxHy + O2→C2H4 + H2O | Paraffins, C3+ olefins, C6H6, C6H5–CH3, coke |
aOCM: Oxidative conversion of methane; bNCM: Non-oxidative conversion of methane; cODH: Oxidative dehydrogenation; dCO2-ODH: Oxidative dehydrogenation with carbon dioxide; eNDH: Non-oxidative dehydrogenation.
The non-oxidative routes are thermodynamically limited, particularly from methane. The calculated equilibrium constant for the formation of ethylene at 1200 °C and 1 atm being just 0.272, very high temperatures are necessary in methane coupling. Moreover, secondary products such as coke, benzene and acetylene are thermodynamically favored at the expense of ethylene at elevated temperatures, while at low temperatures they are less favored. Therefore, the thermochemical routes are much less selective. Two important aspects need to be taken into consideration: (1) inhibition of benzene and coke formation is very important, and radical reactions must be considered for the reactor design and the selection of the operational parameters, since they can significantly alter the product yield and selectivity [5], and (2) for non-oxidative, non-thermochemical and low temperature approaches, such as plasma enhanced and electrochemical non-oxidative coupling, catalysts play a critical role in improving the activity and ethylene selectivity.
From a thermodynamic point of view, the oxidative routes have clear advantages for ethylene formation, but the byproducts, i.e. coke and COx, are still energetically favored even at low temperatures [4]. One way to enhance the selectivity is to use CO2, a softer oxidant, instead of O2 in ethane oxydehydrogenation. However, high temperatures are needed for convenient yields, but in these conditions ethane thermal dehydrogenation becomes favored and side reactions, such as methanation of COx [6], steam reforming or dry reforming of ethane/ethylene [7, 8], can also occur and, therefore, good selectivities are hard to obtain. Sulfur and nitrogen oxides have also been used as soft oxidants in ODH [9, 10, 11, 12] and OCM [13, 14, 15, 16, 17], but they are still in an early development stage and more research is needed.
In conclusion, both oxidative and non-oxidative pathways have thermodynamic limitations regarding ethylene selectivity. Therefore, in order to attain convenient product yields, variables such as process optimization, catalyst design and kinetics control for secondary versus desirable reactions become of crucial importance. A good summary for the conversion of ethane and methane with respect to the reaction temperature correlated with the selectivity/conversion/yield is presented in Ref. [4]. The most convenient feedstock for ethylene production is ethane. Among the possible methods, the thermochemical route—oxidative dehydrogenation of ethane (ODH)—can be performed with membrane, in redox mode or under oxygen co-feed, being the most broadly investigated topic.
1.2. Why oxidative dehydrogenation?
In ethane oxydehydrogenation (ODH), ethane is transformed into ethylene using an oxidant, the most common being oxygen in air. Even though research has been done to develop catalysts for non-oxidative dehydrogenation of ethane, e.g. supported Cr/Al2O3 or supported Pt, ODH processes manage to give better ethylene yields (>30%) [18, 19]. In recent years, several catalysts have been developed for ethane ODH, such as Mo/V/Te/Nb/O mixed oxide with yields up to 78% [20, 21], doped and supported NiO that give up to 60% ethylene yield [22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64], Sn-doped Pt with yields up to 55% [65, 66], modified boron nitride and carbon nanotube with maximum ODH yields around 50% [67, 68], and many others [69, 70, 71, 72]. Conventional oxidative dehydrogenation reactions take place with atmospheric oxygen as oxidant and between 300–700 °C using suitable catalysts. It is noteworthy that ethane ODH was, in the last decade, the object of several review papers discussing the different types of catalysts used and the reaction mechanisms involved [70, 71, 72].
Among the most investigated systems are the commonly called M1 catalysts—mixed metal oxides Mo/V/Te/Nb/O in a 1:0.15:0.16:0.17 atomic ratio—and NiO catalysts, both bulk and supported. The M1 catalysts can achieve almost total conversion coupled with significantly high ethylene selectivity in ethane ODH after being treated at around 600–650 °C [73]. At low temperatures—around 360 °C—the best catalysts exhibit an ethylene selectivity higher than 90% at conversion levels of more than 65% [74]. Such impressive performance can be achieved at low temperatures thanks to the exothermic nature of the ODH reaction, thus leading to great energy savings, i.e., 30%+ in comparison with conventional steam cracking process [75]. Moreover, reducing the number of unit operations is another advantage of the ODH process [76], which is the effect of extremely high selectivities (higher than 98%) and of a sole byproduct (CO2) that can be removed. Furthermore, the coke deposition is limited because the excess oxygen oxidizes coke with formation of CO2, thus eliminating the need of de-coking procedures that are currently implemented in commercial reactors [77].
Unfortunately, oxidative dehydrogenation still cannot be commercially implemented because of several challenges. One of them is the requirement of inert atmosphere to prevent the reaction mixture from reaching the flammable domain, the reaction being exothermic by nature. As summarized by Gao et al. [4], the vast majority of catalysts use relatively low ethane partial pressures (<0.3 atm), being highly diluted with inert gases such as argon, nitrogen or helium. This practice not only presents higher risks of safety hazards, but also imposes an increase in equipment size [77]. Other challenges, regarding ODH process retrofitting in the petroleum industry, include low ethylene selectivity at high pressures and the production of pure oxygen [78]. Fortunately, the staged feeding of O2 at limiting amounts in distinct positions of the reactor both improves the ethylene selectivity and eliminates the need of inert gases, as indicated by experimental studies [69]. Moreover, it improves safety and manages to make the oxydehydrogenation process profitable [77]. But staged feeding is not the only process optimization studied; research is also done with respect to novel reactor configurations—membrane-packed bed reactors that control the oxygen dispersion in the stream—and modifications of the catalyst structure for improving the selectivity [69].
2. Promoted bulk NiO catalysts
NiO-based catalytic materials are among the most active and selective catalysts for the low-temperature (300–500 °C) ethane oxydehydrogenation to ethylene [69]. The good catalytic performance of NiO in the low-temperature ethane ODH was first reported by Ducarme and Martin [22]. Schuurman et al. [23] also showed that the ethylene oxidation activity of NiO is lower than its ethane oxidation activity, thus explaining its interesting catalytic behavior in ethane ODH and demonstrating the potential of NiO-based systems in this reaction. A high number of studies focusing on ethane ODH over both unsupported [24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52] and supported [41, 42, 43, 44, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64] NiO-based catalysts have been published till date and is discussed in detail below.
Chen et al. [79] showed for the first time that the catalytic behavior of bare NiO strongly depends on the amount of nonstoichiometric oxygen in the solid, which can be controlled by its pretreatment temperature under oxygen. More recently, Zhao et al. [80] clearly demonstrated that ethylene selectivity at isoconversion, within a wide range of reaction conditions, decreases with the nonstoichiometric oxygen density, defined as moles of non-stoichiometric oxygen per mole total nickel. The latter is controlled by the treatment temperature of NiO catalyst: the higher the treatment temperature in the range from 400 to 1000 °C, the lower the nonstoichiometric oxygen density.
Abdelbaki et al. [81] succeeded in preparing highly stable and quite selective (73% ethylene selectivity at 340 °C) bare NiO catalysts by optimizing both the amount of oxalic acid in the synthesis gel and the calcination temperature. It has been shown that, on one hand, the optimum oxalic acid/nickel mol ratio is equal to 1, and, on the other hand, the catalysts calcined at 350 °C are more active and more selective than those calcined at 500 °C. Interestingly, the authors pointed out that, not only the crystallite size and the amount of electrophilic oxygen play a key role in the selectivity to ethylene, but they also observed that the catalysts showing a high extent of isolation of non-stoichiometric oxygen and low p-type semiconducting character give the best selectivities.
Since ceria (CeO2) is known for its both remarkable oxygen transport ability and ease of undergoing Ce3+–Ce4+ oxidation-reduction cycles [34, 40], Solsona et al. [34] used it as a dopant for NiO in an attempt to increase both the selectivity towards ethylene and the reaction rate. Indeed, they discovered that by adding only a small amount of ceria, corresponding to a Ni/Ce atomic ratio of 50, the ethylene productivity increased five times. However, the best ethylene productivity (21 g/kgcat/h) and ethane conversion (10.4%) with a reasonable selectivity of 59% were obtained with the system with a Ni/Ce atomic ratio of 12 at a reaction temperature as low as 275 °C. Notably, compared at isoconversion, at a reaction temperature of 300 °C, the systems with Ni/Ce atomic ratios in the range from 2 to 20 showed the best ethylene selectivities of ca. 65%, significantly higher than that of bare NiO.
The enhanced activity of the Ce–NiO systems was attributed to the significant increase of the surface area accompanied by a decrease of the crystallite sizes of NiO (9–12 nm) compared to the unpromoted NiO catalyst (ca. 35 nm). This is due to the so-called “mutual protective effect” that is encountered for many doped NiO catalysts [25, 27, 29, 32, 35, 37, 42, 50, 59]: the crystallization of one oxide hinders the process for the other one. The increase in ethylene selectivity, which takes place at the expense of carbon dioxide, has been related to the modification of the nature of Ni–O species resulting in a faster diffusion of oxygen into the bulk and a lower amount of oxidizing surface oxygen species in the CeNiO mixed oxides compared to bare NiO.
The temperature-programmed 18O2 isotope exchange can be used to determine the nature of the oxygen species involved in the mechanistic pathway of oxidation: if 16O2 is mainly formed, it means a faster incorporation and diffusion of oxygen into the bulk, indicating selective formation of ethylene, while 18O16O is a major product when the surface oxygen exchange is much faster than the diffusion, observed in the formation of CO2. CeNiO mixed oxides were shown to present higher 16O2∕16O18O ratios than NiO, confirming the better C2H4 selectivity of the former.
Tin is an element that prefers the oxidation state +4, higher than +2 that is specific for nickel. Therefore, according to Heracleous and Lemonidou [25], it should act as a donor of electrons, reducing the quantity of non-stoichiometric oxygen species present in NiO, thus favoring a higher selectivity for ethylene. Indeed, Solsona et al. [33] observed that the addition of a small amount of Sn induces a huge increase in the selectivity to ethylene to ca. 90% at 350 °C, from ca. 40% for pure NiO. An increase in the catalytic activity was also noticed. Such high selectivities were found for catalysts with low content of tin (Ni/Sn atomic ratio higher than 3). This has been explained by the decrease in NiO crystallite size and a different local environment for the Ni species in the presence of tin, with different unsaturation degrees depending on the Sn loading. This leads to lower reducibility of the Ni–Sn–O, smaller amounts of electrophilic oxygen species and, therefore, higher selectivity for ethylene. A change in surface acidity was also observed, with increased surface acid strength at high tin content, therefore high Sn loadings are not favorable. These changes in the surface sites lead to the modification of the mechanism of oxygen activation and the reactivity of surface oxygen species, evidenced by temperature-programmed 18O2 isotopic-exchange.
Aluminum is also successfully used as a dopant due to its acidic character. Moreover, because of its higher valence than that of Ni in NiO, it can also serve as an electron donor, thus adjusting the mobility and quantity of active superficial oxygen species. Skoufa et al. [37] recorded an increase in ethylene selectivity (accompanied by a decrease of ethane conversion) with increasing Al content, reaching a maximum of 70% for the best catalyst that contains 30 at.% Al. Further increase of dopant content decreases the selectivity, likely due to the decrease in the number of ethane activating surface sites [82]. The specific surface areas gradually increased with the Al content (partly due to diminishing crystallite sizes), reaching 174 m2/g for the oxide modified with 50 at.% Al, obviously due to the so-called “mutual protective effect”, as explained above. No segregated Al2O3 phase was observed, but small amounts of an Al-rich, amorphous precursor of the Ni–Al spinel were detected for the sample with 50% Al. The reducibility of the Al–NiO systems decreased with the addition of Al up to 30%, indicating a strong interaction between alumina and NiO, as well as the hindrance effect of Al on the NiO reduction. However, at higher Al loadings the easiness of reduction increased again, likely due to the presence of the Al-rich phase that impedes the Al–Ni interaction, letting NiO particles behave more as in pure NiO. Increasing the Al content also influenced the oxygen desorption properties of the Ni–Al mixed oxides: the amount of desorbed oxygen gradually decreased, diminishing the NiO over-stoichiometry, thus explaining the increased selectivity in ethane ODH. Notably, for the Ni70Al30 sample, the modification of the aluminum precursor from nitrate to isopropoxide resulted in ca. 25% higher ethylene selectivity at similar conversion level at 350 °C. This was explained by an important increase in the oxygen exchange activation energy, meaning stronger bound oxygen species, and the decrease of the amount of non-stoichiometric oxygen species obviously due to the reductive nature of the isopropoxide precursor.
Another good way to improve the ODH performance of a catalyst is by using a softer oxidant, like N2O, instead of oxygen. Zhou et al. [39] used this approach for a series of alumina-supported NiO catalysts and Ni–Al–O mixed oxides derived from layered double hydroxide (LDH) precursors, a method proved to create oxide catalysts showing improved dispersion of the cations, larger surface areas and high thermal stabilities with positive consequences on their catalytic performance [83, 84]. Three Ni/Al atomic ratios were used, denoted NixAl–MO (x = 2,3 and 4), while only one supported catalyst, with a NiO loading corresponding to an approx. Ni/Al atomic ratio of 3, denoted NiO/Al2O3, was kept for comparison purposes [39].
One would expect the conversion to increase with increasing the NiO loading as NiO is responsible for the ODH reaction. Ni3Al–MO exhibited the highest ethylene selectivity among all the tested catalysts, ranging from 70% to 98% over the reaction temperature range studied. NixAl–MO performed significantly better than NiO/Al2O3, and the performance of Ni3Al–MO—91% ethylene selectivity at 5% conversion at 340 °C—is better than that of most of catalytic systems tested using N2O, regardless of the reaction temperature [85, 86]. The catalyst was also tested with O2 as oxidant, showing a slight selectivity loss of ca. 5% at constant conversion, approaching the performance of Ni–Nb–O, one of the best catalysts for ethane ODH [36, 38]. Testing the catalyst on stream for 48 h showed no activity or selectivity loss, which suggests a good stability. These results were explained by the good dispersion of Al cations in the nickel oxide lattice, which proves once again that LDH are precursors of choice for the mixed oxides. The presence of aluminum decreases the reducibility of the solids, but also the content of surface electrophilic oxygen (O−) species, leading to increased ethylene selectivity. The reduction temperature increased with decreasing the Ni/Al ratio, reaching a constant value for ratios smaller than 3. XPS measurements confirmed the decrease in surface O− species compared to the alumina-supported nickel oxide, as did O2-TPD results, indicating the decrease of adjacent O− species, which is in agreement with a better isolation into the lattice [87]. A further confirmation comes from the TPSR experiments under C2H6, which demonstrated that under 500 °C only surface O− species are responsible for ethane activation [88] over both types of catalysts. What makes Ni3Al–MO so selective is the isolation of these species, known to promote over-oxidation when numerous.
Tantalum is an “exotic” element not frequently used in catalysis. When tried as modifier for NiO by Heracleous and Lemonidou [25], it showed a poor activity in ethane ODH reaction, with 10% conversion at 450 °C. This is likely due to the segregation of the solid into NiO and NiTa2O6 phases [25]. However, Zhu et al. showed that the method of preparation is critical for a good catalytic performance [28], and, hence, they prepared Ni–Ta–O mixed oxides by two methods: sol-gel (SG) and solid-state (SS) [35]. Surprisingly, the catalysts obtained showed remarkable catalytic properties, both of them giving 10% conversion at temperatures below 300 °C, a feat not achieved even by Ni–Nb–O, one of the best modified NiO catalyst known to date. Tantalum was shown, similarly to Nb, to eliminate electrophilic oxygen species that lead to the formation of CO2, therefore increasing ethylene selectivity. Notably, while for the SG method there is a well-known protocol of synthesis [89], for the SS method the authors used oxalic acid as an additive. They showed that adding and increasing the amount of oxalic acid drastically improves the performance of the catalysts in the ODH reaction, due to the bidentate nature of the oxalate ligand leading to an intimate mixture of Ni and Ta in the lattice, and also to an increased surface area of the final catalysts.
Only crystalline NiO was observed by XRD, together with highly amorphous Ta2O5 at high Ta loadings, without any composite Ni–Ta phase. The solids prepared by the SS method had higher crystallinity. Increasing Ta content led to a strong increase in the specific surface area, coupled with the reduction of the crystallite size, especially for samples prepared by the SG method. Electron energy-loss spectroscopy (EELS) mapping showed Ta being homogeneously distributed into the NiO lattice for mixed oxides prepared by both methods, but dark-field scanning transmission electron microscopy (HAADF-STEM) proved that the solids prepared by SG had a higher concentration of Ta atoms at the crystallite surface. An increase in the reduction temperature with Ta addition was observed, implying the strengthening of Ni–O–Ni bonds.
Addition of Ta increased the ethylene selectivity, from 60% to 90% for the SS catalysts, and from 55% to 87% for the SG systems. The samples obtained from the SS method showed much higher ethylene production rates than those from the SG method, the best results being achieved with Ni0.93Ta0.07O at 350 °C: 55% conversion and 70% selectivity. Notably, the ethylene yield of 38%, and the turnover frequency (TOF) of 0.77 h−1, are higher than those reported for Ni0.85Nb0.15O, at a lower dopant loading (7 at.% Ta, compared to 15 at.% Nb). Tests in higher oxygen content (C2H6∕O2 molar ratio = 1, compared to 2) promoted ethane conversion at the expense of C2H4 selectivity. With a C2H6∕O2 molar ratio = 2, the activity of the Ni–Ta catalysts is high enough to exhaust the oxygen in the feed below 400 °C. The authors concluded that, for the catalysts from the SG method, CO2 is produced mainly by ethylene oxidation, due to lower desorption rates of C2H4 from Ta Lewis acid sites. On the catalysts prepared by the SS route, however, where much less Ta atoms are found at the crystallite surface, the possibility for ethylene over-oxidation decreases and CO2 is directly formed from complete ethane oxidation. Stability tests for 48 h showed that the catalysts prepared by SS method were more stable, and the deactivation was mainly due to the loss of specific surface area.
The ability of iron to easily change its oxidation state (Fe2+ ↔ Fe3+), the acidic character of Fe3+ as well as its similar radius compared to nickel, makes it a good candidate for creating a Ni–Fe mixed oxide. Hurtado Cotillo et al. [43] used Fe as a dopant for NiO with good results: in the ODH reaction the catalysts with 10 and 20 at.% Fe have emerged as the best ones. The Ni–Fe–O with 20 at.% Fe exhibits a selectivity of 84% with 7% conversion at 300 °C. This performance was explained by the formation of a NiFe2O4 spinel, leading to decreased particle sizes and higher reduction temperatures, while the low-temperature TPR peak ascribed to non-stoichiometric oxygen disappeared as Fe loading was increased.
The same Ni–Fe mixed oxides were used by the authors in a different ethane ODH study after being deposited on 𝛾-Al2O3 and TiO2 supports, which have shown very good results in ethane ODH [44] when compared with NiO [41]. Excellent catalytic activity was observed on 𝛾-Al2O3, with NiFe-0.9/𝛾-Al2O3 exhibiting 94% ethylene selectivity with more than 40% conversion at 400 °C. When deposited on TiO2, however, the catalysts showed conversion values below 6% on the entire temperature range, even if the selectivities were above 80%, possibly because of the small specific surface area of this support. The interesting results obtained on 𝛾-Al2O3 were explained by the very good dispersion of the Ni–Fe mixed oxides on the support: no iron oxides separated phases were identified, small crystallite sizes (indicating a strong active phase-support interaction [90]) and high specific surface areas. The supported samples showed a NiO cell parameter larger than that for bulk oxides, confirming the Fe insertion into the NiO lattice. The formation of Ni–Fe mixed oxide caused a shift of the maxima of the H2-TPR reduction peaks to higher temperatures in the case on 𝛾-Al2O3-supported catalysts, but to lower temperatures for those supported on TiO2. The same behavior was observed for other TiO2-supported NiO catalysts [59, 62]. The authors concluded that the high dispersion on the alumina support led to the formation of different surface species, such as Ni and Fe species interacting with tetrahedrally coordinated sites in 𝛾-Al2O3, leading to increased reduction temperature with Fe loading. These modifications result in better catalytic activity. O2-TPD experiments showed two types of electrophilic oxygen species on the surface: (denoted 𝛽, desorbed at 260–360 °C) and O− (𝛾, which desorbs at 400–500 °C, and 𝛿, at temperatures higher than 550 °C), the latter being adsorbed on different sites [27]. These oxygen species were found in much lower concentration on the supported catalysts, inferring a significant reduction of the NiO non-stoichiometry. Moreover, the ratio between the 𝛽 and 𝛾 oxygen species, was considered to play an important role in ethane ODH.
Phosphorous has been shown to have a beneficial effect in ODH reactions, although its mechanism of action depends on the catalytic system used [45, 46, 47, 48]. Ivan et al. [49] studied the effect of phosphorus on the catalytic performance of NiO in ethane ODH, preparing three P-modified NiO samples by wet impregnation of NiO with NH4H2PO4 solutions with three different concentrations, i.e., 0.05, 0.5 or 1.0 M, leading to 0.05P@NiO, 0.5P@NiO and 1.0P@NiO catalysts. The catalytic results clearly showed that phosphorous markedly improved the ethylene selectivity at the expense of ethane conversion, the best system reaching over 70% ethylene selectivity at around 10% conversion, as opposed to pure NiO, which was more active, but less selective (Figures 1 and 2).
Ethane conversion and ethylene selectivity versus reaction temperature in ethane ODH over bare and phosphated NiO catalysts [49].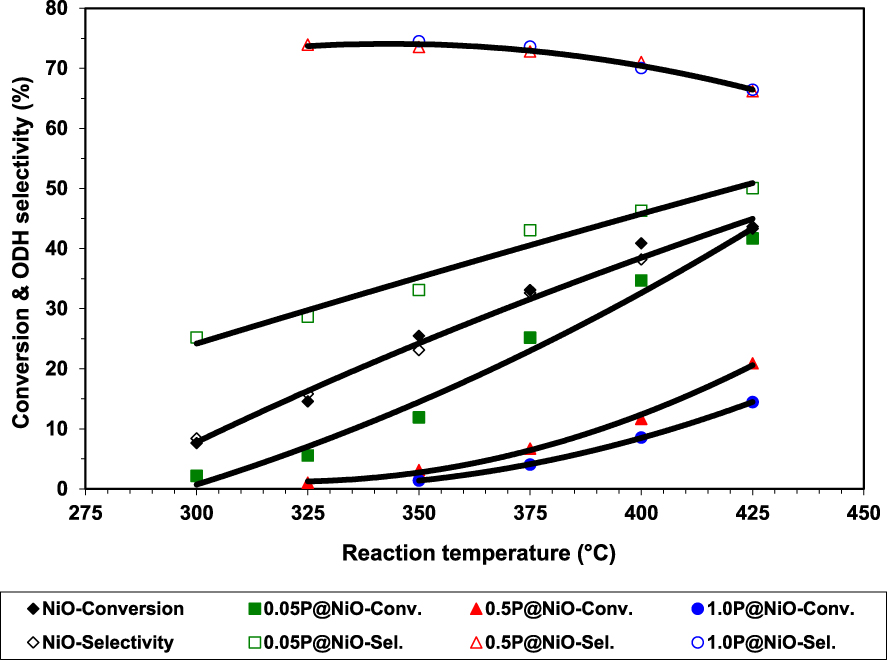
Effect of ethane conversion on the ethylene selectivity in ethane ODH over 0.5P@NiO and 1.0P@NiO catalysts at 400 °C (oxygen-to-ethane mol ratio = 1) [49].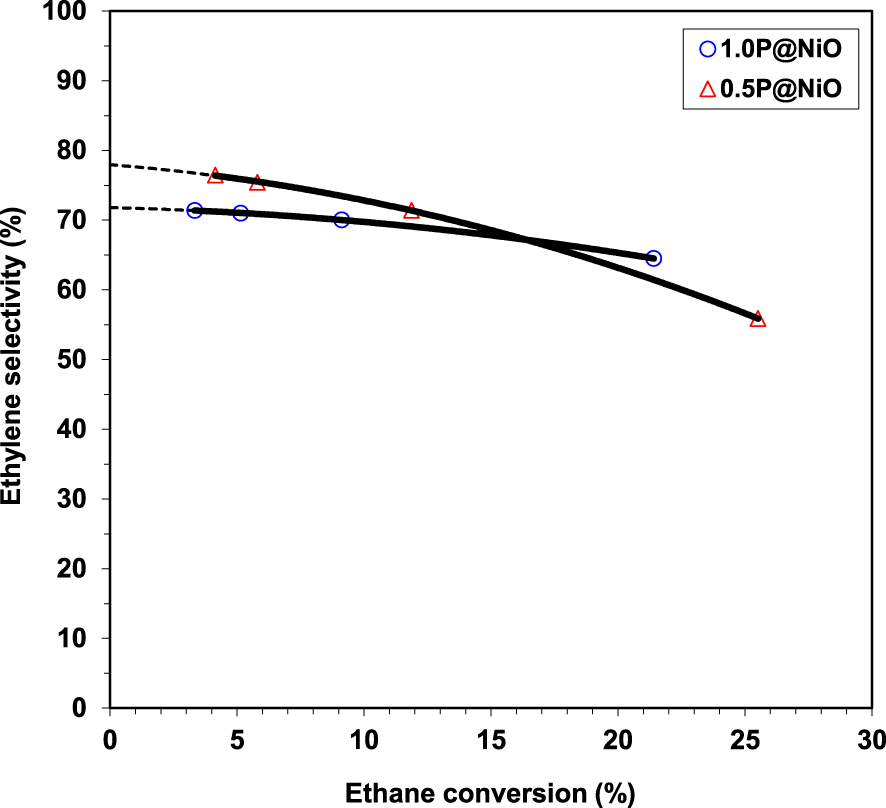
As the P loading increased, the surface coverage with phosphate/pyrophosphate groups (calculated based on ICP-OES data) increased from 4% to 70%. XPS and Raman spectra confirmed the presence of these species on the surface of the catalysts. However, the modifier was very well dispersed, since the XRD patterns showed no peaks corresponding to P-containing phases, and no phosphate clusters were observed in the HRTEM images. XPS results showed the coexistence of two nickel species on the surface of all three catalysts: Ni2+ and Ni3+, the content of the latter increasing with the P loading. Such behavior was also observed for Fe-doped NiO supported on alumina and titania and Ti-containing PCH-supported NiO [44, 62].
Regarding the catalytic performance of the catalysts, NiO and 0.05P@NiO exhibited an increase in selectivity with increasing temperature, attributed to the higher ethylene formation rate compared to the ethylene oxidation rate [23, 55]. 0.05P@NiO behaved similarly to NiO due to its very low P surface coverage. By increasing the P content, the activity decreased. At the same time, the selectivity increased until a phosphorous concentration of 3.0 wt%, then it reached a plateau. This clearly indicated that the presence of P on the surface of NiO diminished the amount of non-selective active sites. This finding was further confirmed by the inactivity of Ni2P2O7 in the temperature range studied, indicating that the phosphate and pyrophosphate groups did not actively contribute to the catalytic performance, but just diminished the density of non-selective sites. By varying the contact time for 0.5P@NiO and 1.0P@NiO it was revealed that with increasing the conversion the selectivity decreases, the effect being more pronounced for 0.5P@NiO than for 1.0P@NiO. However, none of them gave 100% selectivity with extrapolation to 0% conversion (Figure 2), indicating that CO2 is obtained not only by ethylene oxidation, but also by direct ethane oxidation. The stability of the 0.5P@NiO catalyst was monitored, the catalytic performance being found stable for 42 h on stream.
In order to better understand the redox properties of the catalyst, electrical conductivity measurements were performed. All materials behaved as p-type semiconductors, with the conductivity decreasing with increasing the P content. Since the intrinsic ethane conversion rate followed the same trend, it was clear that the charge carriers, i.e., positive holes, are involved in the conversion of ethane. By studying the influence of the partial pressure of oxygen on the electrical conductivity, the value of the exponent for the oxygen partial pressure (PO2) can reveal the nature of the defects in the solid. This value was found to be much higher than 4 or 6, which correspond to single and double ionized nickel vacancies, respectively. This indicates that there are two mechanisms of electrical conductivity, one being independent of the oxygen partial pressure. Since the exponent was found to increase with increasing the phosphorous concentration, this implies that the ability of the solids to exchange oxygen with the gas phase decreased. These results are in perfect agreement with the proposed ODH mechanism in which the chemical equivalent of positive holes, surface lattice O− species, catalyze the ethane activation [26, 27, 91, 92, 93].
By sequentially exposing the catalysts to air, reaction mixture, air, pure ethane and again air, it was demonstrated their ability to fully regenerate under air, as well as their p-type character in the presence of ethane since the conductivity markedly decreased (Figure 3). These observations are consistent with a Mars–van Krevelen type mechanism [94]. Moreover, the degree of reduction of the catalysts under the reaction mixture, expressed as 𝛥lg(𝜎), decreases with increasing the P content, being inversely correlated with ethylene selectivity (Figure 4).
Variation of the electrical conductivity during sequential exposures to air, ethane–air mixture (reaction mixture) and pure ethane for NiO and phosphated NiO catalysts at 400 °C (𝜎 in 𝛺−1⋅cm−1) [49].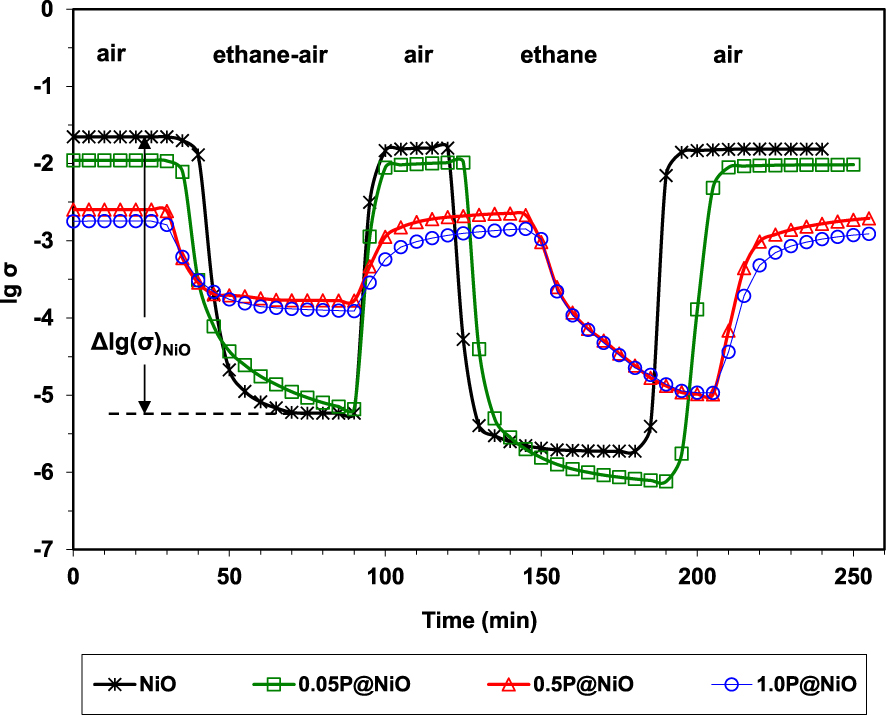
Ethylene selectivity at 400 °C and the number of available oxygen species in the catalyst expressed as 𝛥lg(𝜎) as a function of the P content in the solid [49].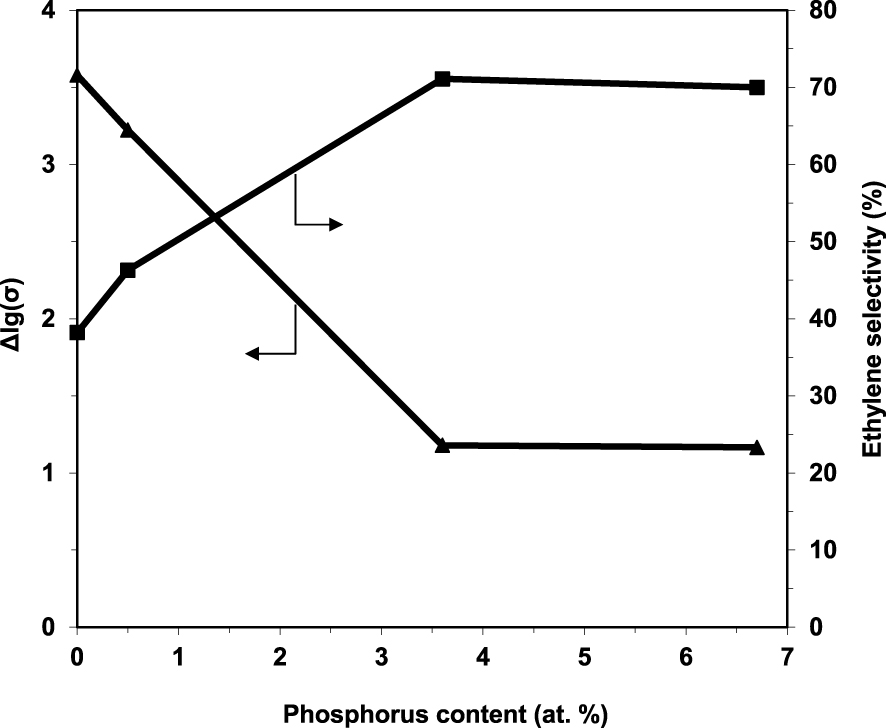
A summary of the experimental results from literature, obtained on promoted NiO catalysts, is presented in Table 2.
Promoted NiO catalysts for ethane oxidative dehydrogenation
| Promoter (catalytic system) | Preparation method | Reaction conditions | Best performance | Ref. |
|---|---|---|---|---|
| Ce (Ni–Ce–O with at. ratio Ni/Ce = 0.2–50) | Evaporation of nitrates solution in ethanol | C2H6∕O2∕He = 9.1∕3∕87.9 (mol); 25 mL/min; T = 200–350 °C | on Ni–Ce–O with Ni/Ce = 12 | [34] |
| Ce (XCeNiNb, with X = 0, 0.5, 2, 4, 15 wt%, and constant 17.6 at.% Nb) | Evaporation of nitrates solution in ethanol | 20% C2H6, 10% O2, 70% N2; T = 250–350 °C; W/Fa = 0.6 g⋅s/mL; 10 mL/min | Smax = 65.4% at Cmax = 14.5%, 300 °C on 0.5CeNiNb | [40] |
| Sn (NiSn-x, with x = Ni∕(Ni + Sn) = 0–100) | Evaporation of nitrates solution in ethanol | C2H6∕O2∕He = 3∕1∕29; 0.5 gcat; T = 200–450 °C; 25 mL/min | Smax = 90% at Cmax = 10%, 350 °C on NiSn-92 | [3] |
| Al (Ni100−xAlx, with x = 1–50 at.%) | Evaporation of nitrates aqueous solution under reduced pressure | 10% C2H6, 5% O2, 85% He; T = 300–425 °C; W/F = 0.24 g⋅s/cm3 | Smax = 70% at C = 10%, T = 350 °C, on Ni70Al30 | [37] |
| Al (Ni70Al30-org) | Evaporation of nitrates solution in ethanol, under reduced pressure; Al from aluminum isopropoxide | 10% C2H6, 5% O2, 85% He; T = 300–425 °C; W/F = 0.24 g⋅s/cm3 | Smax = 82% at C = 14%, T = 350 °C, on Ni70Al30-org | [37] |
| Al (NixAl–MO; x = 2; 3; 4) | Coprecipitation of LDH precursors and calcination to mixed oxides | 2% (vol.) C2H6, 2% N2O in He; T = 260–480 °C; 30 mL⋅min−1; W/F = 0.05–0.6 g⋅s/mL | Smax = 91% at 5% conversion of C2H6 at 340 °C | [39] |
| Ta (at. ratio Ta/Ni = 0.176) | Solution evaporation | C2H6∕O2 = 1∕1; T = 300–425 °C; W/F = 0.02–0.71 g⋅s/mL | Cmax = 10%; Smax = 80% at 400 °C | [25] |
| Ta (Ni1−xTaxO with x = 0.01–0.11) | Sol-gel with citric acid | 10% C2H6∕5% O2 or 10% C2H6∕10% O2 in He; T = 250–400 °C; W/F = 0.05–0.6 g⋅s/mL | S = 90% at 330 °C, C2H6∕O2 = 2, for Ni0.89Ta0.11O with H2C2O4∕(Ni + Ta) = 0.75 | [35] |
| Ta (Ni1−xTaxO with x = 0.01–0.20) | Solid-state method, with oxalic acid added (H2C2O4∕(Ni + Ta) = 0.25–1 | 10% C2H6∕5% O2 or 10% C2H6∕10% O2 in He; T = 250–400 °C; W/F = 0.05–0.6 g⋅s/mL | C = 55%, S = 70% at 350 °C, C2H6∕O2 = 2, for Ni0.93Ta0.07O with H2C2O4∕(Ni + Ta) = 0.75 | [35] |
| Nb (NixNb1−xO, x = 0.8–0.99) | Citrate method | 10% C2H6∕5% O2 or 10% C2H6∕10% O2 in He; T = 200–400 °C; W/F = 0.05–0.6 g⋅s/mL | Ethylene yield = 22% at 350 °C for Ni0.97Nb0.03O and Ni0.96Nb0.04O | [28] |
| Fe (NixFe1−xO, x = 0.5, 0.8, 0.9 1.0) | Coprecipitation | 0.5% C2H6 and 0.5% O2 in N2; T = 250–400 °C; W/F = 0.48 g⋅s/mL | S = 84% at 7% conversion on Ni0.8Fe0.2O | [43] |
| Fe (NiFe-a, where a = Ni∕(Ni + Fe) = 0.8 and 0.9, deposited on 𝛾-Al2O3 and TiO2) | Coprecipitation and deposition on 𝛾-Al2O3 and TiO2 | 0.5% C2H6 and 0.5% O2 in N2; T = 300–400 °C; W/F = 0.48 g⋅s/mL | S = 93% at 43% conversion on NiFe-0.9/𝛾-Al2O3 at 400 °C | [44] |
| P (xP@NiO, with x = 0.05; 0.5; 1.0) | Wet impregnation of NiO (obtained by precipitation with NaOH) with NH4H2PO4 solution | O2∕C2H6 (mol) = 0.5–3.0; T = 300–425 °C; W/F = 0.18–1.09 g⋅s/mL | S = 72% at 17% conversion on 0.5P@NiO at 400 °C | [49] |
| Nb (NixNby, y∕x = 0–0.67; x, y = atomic content of Ni and Nb) | Evaporation of aqueous solution of precursors | 9.1% C2H6, 9.1% O2, 81.8% He; T = 300–425 °C; W/F = 0.02–1.73 g⋅s/mL | S = 90% at 20% conversion on Ni0.85Nb0.15 at 350 °C | [38] |
| Nb (Ni1−xNbxO, x = 0.03–0.19) | Evaporation of aqueous solution of precursors + oxalic acid | C2H6∕O2∕N2 = 9∕9∕82; T = 300–400 °C; W/F = 0.54–1.73 g⋅s/mL; 0.1–1 gcat | S = 78% at 33% conversion on Ni0.85Nb0.15 at 350 °C | [27] |
| Nb (xNb2O5/NiO/Ni-foam, x = 1, 3, 5, 7 wt%) | NiO/Ni-foam obtained by hydrothermal synthesis using H2C2O4, NH4Cl and Ni foam, then wet impregnation with ammonium niobium oxalate | C2H6∕O2∕N2 = 1∕1∕8; 30 mL⋅min−1; T = 250–450 °C; GHSVb = 1500–45,000 mL⋅g−1⋅h−1 | S = 68% at 60% conversion on 5Nb2O5/NiO/Ni-foam (calcined at 450 °C) at 400 °C | [52] |
| Nb (Ni–Nb–O with 0–30 at.% Nb) | Hydrothermal method with PEG 4000 | 10% C2H6, 10% O2 in N2; 30 mL⋅min−1; T = 250–400 °C; 3 g catalyst | S = 72% at 66% conversion on Ni0.85Nb0.15O at 400 °C | [29] |
aThe contact time defined as the ratio between the catalyst weight (W) and the flow rate (F). bGas Hourly Space Velocity defined as the ratio between the gas flow rate and the catalyst weight.
The beneficial effect of high valence elements, combined with a dry solid grinding synthesis method, inspired Zhu et al. [51] to prepare Sn, Ti and W-doped NiO catalysts (2.5–20 at.% dopant) for ethane ODH. The results were very satisfactory, selectivities higher than 70% being obtained for the best catalysts in each dopant series at conversion levels above 30%.
While all the catalysts showed diffraction lines belonging to NiO, the doping oxides were observed as separated phases from different dopant loadings: TiO2 at 20 at.%, but WO3 from 5 at.% and SnO2 even at 2.5 at.%. As the dopant loading increased, a decrease in the particle size was observed. It was explained by the “mutual protective effect”: one oxide inhibits the crystallization process of the other one; therefore, the apparition of large particles is obstructed. All the samples exhibited much larger surface areas than pure NiO, and also compared to catalysts synthesized by typical evaporation method. This was attributed to the presence of a doping metal amorphous layer on the surface of the mixed oxides, as previously described [28]. At high dopant amounts the surface area decreased, especially in the case of Sn and W, likely due to the segregation of SnO2 and WO3 phases with higher densities than NiO.
H2-TPR measurements revealed in all the samples a small peak at around 200 °C, attributed to the reduction of non-stoichiometric electrophilic O∙− species, usually noted O− [95]. These species are associated with the ODH rate-determining step, first C–H bond breaking in ethane, but inversely correlated with the ethylene selectivity [96, 91]. The intensity of this peak gradually diminished with increasing the amount of dopant, clearly demonstrating that doping nickel oxide with another metal can decrease the quantity of non-stoichiometric oxygen species, thus altering its catalytic performance. The XPS spectra and DFT calculations confirmed these findings.
Finally, it can be concluded that even a small amount of dopant (2.5 at.%) can significantly improve the catalytic performance of NiO, with W having the largest effect due to its oxidation state that strongly favors the decrease of the number of non-stoichiometric oxygen sites.
In order to better understand and confirm the relation between the chemical properties of dopants and their catalytic performance, Heracleous and Lemonidou [25] used several metals with similar ionic radii to Ni2+ and valences spanning from +1 (Li) to +5 (Nb and Ta). The best catalyst they found was Ni–Nb–O which exhibited a remarkable ethylene yield of 46% at 400 °C, while the worst performance was demonstrated by Ni–Li–O with a poor yield of ca. 8.5%. Niobium was also the only dopant to ease the reduction of NiO, all the others shifting the reduction temperature to higher values.
All dopant cations (except for Ta+5) easily formed solid solutions in the NiO lattice. The substitution of Ni ions with foreign cations modulated the nickel oxide non-stoichiometry and, therefore, the activity in ethane ODH. The lower valence cations (Li+, Mg2+) increased the non-stoichiometric oxygen content [25], determined by O2-TPD, in line with previously reported results [97]. On the contrary, those with higher valences caused a decrease of the non-stoichiometric oxygen by acting as electron donors, thus reducing the concentration of electrophilic O− species that lead to non-selective oxidation [25]. The higher valence cations form strongly bonded, less mobile oxygen species that selectively convert ethane: the higher the valence, the higher the initial selectivity to ethylene, at the expense of conversion.
There is a wide range of conversions exhibited by the catalysts, spanning from 2% for Ti-doped NiO to 66% for Nb-doped NiO at 400 °C [25]. The descending order is as follows: Nb ≫ Mg > Li > Ga ≈ pure NiO ≫ Al ≫ Ti ≫ Ta; however, if one accounts for the specific surface areas, then the order of activity becomes Li ≫ Mg > pure NiO > Nb ≈ Ti > Ga > Al ≫ Ta. Moreover, for most of the catalysts the ODH selectivity remains almost unchanged over the conversion range studied.
López Nieto et al. [24] also tested several metal dopants, including K, Ce, Zr and Nb. The selectivities obtained are in agreement with what was reported by Heracleous and Lemonidou [25]: a lower valence metal dopant has a negative effect on the ODH selectivity, while the opposite is true regarding higher valence elements. Low valence dopants push the valence state of the main element to its highest accessible value, while the opposite is true for acidic, high valence promoters. Therefore K+ will favor the formation of Ni3+ species, while Sn4+ and Nb5+ will keep Ni in its +2 state. This clearly is a critical factor affecting the NiO catalytic performance. So, by promoting NiO with acidic, high valence elements the amount of non-stoichiometric species, found as Ni3+ and O−, is significantly reduced, markedly increasing the ODH performance. Therefore, unsurprisingly Sn and Nb-promoted catalysts exhibited the highest ethylene selectivities, while Ni–K–O performed very poorly.
The authors [24] found that not only the valence of the dopant is altering the catalytic performance, but also the crystallite size. XRD measurements revealed that doping resulted in significantly smaller particle sizes than in NiO. By plotting the ODH selectivity versus crystallite size it became evident that the smaller the crystallite size, the higher the selectivity. The trend takes an exponential shape with a steep increase at very small dimensions.
By studying the XPS Ni 2p signals, the authors highlighted the presence of the characteristic pair main peak—satellite I peak. The satellite I peak is generally correlated with the degree of NiO non-stoichiometric Ni3+ sites involved in the formation of COx, meaning that a more intense satellite peak will usually be observed for catalysts that exhibit a lower ODH selectivity.
Another very important factor for the catalytic performance was considered to be the acidic/basic character of the catalysts, due to influences on the adsorption/desorption properties of ethane and ethylene, and also on the nature of the Ni active sites. These acidic/basic properties were estimated by measuring the isoelectric point (IEP). The authors thus revealed that the most selective catalytic systems were the most acidic ones, having the lowest IEP. This holds true even in a series of dopants with the same valence. Quantitative measurements by FTIR monitoring of CO desorption showed that the catalysts which exhibited the best ODH selectivity also presented the highest amount of acid sites, a clear trend being established. Therefore, it has been concluded that both the number of surface acid sites and their strength play a critical role in regulating the ethylene selectivity.
Electrical conductivity measurements represent a powerful tool for investigating the redox properties of the catalysts and thus can help to elucidate the differences in their catalytic behavior [98]. Bearing this in mind, Popescu et al. [26] used this technique to characterize the M-doped NiO (M = Li, Mg, Al, Ga, Ti, Nb and Ta) catalysts described in Ref. [25], where it has been shown that the activity slowly decreased with increasing the valence of the doping element except for Nb-doped NiO, which exhibited the highest conversion, while the selectivity gradually increased with the increase in the oxidation state, suggesting the progressive removal of non-selective active sites that promote total oxidation.
By measuring the electrical conductivity (𝜎) as a function of temperature it was observed that all the solids behave as semiconductors, with 𝜎 varying exponentially with temperature. The electrical conductivity at constant temperature and, hence, the amount of charge carriers decreases with increasing the valence of the dopant (Figure 5), except for Nb whose electrical conductivity is lower than expected indicating that Nb oxidation state decreases during doping to +4 and, even, +2. A very high activation energy of conduction was observed for Ni–Ti–O, and it was explained by the electron transfer from TiO2, an n-type semiconductor, to NiO, a p-type semiconductor [99]. The p-type semiconducting character of all the solids was confirmed from log(𝜎) versus log(PO 2) plots at constant temperature (320 °C), whose slopes were, in all cases, positive. Moreover, it has been shown that the main point defects in these solids are doubly ionized cationic vacancies.
Variation of the electrical conductivity during sequential exposures to air, an ethane–air mixture (reaction mixture with an oxygen-to-ethane mol ratio of 1) and pure ethane for pure and doped NiO catalysts at 400 °C (𝜎 in 𝛺−1⋅cm−1) [26].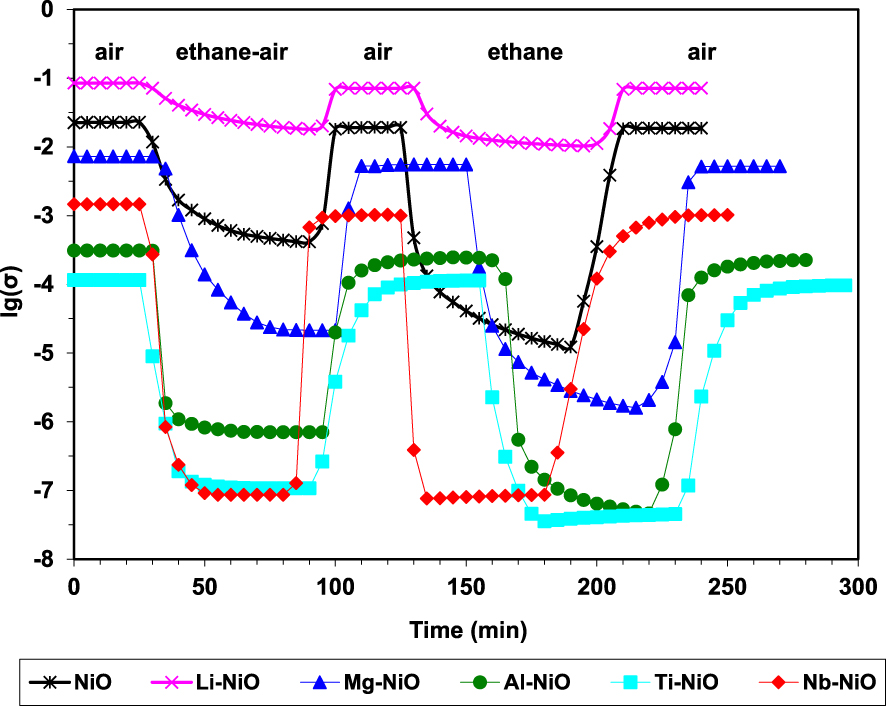
In order to study the electrical behavior of these catalytic systems in conditions closest to those of catalysis, they were kept at a convenient reaction temperature and the composition of the gaseous mixture was sequentially changed from air to reaction mixture to pure ethane. It was observed that during each sequence of ethane-containing mixture the conductivity decreased and, then, it rose back to its initial value under air, and that under pure ethane the conductivity reached its lowest value (Figure 5). This indicated that ethane was transformed by consuming the positive holes from the p-type solids. Therefore, it was suggested that the C–H bond cleavage, which is the first step in the ODH mechanism, is done by a positive hole attack.
The electrical conductivity under the reaction mixture was observed to decrease with the increase of the valence state of the doping cation. This implies that the number of positive holes decreased following the same order. Since the chemical site of the positive holes corresponds to lattice O− species, which are known to promote total oxidation at the expense of oxydehydrogenation when too numerous, this perfectly explained the observed increase of the ODH selectivities with the valence state of the promoters. Nonetheless, this correlation was not linear, implying that the ethylene selectivity is not modulated only by the electrical properties of the solid (Figure 6). This is in line with the findings reported by López Nieto et al. [24] and discussed above.
Variation of the ethylene selectivity at isoconversion at 400 °C as a function of the electrical conductivity in the reaction mixture for pure and doped NiO catalysts (𝜎 in 𝛺−1⋅cm−1) [26].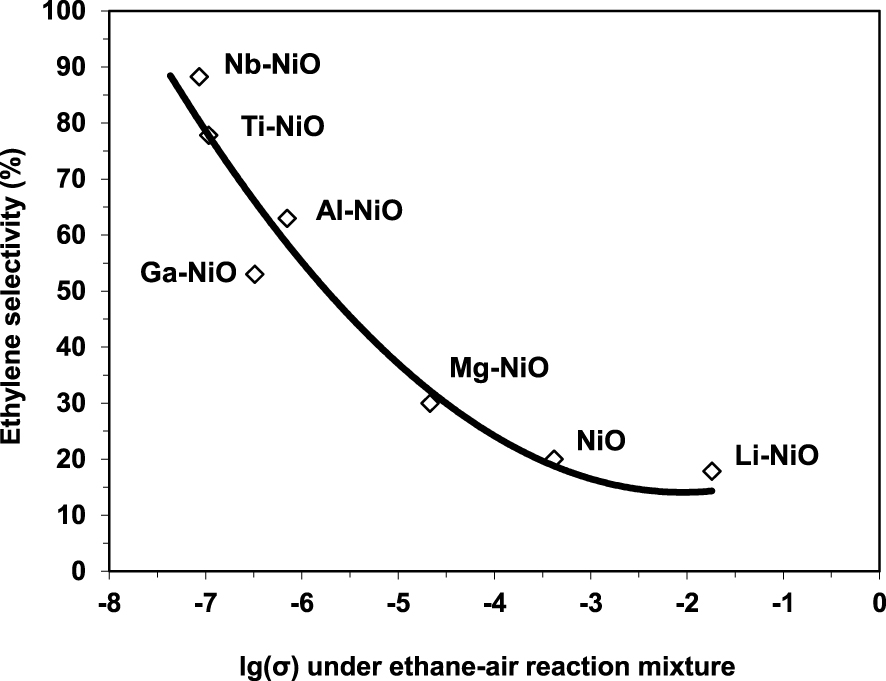
The difference 𝛥lg(𝜎) between the value of 𝜎 in air (corresponding to a fully oxidized solid) and under the ethane–air reaction mixture was considered by the authors a measure of the number of oxygen species in the solid lattice that were removed during catalysis. This difference correlated with the amount of exchanged oxygen in temperature-programmed 18O2 experiments from Ref. [25], indicating that the exchanged O atoms are in fact the same as the ones removed during the ODH reaction. Even more, a linear correlation was obtained by plotting the oxygen exchange activation energy as a function of the activation energy of conduction, which suggests that the same oxygen species are involved in both the oxygen exchange and the conduction mechanism and confirms that the positive holes are associated to the O− species from the solids [26].
Zhu et al. [32] tried to improve the catalytic performance of NiO by doping it with Ti, W, V, Zr and Mo at a molar ratio of M/(Ni + M) of 0.05. Lower activity compared to pure NiO was observed for all doped catalysts. However, ethylene selectivity was markedly improved, the Ti and W-doped catalysts exhibiting up to 90% selectivity in certain reaction conditions.
The XRD spectra presented only lines corresponding to well-crystalized bunsenite-type NiO for all the catalysts, except Ni–V–O. In a previous article [28], the authors observed that in the absence or at small concentrations of Nb, metallic Ni formed during the preparation of the catalyst. However, except for the V-doped sample, all the other transitional metals inhibited the formation of metallic Ni. This finding is consistent with the H2-TPR experiments, which showed a systematic shift of the reduction peaks to higher temperatures (confirming the inhibiting effect of the doping oxides on the NiO reduction), limiting the formation of Ni0. For Ni–Mo–O and Ni–W–O oxides, high-temperature shoulders are present, indicating the reduction of WO3 and MoO3 [100, 101].
As already stated, doping with these transitional metals negatively impacted the catalytic activity compared to pure NiO synthesized by citrate method, all the mixed oxides presenting lower conversions than pure nickel oxide and decreasing in the following order: Ni–Zr–O > Ni–Ti–O > Ni–W–O > Ni–Mo–O > Ni–V–O. Since all the doped catalysts presented higher reduction temperatures than pure NiO, this means a lower reducibility would explain their lower activity. On the other hand, it is worth noting that only titanium and wolfram managed to significantly improve the ethylene selectivity over the investigated temperature range.
Since Ti and Nb proved to be among the best promoters, Delgado et al. [50, 62] used them for synthesizing NiO-based catalysts for ethane oxydehydrogenation. A novel idea was to support NiO on TiO2 and Nb2O5 and to compare with mixed oxides. Therefore, two series of catalysts, supported on and promoted with the appropriate amount of Nb or Ti (5, 20, 50, 80, 92 and 97 wt% NiO) were prepared and studied in ODH reaction. Surprisingly, the catalytic performances of Nb-doped and Nb2O5-supported NiO are markedly different: while in the promoted series the ethylene selectivity increased with increasing the Ni content, reaching a maximum of ca. 90% for 92% Ni, the Nb2O5-supported catalysts exhibited much poorer results (possibly due to lower specific surface areas), presenting selectivities between 48–64% regardless of the NiO loading. Nevertheless, the ODH selectivity was clearly improved compared to pure NiO (ca. 33%).
Regarding Ti-containing systems, impressive results are obtained using both supporting and doping approaches. An ethylene selectivity of up to 90% was obtained on the best catalysts from each class—8% Ti for the doped NiO, and 20% NiO loading for the supported catalyst, respectively. The trend followed by the selectivity as a function of Ti loading in the promoted series is similar to the Nb-promoted one, in both cases the best catalyst being obtained at 8% of promoter. For TiO2-supported systems, the ethylene selectivity progressively increased with decreasing the NiO loading, reaching a maximum for 20% NiO. More important, the selectivity remains almost unchanged when the conversion is increased, an effect that can be explained by the lack of non-selective sites that are responsible for overoxidation. In conclusion, the preparation method, as well as the NiO or promoters loading, play a pivotal role in modulating the active sites and, consequently, the catalytic performance.
By using the XPS analysis the authors managed to correlate the selectivity with the Ni 2p3∕2 and O 1s signals. As usual, for Ni there are 2 peaks of interest: the main peak and the so-called satellite I peak, the latter being associated to species such as Ni3+ or Ni2+ vacancies, in other words NiO non-stoichiometry. By promotion or supporting, the satellite I/main peak ratio significantly decreased, the effect being more obvious for the promoted catalysts, which indicated the dopants strength to alter the surface chemistry. From the O 1s spectra there are 2 types of oxygen species that can be discriminated: nucleophilic (O2−), responsible for the selective oxidation, and electrophilic ( and O−), prone to overoxidation [102]. Since the intensity of the signal attributed to electrophilic oxygen species is decreasing upon doping or dispersion, it could be concluded that the O 1s spectra come in complete agreement with the catalytic results. A further confirmation of the reduced non-stoichiometry came from XANES measurements which showed a decrease in the intensity of the “white line” for both Ni–Nb–O with 8% Nb and NiO/TiO2 with 20% NiO loading associated to a decrease of the average Ni oxidation state in the samples [103].
In line with the aforementioned results, the reducibility measured by H2-TPR is lower for the promoted series as the reduction peak shifts to significantly higher temperatures, meaning that the O atoms are not as reactive as in pure NiO, favoring selective oxidation rather than conversion to CO2. However, the same correlation could not be made for the supported catalysts as the reduction temperature varied insignificantly.
Comparative results from literature of the ethane ODH on NiO catalysts modified with different dopants can be found in Table 3.
Comparative results of the ethane ODH on NiO catalysts modified with different dopants
| Doped catalyst | Preparation method | Reaction conditions | Best performance | Ref. |
|---|---|---|---|---|
| M–NiO (M = Li, Mg, Al, Ga, Ti, Ta, with at. ratio M/Ni = 0.176) | Solution evaporation | C2H6∕O2 = 1∕1; T = 300–425 °C; W/F = 0.02–0.71 g⋅s/mL | Cmax = 66%; Smax = 90% for Nb–Ni–O at 400 °C | [25] |
| M–NiO (Al, Sn, Nb, La, Ce, Zr, K, with Ni/(M + Ni) = 0.92) | Evaporation of nitrates solution in ethanol | C2H6∕O2∕He = 3∕1∕29; T = 300–450 °C; 25 mL⋅min−1; 0.5 g cat | Smax = 87% for NiSn at 10% C2H6 conversion, 300 °C | [24] |
| Ni–M–O (M = Sn, Ti, W; 2.5, 5, 10, 15 and 20 at.%) | Solid state grinding | 10% C2H6, 5% O2 in He; T = 200–400 °C; W/F = 0.05–0.6 g⋅s/mL; 100 mg catalyst | Smax = 78% for Ni0.80Ti0.20 at 32% C2H6 conversion, 350 °C | [51] |
| Ni–M–O (M = Zr, Ti, V, Mo, W) with M/(Ni + M) = 0.05 | Citrate method | 10% C2H6, 5% O2 in He; T = 200–400 °C; W/F = 0.05–0.6 g⋅s/mL; 100 mg catalyst | S = 75% for Ni–Ti–O at 32% C2H6 conversion, 400 °C | [32] |
| NiO/TiO2 (5–92% NiO); NiO/Nb2O5 (5–98% NiO); xNi–Ti–O; xNi–Nb–O (x = 20–98% Ni) | Evaporation of ethanolic solution of precursors + oxalic acid | C2H6∕O2∕He = 3∕1∕26; T = 350–450 °C; 30 mL⋅min−1; GHSV = 1500–45,000 mL⋅g−1⋅h−1 | Smax = 84% for 80% NiO/TiO2 at 18% C2H6 conversion, 450 °C | [50] |
| 5MOx/NiO/Ni-foam (MOx = Li2O, MgO, Ga2O3, CeO2, ZrO2, MoO3, WO3, Nb2O5) | NiO/Ni-foam obtained by hydrothermal synthesis using H2C2O4, NH4Cl and Ni foam, then wet impregnation with metal salt solution | C2H6∕O2∕N2 = 1∕1∕8; T = 250–450 °C; 30 mL⋅min−1; GHSV = 1500–45,000 mL⋅g−1⋅h−1 | S = 68% for 5MOx/NiO/Ni-foam calcined at 450 °C, at 60% C2H6 conversion, 410 °C | [52] |
After the discovery, using combinatorial methodologies, of low-temperature Nb–NiO mixed oxide catalysts for ethane ODH by Liu et al. [30], Heracleous and Lemonidou [38] studied in great detail and explained the effect of Nb content on their catalytic performance. A high valence element as niobium leads to a significant decrease in NiO non-stoichiometry, while its close ionic radius to Ni2+ permits its isomorphous substitution into the NiO lattice. The results obtained were astonishing, the best mixed oxide, which corresponded to a Nb/(Ni + Nb) ratio of 0.15, exhibited a remarkable 90% ethylene selectivity at a huge 66% conversion. In order to explain such a fabulous catalytic performance, multiple characterization techniques were used.
The surface area increase recorded for the mixed oxides was attributed to the use of niobium oxalate, in agreement with recent results which pointed to the beneficial effect of oxalate anion in the synthesis route [27, 33, 40]. Also, the “mutual protective effect” can also play a significant role. The surface area increased until an optimum Nb loading (Ni0.85Nb0.15), then it gradually decreased mainly due to phase segregation and formation of large niobia crystallites. Indeed, for low Nb concentration (<15%) only diffraction lines corresponding to NiO together with a broad band ascribed to an amorphous Nb2O5 phase were observed in the XRD patterns. The increase of Nb/(Nb + Ni) ratio from 0.15 to 0.2 led to the appearance of intense lines characteristic of NiNb2O6 phase. By further increasing the Nb content, lines corresponding to crystalline niobia appeared and increased in magnitude. A gradual decrease of the lattice constant with the increase in Nb amount up to 15% was observed likely due to the substitution of Ni2+ by Nb5+. This is accompanied by a charge imbalance due to their different valence, which significantly influenced the properties of the solids. For higher Nb content the lattice constant remained unchanged, indicating an upper limit for the Nb content in the Ni–Nb solid solution.
The crystallite sizes were smaller for the mixed oxides compared to NiO, due to the “mutual protective effect” and an enhanced crystallographic disorder in the promoted catalytic systems. At higher Nb concentrations, the segregation of phases allows the particles to grow more freely leading to larger crystals. The same effect was reported for other mixed oxides [34, 51, 62].
The reducibility of mixed oxides was higher than that of pure NiO, as the main reduction signal shifted to lower temperatures, with the amount of consumed H2 linearly decreasing with increasing the Nb content. This is in line with the known irreducibility of Nb2O5 in the temperature range studied. Therefore, it was concluded that promoting with Nb leads to weaker Ni–O–Ni bonds, thus easing the reduction process.
NH3-TPD measurements revealed a strong increase in the total acidity of the mixed oxides upon doping, up to 15% Nb, then it slightly decreases. The acidic nature of Nb5+ promoter generates acidic sites of weak and moderate strength. This was explained by the Tanabe model [104] which states that by interchanging the host cation with a different valence one results in the formation of acidic centers because of charge asymmetry along M1–O–M2 bonds.
Raman spectra indicated the presence of NiO non-stoichiometric species associated with Ni3+ ions and Ni2+ vacancies [105], while the O2-TPD experiments showed the reduction of NiO non-stoichiometry upon Nb addition. XPS results confirmed the non-stoichiometry and showed similar surface compositions for solids with Nb/Ni from 0.11 to 0.43. This similarity of surface compositions, independent of the nominal content, was explained by the most energetically favorable arrangement of the surface.
In line with their physicochemical characteristics, all the mixed oxides presented a much higher selectivity to ethylene than pure NiO, with an enormous increase for just a small Nb amount present in the catalyst (from 20% to 80%). However, both selectivity and conversion increased until the Nb/(Nb + Ni) ratio of 0.15 after which the selectivity slightly decreased while the conversion plummeted to values even smaller than for unpromoted NiO, as phase segregation, reduced surface area and morphology changes occurred. Notably, the Ni0.85Nb0.15 catalyst proved to be stable on stream for almost 24 h with regard to both conversion and ethylene selectivity.
Electrical conductivity measurements represent a powerful tool able to provide valuable information about the structural defects and non-stoichiometric character in general, oxidizing species, etc. [98]. Due to oxygen vacancies in its lattice, pure Nb2O5 is a n-type semiconductor. Therefore, by doping NiO with just a low amount of Nb leads to a strong decrease in its electrical conductivity [93] as Nb acts like an electron donor, confirming the insertion of Nb into the NiO lattice and the formation of a Ni–Nb solid solution. Further concentration increments lead to a less significant decrease in conductivity (Figure 7).
Variation of the electrical conductivity (𝜎) at 350 and 400 °C of the Nb(x)NiO catalysts as a function of the Nb content x [93].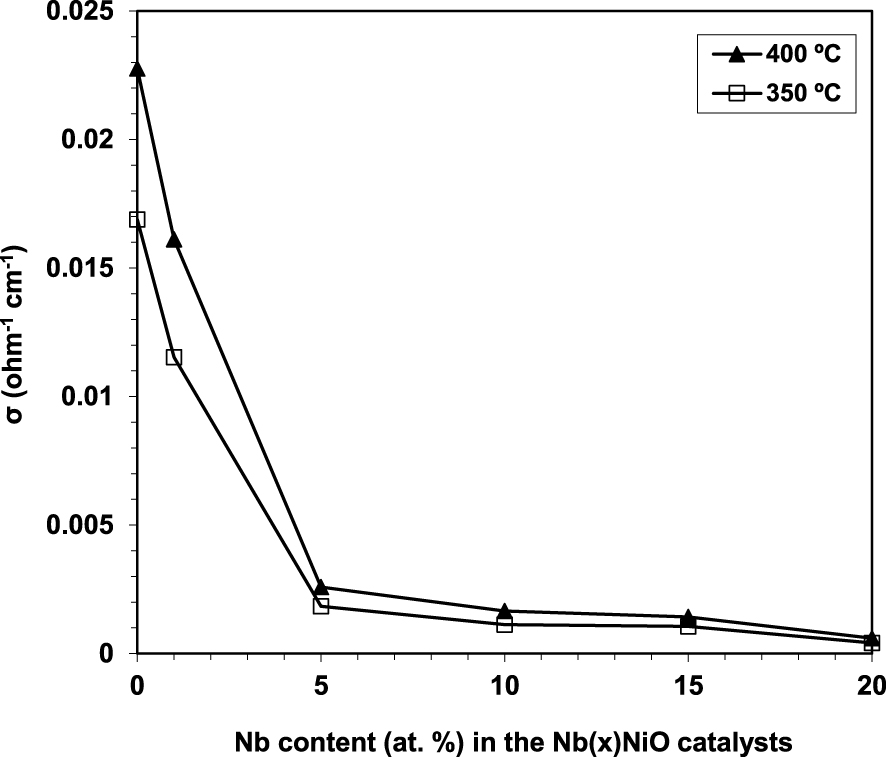
At the same time, a good correlation between the rate of ethane transformation and the electrical conductivity of the solid under air was observed (Figure 8), suggesting that the charge carriers, i.e., the positive holes associated to the lattice O− species, are involved in the catalytic oxidative conversion of ethane. However, high concentration of positive holes, i.e., lattice O− species, promotes ethane combustion rather than oxidative dehydrogenation and vice versa, as suggested by the ethylene selectivity versus the electrical conductivity under the reaction mixture dependency (Figure 9).
Rate of ethane consumption at 400 °C versus electrical conductivity under air for NiO and Nb-doped NiO catalysts (𝜎 in 𝛺−1⋅cm−1) [93]. The solid line is shown as a guide to the eye.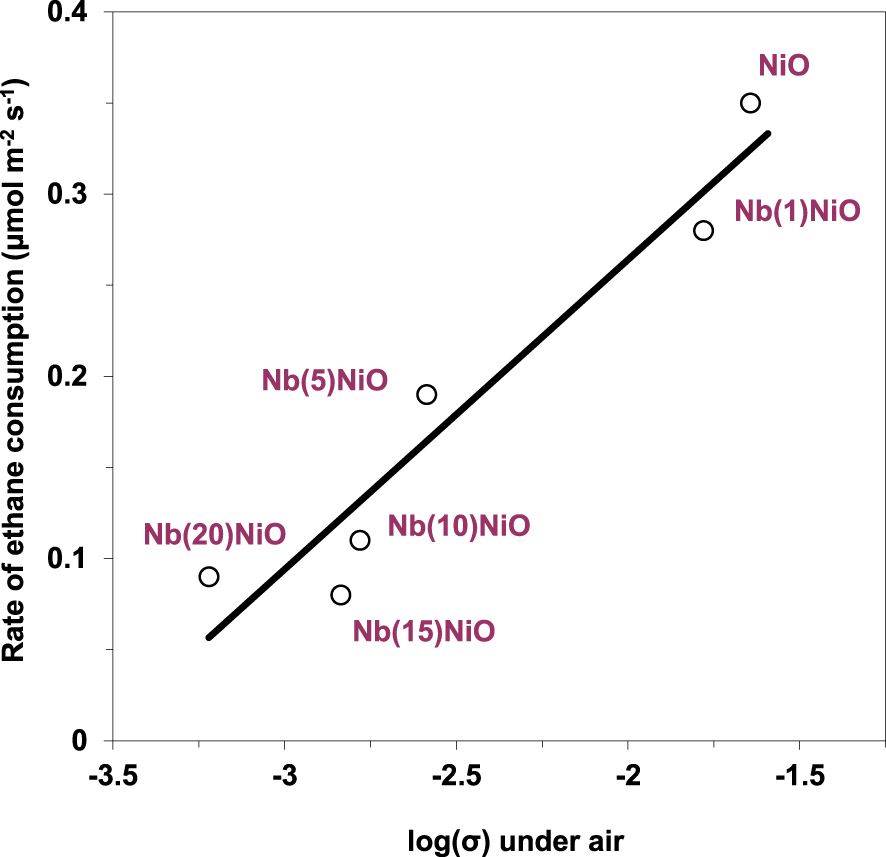
Ethylene selectivity at isoconversion at 350 °C versus electrical conductivity under the reaction mixture for NiO and Nb-doped NiO catalysts (𝜎 in 𝛺−1⋅cm−1) [93]. The solid line is shown as a guide to the eye.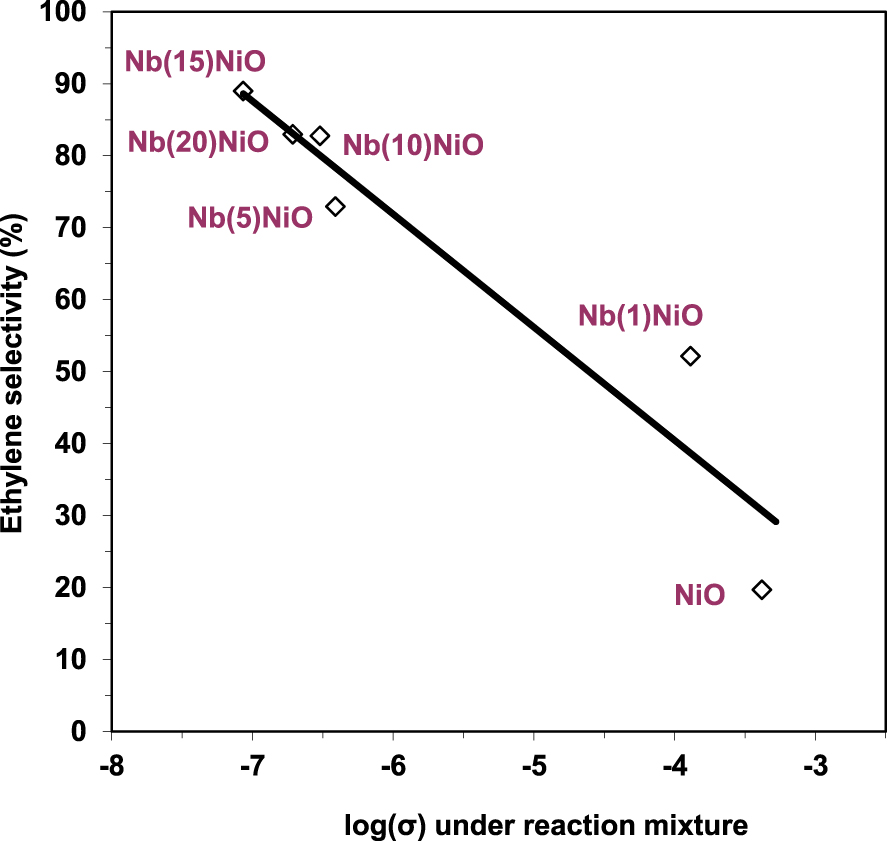
The Ni–Nb–O systems were, thus, shown to be very active and selective in ethane ODH reaction, with the selectivity almost invariant regarding the conversion increase. Similar conclusions were reached by Savova et al. [27], who synthesized Ni–Nb–O catalysts with Nb concentrations spanning from 3 to 19 at.%. The results confirmed that the catalyst with 15 at.% Nb exhibited the best activity and ethylene selectivity (33% and 78%, respectively, at 350 °C). However, EDX analyses on the Ni0.85Nb0.15 catalyst could not evidence the Ni–Nb solid solution formation in this case, and the calculated lattice constants showed no significant variation as opposed to the decreasing trend with increasing Nb content reported by Heracleous and Lemonidou [38].
The O2-TPD patterns showed at low Nb concentrations both (𝛽) and O− (𝛾 and 𝛿) species, while increasing the Nb content leads to the disappearance of the former. The Nb incorporation seemed to affect both the desorption temperature and the amount of oxygen desorbed; adding just a small amount of Nb leads to a significant drop in the desorbed amount while significantly decreasing the 𝛿∕𝛾 ratio. The authors concluded that 𝛿 O− species are responsible for the selective oxidation as they become more labile when their concentration is reduced.
Notably, contrary to the conclusions in Ref. [38], a significant decrease in conversion coupled with a slight increase in ethylene selectivity were recorded for both Ni0.93Nb0.07 and Ni0.85Nb0.15. However, the catalysts have been maintained for a much longer time on stream at 380 °C compared to Ref. [38] (over 325 h versus ca. 24 h). After about 290 h, the authors tried to regenerate the samples by passing only air at 450 °C for 30 min, but the attempt was unsuccessful. Therefore, the used catalysts were subjected to various characterization techniques, leading to the conclusion that NiNb2O6 formation and the disappearance of O− active species [106, 107] were responsible for the deactivation.
Zhu et al. [28] proposed the citrate method as an alternative preparation way for the Ni–Nb–O mixed oxides. For NiO, three protocols of synthesis were used: precipitation with oxalic acid from Ni(NO3)2 solution (Protocol 1), with ammonia from NiCl2 solution (Protocol 2) and using citric acid from Ni(NO3)2⋅6H2O (Protocol 3). The mixed oxides needed only a small amount of Nb (4–5 at.%) to peak the conversion values (ca. 31% at 350 °C), while the selectivity gradually increased until the already-established [38] optimum amount of Nb, 15 at.% (86% for a C2H6:O2 ratio = 2:1), and then it slightly decreased.
Interestingly, NiO prepared by Protocol 3 presented a smaller surface area than the other NiO catalysts, due to the presence of a denser metallic Ni phase under the form of a layer of metal-like material over the black NiO powder. This was explained by the incomplete oxidation of NiO due to the remaining citric acid, as reported elsewhere [108]. A strong increase in the catalytic performance of the citrate-prepared NiO was observed, with 57% selectivity at 42% conversion [28], much higher compared to NiO catalysts prepared using the evaporation method (20–30% selectivity at ca. 12% conversion) [27, 38] tested in similar conditions, i.e., at 350 °C and ethane-to-oxygen mol ratio of 1.
The surface area of the Ni–Nb–O mixed oxides increased up to 15 at.% Nb, for which it reached 225 m2⋅g−1, among the biggest value reported in the literature for this mixed oxide [28, 33, 37, 51, 62]. The crystallite dimensions did not vary with the Nb content, but the surface area was larger than that calculated due to the presence of a less dense, amorphous NixNbyO phase, whose concentration increases with the Nb amount. This was confirmed by EFTEM, EELS and XPS measurements.
H2-TPR measurements revealed the gradual shift to higher temperatures of the main Ni2+→Ni0 peak, indicating that the rate-limiting step in ODH, i.e., the first C–H bond breaking, becomes more and more difficult and, thus, explaining the activity loss as the Nb content increases. At the same time, the total amount of consumed H2 decreases with increasing the Nb content. This is partly because of the lower amount of NiO, but also due to fewer reducible/active oxygen species. Another explanation proposed was the limited access due to the Nb-rich phase covering the NiO crystallites, as already shown by Savova et al. [27].
Unfortunately, the solids lost their activity with time on stream: the higher the niobium loading, the higher the degree of deactivation. Therefore, the loss of activity in time was not correlated with the initial activity, but rather with the Nb content. However, at low Nb content the deactivation was well correlated with the decrease in surface area, suggesting that these catalysts retained their intrinsic activity.
Zhang et al. [52] synthesized a series of nickel foam-structured composite oxide catalysts using various metals, such as Li, Mg, Ga, Ce, Zr, Mo, W and Nb. The catalytic systems were prepared by hydrothermal growth of nickel oxalate over Ni-foam, followed by impregnation with an aqueous solution containing the salt of the required metal and subsequent calcination. The Nb-containing one, denoted as Nb2O5–NiO/Ni-foam, exhibited the best activity and ethylene yield, performing far better than the rest of the catalysts. Only W, Mo and Zr-containing catalysts managed to show a similar ODH selectivity, but at significantly lower conversion values.
The Nb2O5 concentration (within the range from 0 to 7 wt%) and, for a fixed niobia amount, the final calcination temperature were also investigated. The optimum concentration was found to be 5% Nb2O5: it showed the best selectivity values for all the temperature range and, above 400 °C, also the best conversion levels. Below 400 °C all the catalysts were almost equally active. The poor performance of the systems with very low amounts of Nb2O5 could be ascribed to total O2 conversion due to overoxidation. The ODH selectivity and ethane conversion were strongly influenced by the calcination temperature. For a fixed conversion and reaction temperature the ethylene selectivity increased with increasing calcination temperature. However, by keeping the reaction temperature constant, the conversion levels dropped with the increase in calcination temperature. This behavior was attributed to Nb2O5 aggregation and decreasing amount of active sites. The best results were observed on the catalyst calcined at 450 °C. Therefore, it was suggested that there is a synergistic interaction between NiO and Nb2O5 which is maximized by both a carefully chosen niobia loading and a proper calcination temperature. The stability on stream was studied for 50 h and, unlike previous studies [27, 28, 38], the best catalyst, 5Nb2O5–NONF-450, showed a small conversion and selectivity loss in the first 10 h, and then both were stabilized. BET, XRD and H2-TPR characterization of the used catalyst showed similar patterns compared to the fresh systems, indicating excellent structural and textural stability.
The reducibility of the catalysts was similar regardless of the niobia loading. On the contrary, when the calcination temperature was increased the reduction peaks shifted to higher temperatures, this behavior being explained by the oxygen diffusion hindrance in larger particles. Another interesting aspect was the strong decrease of the low-temperature reduction peak, associated with NiO non-stoichiometry, at high calcination temperatures, practically disappearing at more than 400 °C, while it seems independent on the Nb2O5 loading. Therefore, for a given dopant, the concentration of non-stoichiometric nickel oxide can mainly be controlled by an optimum calcination temperature.
Finally, it is worth noting that the presence of both and O− species has been highlighted by O2-TPD measurements, species considered to be responsible for over-oxidation [27, 38]. However, while the amount of O− species remained almost constant, the amount of greatly diminished with increasing the Nb2O5 loading, in perfect agreement with the increased selectivity observed which suggests that species are responsible for over-oxidation, while O− species are selective. The increase in calcination temperature decreases the amount of both species, but maintains their ratio constant. Again, a linear trend was observed relating the diminishing total oxygen area to the improved selectivity. XPS measurements confirmed the decrease of non-stoichiometry with increasing the calcination temperature [52].
Kong et al. [29] used a polyethylene glycol-assisted one-pot hydrothermal method to synthesize Ni–Nb–O mixed oxides (0–30 at.% Nb). The best catalysts, corresponding to 15 and 20 at.% Nb, exhibited very good ethylene yields of about 47% at 400 °C for ODH selectivities higher than 70%.
Only crystalline NiO phase has been observed, indicating that Nb was successfully integrated into the NiO lattice and/or is highly dispersed on the NiO surface. Smaller NiO particles were obtained upon Nb loading, partially explaining the higher activity by exposing more active centers. The lattice constant gradually decreased with Nb addition until 20 at.%, after which a small increase took place, probably due to lattice saturation in niobium ions that led to phase segregation.
Raman spectra showed bands corresponding to Ni–O and, upon Nb loading, a band with the vibration modes of bridging Ni–O–Nb bonds [109]. Furthermore, a shift of the Ni–O vibration band to higher wavenumbers was registered, pointing to a strong Ni–Nb interaction. For Nb contents higher than 10 at.%, another weak band appeared that was ascribed to Nb–O–Nb linkage vibrations, demonstrating the presence of Nb2O5 nanoparticles. Meanwhile, the band assigned to non-stoichiometric Ni–O vibrations decreased in intensity, suggesting that Nb led to a reduction of these species. This finding was confirmed by the H2-TPR profiles. Another Nb effect was the gradual shifting of the reduction peak to lower temperatures, indicating that Nb weakens the Ni–O bonds, in agreement with previous studies [38]. For the catalyst with 30% Nb, a new reduction peak appeared at 450 °C, indicating the presence of a possible NiNb2O6-like precursor as the pure material presents a close reduction temperature (480 °C) [110]. O2-TPD results confirmed the reducing amount of non-stoichiometric O− species in NiO [111]. Indeed, as Nb was incorporated, the desorption peak decreased and shifted to lower temperatures, indicating the lower amount of non-stoichiometric species.
The activity of the catalysts gradually and significantly improves with Nb addition, reaching a maximum of 67% ethane conversion for Ni0.8Nb0.2 and Ni0.7Nb0.3 catalysts at 400 °C, highlighting the potential of the hydrothermal method of preparation. On the other hand, ethylene selectivity reaches a maximum of ca. 88% at 300 °C for the Ni0.85Nb0.15 system, in agreement with previous studies [27, 28, 38]. The selectivity only slightly decreases with the increase in conversion by varying the contact time, implying that the vast majority of COx is coming from the direct oxidation of ethane. Notably, the best catalyst exhibited a maximum ethylene space-time yield greater than all the other Ni–Nb–O mixed oxides reported in the literature for the same GHSV [27, 28]. The authors tested the stability of the Ni0.85Nb0.15 catalyst for almost 60 h at 350 °C and revealed a slight decrease in conversion from 35.4% to 30.4%, coupled with a very small decrease in selectivity (from 84 to 83%) which happened after 50 h. Therefore, the hydrothermal method proved to be capable of producing more stable Ni–Nb–O mixed oxides.
Finally, it is noteworthy that polyethylene glycol (PEG) promotes a higher surface area and a better dispersion of Nb, at the same time inhibiting the formation of NiNb2O6 which is present in the catalyst without PEG [29]. Therefore, the catalyst prepared in the presence of PEG showed both better ethane conversion and selectivity towards ethylene.
The performance of Ni–Nb–O mixed oxides can be improved based on the synergy between three different metal oxides. This has been unambiguously shown by Liu et al. [30]. Indeed, using the high throughput combinatorial technology they clearly showed that the addition of small amounts of Co or Ta significantly improves the performance of NixNbyO catalysts. For the ternary NixNbyTazO mixed oxides tested in a multi-channel fixed bed microreactor, the optimum Ta content was found to be z ≈ 0.1 for a wide range of Nb/Ni ratios. In line with the expected synergistic effect, the Ta-containing ternary system was both more active and more selective than the binary Ni–Nb–O mixed oxide. Notably, the ternary mixed oxide with the composition Ni0.62Ta0.1Nb0.28O confirmed its performance in a bench scale reactor and was shown to be highly stable on stream during a few hundred hours. In spite of these excellent results, the addition of the third element to the Nb–NiO system was not much exploited to date.
Park et al. [40] used Ce (0.5, 2, 4 and 15 wt%) as promoter for the Ni–Nb–O mixed oxide (with constant 17.6 at.% Nb). As was shown in Ref. [34], Ce lowers the reaction temperature by improving the oxygen uptake. Indeed, the results showed an ethylene production rate increase of ca. 40% due to the remarkable ability of ceria to transport oxygen in a fast and efficient way to nickel oxide active sites.
Small NiO crystallites were obtained, i.e., 5–6 nm for all the CeNiNb catalysts, likely due to the low calcination temperature used. Large surface areas were obtained for all samples (130–160 m2⋅g−1), but no correlation between the Ce amount and the catalysts surface area was noticed. Since Ce is not compatible with the NiO lattice, a possible explanation is that Ce is forced to remain at the surface of the catalyst particles or in their pore network, and the surface area is mainly controlled by the Ni–Nb interaction. Segregation of CeO2 phase was confirmed for the sample with 15% Ce, with ceria particles having ca. 3 nm diameter. For lower Ce contents, it is present as highly dispersed amorphous ceria. XPS measurements suggested that cerium pushes Ni towards its higher valence state either by lattice restructuring or by directly acting upon Ni itself.
At 250 °C, the Ce-doped catalysts presented almost the same activity as Ni–Nb–O, but the selectivity significantly increased for the samples with small Ce concentrations [40]. As the amount of cerium increased, the selectivity linearly decreased, in line with previously reported results [33]. This is likely due to Ce blocking the NiO active sites when present in high amounts. For the system containing 0.5% Ce, a slight conversion increase was noticed compared to Ni–Nb–O, suggesting a different activation pathway at the catalyst surface because of NiO sites being surrounded by small amounts of CeO2. This would come in agreement with a recent study which highlighted the limited NiO reducibility when supported on Ce–Zr–O due to rapid oxygen transfer from ceria to nickel oxide [112].
By increasing the ethane-to-oxygen ratio, a significant increase in selectivity was observed, coupled with a loss in conversion, confirming the hypothesis that Ce is actually transporting the oxygen towards Ni active sites and, thus, ethane being the limiting reagent at the surface [40]. The activation energy for C2H4 formation was calculated to be significantly lower than for CO2, showing that at small cerium loading ethylene formation is thermodynamically favored. However, for the catalyst with 15% Ce the two activation energies have very similar values, indicating that ODH selectivity should not significantly vary with temperature for high cerium concentrations. The catalyst containing 0.5% Ce emerged as the best in terms of ethylene production rate, which was almost three times higher compared to that reported in Ref. [38].
3. Supported NiO catalysts
In an attempt to find a good support for NiO, Nakamura et al. [55] studied several oxides as potential supports: MgO, Al2O3, SiO2, Y2O3, La2O3 and TiO2, using a NiO concentration corresponding to 5 wt% Ni. The selectivity towards ethylene was very low for all the supports with the exception of a high surface area MgO (HS-MgO) that reached a selectivity of 54.3% at 600 °C for a conversion value of 32%. The low selectivities were explained by the reduction of NiO to metallic Ni for all the other supports, thus promoting the partial oxidation of ethane to CO and H2. Indeed, a pre-reduced NiO/HS-MgO catalyst showed increased activity but exhibited 0% selectivity towards ethylene. Therefore, maintaining Ni2+ species on the catalyst surface is of crucial importance to obtain ethylene and, for this, a high surface area support was considered essential. The authors concluded that NiO phase surrounded by NiO–MgO solid solution was the active phase for ethane ODH.
Very interestingly, the calcination temperature had a huge impact on the performance of the catalyst. Thus, calcination at 400 °C resulted in a performance comparable with the other supports, indicating a partial in-situ reduction to metallic Ni. Increasing the calcination temperature to 600 °C gave better results, while further increase to 950 °C decreases both the activity and the selectivity. These results were explained by a weak NiO–MgO interaction in the low-temperature calcined catalysts, thus being prone to a more facile reduction to Ni0, while for the optimal calcination temperature the NiO particles on the MgO support could form a NiO–MgO solid solution, but with the central part of the particles remaining NiO. By further increasing the calcination temperature, the diffusion of NiO into the support lattice to form the NiO–MgO solid solution is strongly favored.
Multiple Ni loadings have been used in order to find the optimum composition. By increasing the Ni amount from 1 to 3 wt%, the performance, in terms of both ethylene selectivity and activity, significantly increased. If the Ni amount is too high (15 wt%), the selectivity drops to 0% due to higher NiO particle sizes that prevent a more intimate NiO–MgO interaction, therefore favoring their reduction to metallic Ni under the reaction mixture. The optimum concentration was found to be 5 wt% Ni.
This catalyst, however, is not suitable for the low-temperature ODH of ethane since the reaction occurred only at temperatures above 475 °C. By increasing the temperature up to 625 °C, the activity and ethylene selectivity increased at the expense of carbon dioxide selectivity, suggesting a C2H4 formation rate higher than that corresponding to its oxidation. Notably, at low oxygen-to-ethane ratio, the ODH selectivity unexpectedly decreased, with an increase in CO and H2 selectivities. This was due to the formation of Ni0 since there was not enough oxygen to maintain the Ni species in an oxidized state. Finally, it is noteworthy that the conversion slightly decreased with a small increase in selectivity after 8 h on stream.
Al2O3 is a widely used support for various catalysts. Zhang et al. [54] used alumina obtained by six different methods (denoted S1–S6) as support for NiO in an attempt to improve the catalyst performance. Amidst these six supports, catalysts using S1 (prepared using high purity aluminum) and S2 (obtained by precipitation of Al2(SO4)3 with (NH4)2CO3) exhibited high selectivities to ethylene, the highest recorded being at 350 °C for 12% NiO loading—83.4% and 83%, respectively. The authors found that the ethylene yield was approximately proportional to the ratio between the pore volume and the surface area. They also concluded from XRD and TEM measurements that larger NiO crystals exist on the Al2O3 supports with lower pore volume (S5, prepared by thermal decomposition of Al(NO3)3, and S6, obtained by precipitation of Al(NO3)3 with urea), while highly dispersed NiO was formed on the Al2O3 supports with higher pore volume, S1–S4 (S3 was prepared by precipitation of Al2(SO4)3 with ammonia, S4 was obtained from aluminum isopropoxide by hydrolysis).
By testing two different NiO loadings, i.e., 0.12 and 0.24 g NiO/100 m2 support, respectively, the authors showed that although the higher loading samples are more active, the selectivity remains almost unchanged, therefore concluding that the selectivity was mostly controlled by the support. The catalysts with large crystal NiO had low selectivities and yields for ethylene.
H2-TPR measurements showed that there are two reduction peaks present: one at temperatures lower than 500 °C, which corresponds to the reduction of more active species, such as those found in large NiO crystallites, and another one that appears at higher temperatures that can be attributed to well-dispersed NiO strongly interacting with the support [90, 113]. The catalysts using supports S1–S2 showed only the high temperature reduction peak, confirming the well-dispersed nature of NiO and exhibiting a significantly higher selectivity to ethylene. The samples presenting the low temperature reduction peak (S5, S6) exhibit poor selectivity, while those showing both reduction peaks (S3, S4) give moderate selectivity.
The dispersion, therefore, is controlled by the pore volume of the supports. If the pore volume is low, then they are rapidly filled with the precursor salt and all the excess is subsequently transformed in large NiO crystals. Conversely, for high pore volume supports, the NiO is well dispersed in all the pores, therefore favoring better catalytic performances. So, the authors proposed that the pore volume is the determining factor for the level of highly dispersed nickel oxide, calling it “pore volume confinement effect”.
Smolakova et al. [41] used Al2O3 as support for NiO to investigate the effect of tetrahedral and octahedral Ni species in NiO/Al2O3 catalysts on the activity in ODH. Three different Ni precursors were investigated: nickel nitrate (at three different Ni concentrations, noted Ni–Al–NO3-x, with x being the Ni amount), nickel acetate (also at three different Ni contents, noted Ni–Al-ac-x) and NiO mechanically mixed with alumina, noted Ni–Al-mm-x. Indeed, a significant difference regarding the ethylene selectivity was observed among these samples, the best catalysts coming from the nickel nitrate precursor and exhibiting a remarkable 86% ethylene selectivity at 10% conversion and a reaction temperature of 500 °C. The XRD patterns for these catalysts showed only very week diffraction peaks assigned to NiO, therefore a high dispersion of Ni, while the other solids exhibited NiO with good crystallinity and large particles (8–25 nm, increasing with Ni loading).
The UV–Vis spectra highlighted the presence of tetrahedrally coordinated Ni species, Ni–Al–NO3 presenting a higher concentration than the other catalysts. Moreover, bands assigned to Ni in tetrahedral and octahedral sites [114] were also evidenced, with Ni–Al–NO3 presenting octahedrally coordinated nickel species inside the alumina lattice, while Ni–Al-ac exhibited bands specific for Ni in octahedral sites in the NiO lattice. Also, the UV–vis spectra indicated the presence of non-stoichiometry in NiO. These findings were also supported by the H2-TPRmeasurements, the presence of tetrahedral Ni species in NiO lattice and octahedral Ni species in Al2O3 lattice being confirmed from the high temperature reduction peaks.
The catalytic performance of these systems was strongly influenced by the preparation method. The solids prepared by mechanical mixing showed low activity and selectivity. The Ni–Al-ac samples were more active, but less selective than Ni–Al–NO3. The activity of Ni–Al–NO3 increased with increasing the Ni content, reaching ethylene selectivity values around 84–86%. The catalytic behavior of Ni–Al–NO3 with increasing Ni concentration was explained by the increasing amount of tetrahedrally coordinated Ni and non-stoichiometric species. On the other hand, the decreasing trend observed for Ni–Al-ac is ascribed to the increased presence of crystalline NiO particles of larger sizes.
Since alumina-supported NiO exhibited promising catalytic performances, Park et al. [63] used silica as a dopant for Al2O3 in order to tune the acidity and surface area of the catalysts, thus modulating the activity and ODH selectivity. The modified alumina, named SDA, contained 5% silica. Among the catalysts tested, the best performance was exhibited by the 16% NiO supported on SDA calcined at 700 °C, reaching 80% selectivity to ethylene, but at only 5% conversion at 400 °C.
The XRD patterns of the solids calcined at 400 and 500 °C indicated a good dispersion of NiO on the surface, as well as very small crystallite sizes. However, for NiO supported on SDA calcined at 1100 °C, very weak peaks corresponding to NiO are visible for 16% NiO loading, with crystallites smaller than 5 nm, much smaller than those reported in other similar studies [41, 54].
Characterizing the catalysts by nitrogen adsorption isotherms it was observed that there are large changes regarding the pore volume corresponding to NiO supported on alumina and SDA calcined at 700 °C, indicating that the vast majority of NiO particles were formed in the pores of the support. However, for NiO supported on SDA calcined at 1100 °C the pore volume remains almost the same after NiO deposition, implying that a part of NiO is deposited on the external surface. This can also explain why for this particular catalyst NiO diffraction lines were observed.
Using H2-TPR, the authors highlighted the presence of weakly bound and non-stoichiometric NiO sites corresponding to the low temperature (around 280 °C) reduction peak [95, 115]. The second peak, however, shifted to higher temperatures with the addition of silica to Al2O3, the hardest to reduce being NiO supported on SDA calcined at 700 °C. Since the ODH mechanism relies on the reducibility of the active species, this explains why this catalyst presented the lowest activity coupled with the highest selectivity. The third peak present, at around 750 °C, was assigned to the presence and reduction of a nickel aluminate phase.
NH3-TPD revealed that SDA support calcined at 700 °C is the most acidic, followed by alumina and SPD calcined at 1100 °C. The difference in acidity between the SDA calcined at different temperatures is in accordance with the reduction of acidity with increasing the calcination temperature [116]. NiO deposition did not influence acidity, except for the SDA calcined at 1100 °C for which the acid site concentration increased by 17%, due to easily accessible acid sites on the surface, in accordance with the findings of Stanislaus et al. [117].
As stated earlier, NiO supported on SDA calcined at 700 °C presents the highest ODH selectivity, but the lowest activity and is characterized by highest acidity, surface area and amount of NiAl2O4 phase. On the other hand, NiO supported on SDA calcined at 1100 °C presents the highest conversion rate, but the lowest ethylene selectivity. This was explained by the presence of a NiO phase with larger particles that are more active, but less selective. Very interestingly, these catalysts have activities that are not correlated with the surface area; they rather present an inverse trend than usually observed [54].
The authors concluded that the concentration of the acid sites and the surface area play the biggest role in modulating the catalysts performances. By Si-doping the alumina, Ni species become harder to reduce, but their tendency to form superficial NiAl2O4 phase increases. These newly formed sites reduce the concentration of unselective electrophilic oxygen species, favoring the ODH selectivity [37]. However, for a 10% selectivity increase the activity halves, which is, probably, the effect of the increased support acidity. This strengthens the Ni–Al interactions, further reducing the reactivity. Therefore, high selectivity correlates with high acidic sites concentration and large surface area, which favor the apparition of spinel-like type sites.
An interesting explanation for the spinel-like phase influence was proposed by Zhang et al. [118], implying that Si atoms may fill the Al2O3 lattice vacancies. This way, Ni can no longer access the support vacancies, thus forming NiO on the surface and leading to a larger amount of NiAl2O4 spinel phase present at the surface of the catalyst.
Since the morphology and topology of the support play an important role regarding the catalytic performance of a catalyst, Solsona et al. [58] used silica with a Pillared Clay Heterostructure (PCH) as a support for NiO. PCH were obtained by increasing first the interlayer spacing through ion exchange of the clay cations with bulkier ones, then adding a silicon source to form the pillars between the clay layers, resulting in porous, high surface area materials.
After testing a typical SiO2-supported NiO catalyst (5% NiO), for which poor ODH selectivity (20%) was recorded, the authors prepared pillared clay silica that presents a higher surface area and favors a better NiO dispersion on the surface, leading to smaller NiO crystallites which have been recently shown to favor ethylene selectivity [24, 28]. Two NiO loadings where chosen: 5% NiO for having the same NiO concentration as the non-PCH catalyst and 17% NiO for the same NiO surface coverage as the non-PCH catalyst. The results showed a significant increase in both activity and ODH selectivity for both catalysts (55–60% compared to 20%) and, more importantly, its invariance with conversion, as was the case for the non-PCH catalyst.
Due to titania being acidic and capable of maintaining the PCH structure if inserted into it, a catalyst with 17% NiO and titania was synthesized, which exhibited a remarkable 80% ethylene selectivity. Therefore, NiO loadings from 1 to 80% were used with this support. The selectivity to ethylene increased from 60% for 5% NiO to 79% for 17% NiO, but further NiO content increase resulted in lower ODH selectivities. The results were partially explained by the reduced NiO particle size (7 nm for PCH and Ti-containing PCH-supported NiO catalysts) compared to the typical SiO2-supported catalyst (13 nm). However, a clear correlation between ethylene selectivity and crystallite sizes was not attainable, suggesting that other aspects must be taken into consideration, such as various morphological parameters and structural defects that can influence the nature of active O species [119].
XPS measurements showed the presence of non-stoichiometric Ni species, as well as ligand-metal charge transfer [120], a linear correlation being found between the non-stoichiometric species and ethylene selectivity [58]. This clearly shows the positive effect of the support by reducing the NiO non-stoichiometry and, thus, promoting ODH selectivity.
The PCH-supported catalysts were shown to be less reducible than NiO supported on typical silica, with Ti-containing PCH systems exhibiting the highest reduction temperatures in H2-TPR experiments and, in line with this, the most selective. Even more, the reduction temperature shifts to higher values with increasing the NiO content up to 17%, the sample that exhibited the highest ODH selectivity, after which further addition of NiO leads to a slight increase in reducibility. This was attributed to the strong NiO-support interaction due to small particle sizes and high dispersion, which significantly hampers the reduction process and results in higher ethylene selectivity.
A further confirmation of the reduced non-stoichiometry comes from 18O2 isotopic exchange. A high amount of 16O18O accounts for a high concentration of surface electrophilic oxygen species (, O−), while the formation of 16O2 is associated with active lattice oxygen species. All the PCH-supported NiO catalysts formed a higher amount of 16O2 than non-PCH ones and the release temperature for both 16O2 and 16O18O was higher, indicating less reactive, but more selective oxygen species. This comes in good correlation with the ODH mechanism, known to be of Mars-van Krevelen type, which proceeds using mainly the lattice oxygen species. It is important to mention that the ethylene selectivities exhibited by the Ti-containing PCH-supported NiO are very close to those presented by Ni–Nb–O system [36, 96].
For a better comparison, Delgado et al. [62] used not only SiO2 and Ti-containing PCH, but also anatase TiO2 and a high surface area TiO2 from Degusa (P25) as supports for NiO. The optimum amount of nickel oxide was chosen for each support in accordance with previous studies [58]. The results were in good agreement with data from literature, the most selective catalysts being the Ti-containing ones. NiO/TiO2-P25 exhibited the highest selectivity (ca. 90%) while NiO/PCH-Ti was the most active, although not as selective (around 80%). Unsupported and silica-supported NiO performed very poorly. The selectivities were almost constant when the conversion varied at a fixed temperature, indicating that CO2 comes directly from ethane but not from ethylene oxidation.
With the exception of NiO/SiO2, all the catalysts were less reducible than NiO. However, the particular phase distribution characteristic for each catalyst can play a major role in the shape of the reduction pattern. This is illustrated by NiO/PCH-Ti catalyst for which particles found in the interlayer pores are harder to reduce than the surface exposed ones. Peaks found at higher temperatures, absent from the unsupported NiO profile and especially present for NiO/TiO2-P25 and NiO/SiO2, are ascribed to NiO particles well interacting with the support. For pure NiO, NiO/PCH-Ti and NiO/SiO2 catalysts a small, low temperature peak is also present, indicating the presence of non-stoichiometric Ni species that are usually correlated with over-oxidation to CO2. Among Ti-containing catalysts a low reducibility seems to be correlated with high ethylene selectivity, an inverse trend compared to other data from literature regarding Al2O3-supported NiO [54, 63].
By evaluating the reaction kinetics of reduction in hydrogen using XANES measurements it was found that Ti-containing materials presented the slowest reduction kinetics, in line with H2-TPR results and with the best selectivity exhibited. This was explained by a more restrained oxygen supply that would avert deep oxidation.
From the information offered by XPS regarding the Ni 2p3∕2 spectra, the intensity ratio between satellite peak I (Sat-I) peak and main peak is generally used to obtain information about the surface species [102, 121]. As could be expected in the light of the aforementioned results, this ratio is much smaller for the supported catalysts compared to pure NiO. However, for the Ti-containing catalytic systems the selectivity to ethylene seems to be favored by higher Sat-I/main peak ratios, a linear correlation being obtained. This again is opposed to what is reported in the literature [24, 28, 33, 39, 40, 52]. The authors considered that the defects play a critical role in the activity of those materials.
Using EXAFS measurements, the Ni–O and Ni–Ni coordination numbers, corresponding to the first and second coordination shells, respectively, were calculated. It was highlighted that the supported catalysts exhibit lower coordination numbers than unsupported NiO, the effect being more significant for Ti-containing systems, probably due to a higher NiO–TiO2 interaction that may generate more Ni and O vacancies. Moreover, a linear trend was obtained for the Ti-containing catalysts regarding the increased ODH selectivity with a decrease in the second shell coordination number, pointing out the correlation of ethylene selectivity with the disappearance of non-selective sites by the creation of Ni and O vacancies.
Recent research involving the NiO catalyst used titanium either as a promoter or as support in the form of titania. Sanchis et al. [59] used TiO2 as support from three different sources (pure anatase, A; Degussa P25, B; nanocrystalline anatase, C) and exhibiting low (A), intermediate (B) and high (C) surface areas in order to optimize NiO catalytic properties. The results were remarkable, NiO supported on the B and C supports reaching around 90% selectivity to ethylene at more than 10% ethane conversion at 450 °C for the optimized NiO loadings. Nevertheless, the ODH selectivity strongly varies as a function of the characteristics of the catalysts: the best selectivity for each support was achieved for a different amount of supported NiO, according to the surface area of the support: for low and intermediate surface area the optimum loading was below and around 20% NiO, respectively, while for the high surface area support it was at 50% NiO. However, these results were obtained for an ethane/oxygen ratio of 3/1 and, therefore, the risk of running out of oxygen and, thus, modifying the nature of NiO active species and the reaction mechanism, was real. By using an ethane/oxygen ratio of 1/1 the activity strongly increased, while the selectivity decreased to ca. 74% for the catalyst with 50% NiO deposited on support B. In this way, an impressive ethylene yield of ca. 41% was achieved. Notably, the stability of the catalyst containing 20% NiO supported on support B was tested on stream for 26 h. A small decrease in activity was recorded in the first couple of hours, together with a small increase in ethylene selectivity, then the catalytic performance became stable.
Using XPS analysis it was shown that the surface amount of NiO is higher than the theoretical one and increases with the increase of the support surface area, the Ni/(Ni + Ti) ratio reaching 0.54 for 20% NiO on high surface area titania. Also, for the higher surface area supports (B and C), different concentrations of surface cation vacancies, as well as particle sizes and even structural transitions were observed [119]. H2-TPR measurements showed a low temperature (<300 °C) reduction peak that was attributed to poorly dispersed NiO particles, and a high temperature (>300 °C) one that was assigned to particles presenting an intimate contact and interaction with the support [90]. The reduction temperature decreases with increasing the surface area of the support, indicating a higher reducibility of the NiO particles.
By correlating all this information, the low selectivity of low surface area catalysts is due to a low amount of NiO interacting with titania, the large crystallites acting more as in pure NiO. Also, in the higher surface area samples, an excess of NiO loading relative to the surface area leads to free, non-interacting NiO particles. The optimum performance was obtained with catalysts having NiO with some interaction with TiO2, showing core/shell agglomerations with TiO2 particles inside and NiO particles outside.
TiP2O7 was demonstrated to be among the best catalysts for butane ODH [122, 123]. Taking this into consideration, as well as the benefic effect of P seen in Ref. [49], Ivan et al. [60] used titanium pyrophosphate as support for NiO. Five different Ni(x)TiP catalysts were used, with x being the Ni loading ranging from 10 to 26 wt%. The catalysts exhibited good performances, with Ni(15)TiP being the most selective (ca. 86%) at ca. 5% conversion (Figures 10 and 11). However, as NiO loading increased the starting reaction temperature decreased while the conversion and selectivity at lower temperatures increased.
The catalytic performances in ethane ODH of bulk NiO and Ni(x)TiP catalysts. Closed symbols and dotted lines: ethane conversion; open symbols and continuous lines: ethylene selectivity [60].
Ethylene selectivity versus conversion variation in ethane ODH over Ni(x)TiP catalysts (x = 18, 22 and 26) at 400 °C (O2-to-C2H6 mol ratio = 1) [60].
XRD patterns showed that once Ni was added to the support the NiO characteristic lines began to appear and increase in intensity, suggesting that the NiO particle size increased. However, the crystallite dimensions remain significantly lower compared to other supported NiO catalysts at similar loadings.
The basicity of the catalysts was determined by CO2-TPD measurements. Each characteristic peak corresponds to a specific basic site strength, while the area is indicative of the amount of basicity [124]. For all the materials, except Ni(26)TiP, only two bands appeared, one attributed to medium-strength basic sites, leading to bidentate carbonate, and another one for strong basic sites, with O2− being their chemical equivalent that led to monodentate carbonate. Ni(26)TiP exhibits two additional peaks, one for weak basic sites which are found as superficial hydroxyl groups that led to bicarbonate species, and another one for very strong basic sites, most probable found as unsaturated and more reactive O2− species and maybe even O− species [125]. Since the support was shown to be an acidic material [123], all the basicity came from NiO, a fact confirmed by the linear trend of total basicity as a function of NiO loading. Notably, for the used Ni(26)TiP catalyst, the amount of basic sites dramatically decreased. H2-TPRmeasurements showed that the starting reduction temperature decreases with increasing the NiO loading suggesting an increased reducibility of NiOx species. However, the reduction temperatures were higher than for silica-supported NiO [126], suggesting stronger metal-support interactions in Ni(x)TiP systems. The hydrogen consumption corresponding to the first two peaks was perfectly correlated with the theoretical amount necessary for full Ni2+→Ni0 reduction, while the amount corresponding to the rest of high temperature peaks were attributed to different degrees of Ti4+ reduction, the latter increasing with increasing the NiO loading. This was explained by the ability of the newly formed metallic Ni to favor the dissociation of molecular H2. Nevertheless, the reduction of the support starts at much higher temperatures than those used for catalysis, therefore it did not affect the catalytic performance.
Since the support becomes catalytically active at temperatures above 450 °C, all the activity of the studied materials came exclusively from supported NiO. As NiO loading increased so did the conversion, for Ni(26)TiP reaching levels comparable with pure NiO at 425 °C. The selectivity was only slightly affected by the temperature increase for Ni(15)TiP and Ni(18)TiP, while for the solids with higher amounts of NiO the decrease was more evident (Figure 10). The increase in intrinsic rate of ethane conversion with increasing Ni concentration was perfectly in line with the H2-TPR findings, since the reducibility of NiO particles, as well as their density, increased.
By varying the contact time for the catalysts with the three highest NiO loadings it was observed that ethylene selectivity became less affected with increasing NiO content. Notably, by extrapolating the linear trend to 0% conversion none of the catalysts exhibited 100% ethylene selectivity, clearly indicating that CO2 still is a primary product of ethane oxidation (Figure 11).
Interestingly, the selectivity increased with increasing the basicity as opposed to what Lopez-Nieto et al. [24] found, demonstrating a correlation between the surface acidity of doped NiO and ethylene selectivity. However, the reasoning behind this correlation was the blocking of non-selective sites by ethylene strong adsorption, and this worked only for the sites responsible for CO2 formation as a primary product [24]. In the case of Ni(x)TiP catalysts, a higher basicity favors a higher ethylene desorption rate, thus preventing it from being overoxidized. Compared to other NiO-supported catalysts, with 78% ethylene selectivity at ca. 10% ethane conversion, the Ni(26)TiP system shows comparable selectivity at isoconversion with NiO supported on alumina [53] and on a porous clay heterostructure with silica-titania pillars [62].
The stability of the best catalyst, Ni(26)TiP, was tested for 900 min at two temperatures, 400 and 450 °C, the latter also being the calcination temperature. If at 400 °C no change was observed during the entire period, at 450 °C the conversion dramatically decreased, accompanied by a small increase in ethylene selectivity. The strong metal-support interaction effect was eliminated by the lack of effect of air treatment for 4 h at the same temperature. Since the small decrease in surface area after the catalytic treatment cannot solely explain this activity loss, surface diffusion of excess phosphorous [122, 123] from the support onto the NiO surface is the most probable explanation. This hypothesis was supported by the huge drop in basicity observed.
Sakitani et al. [61] compared low (LSZ) and high (HSZ) surface area zirconia as support for NiO catalyst. A weak NiO-support interaction leading to a less stable active phase, which can be reduced to metallic Ni during the ODH reaction, was observed for the catalyst supported on LSZ, with negative consequences on the ODH performance: complete oxidation prevails and, with increasing reaction temperature in the range 400–500 °C, a significant decrease of ethylene selectivity to the benefit of CO and H2 takes place. On the other hand, NiO/HSZ catalyst was characterized by a stronger NiO-support interaction being, thus, more stable compared with NiO/LSZ. Therefore, it was active in ethane ODH, both ethane conversion and ethylene selectivity increasing with the reaction temperature in the range 400–500 °C. At 500 °C, the conversion level and ethylene selectivity were 30 and ca. 60%, respectively. Although stable on stream, NiO/HSZ contained partly reduced metallic Ni after reaction. To further improve the NiO/HSZ system, different modifiers, such as Mo, V, Sb, Bi, Cu, Co, Ce, La, Nd and P, were added by co-impregnation. The addition of Mo, V, Sb or Bi led to the loss of activity (conversion levels lower than 2.5% at 450 °C), although accompanied by an increase of ethylene selectivity in the case of Mo and Sb. The addition of Cu resulted in a decrease of conversion from ca. 25% for NiO/HSZ to ca. 18% at 450 °C, but with a significant increase of the selectivity to CO2 at the expense of ethylene. Co, Ce or La did not produce significant changes in the catalytic performance, while Nd and P had a positive effect on both ethane conversion (which increased from ca. 25% to 32 and 29% for Nd and P, respectively) and ethylene selectivity (which increased from ca. 56% to 58 and 62% for Nd and P, respectively). With this, the benefic effect of phosphorus as additive for ODH reaction has been again confirmed. An optimal loading of 0.52 wt% P (P/Ni = 0.04) was found, which gave 32% ethane conversion with ca. 64% ethylene selectivity at 450 °C. The authors showed that the addition of P to NiO/HSZ catalyst enabled the Ni species to remain in an oxidized state during the ODH reaction, their reduction to metallic Ni being, thus, avoided.
Bortolozzi et al. [64] used ceramic papers comprised of silica-alumina fibers as a novel support for unpromoted and Ce or Zr-promoted NiO. In order to bind the fibers and the active phase together, several binders were used in the form of CeO2, ZrO2 or Y2O3-stabilized ZrO2 nanoparticles. The papers presented an interconnected 3D network composed of ceramic fibers formed during calcination, with pore-resembling spaces spanning from 10 to 100 μm. The addition of active phase produced insignificant morphological changes.
After testing the plain papers as well as ceramic paper-supported Ni–M–O in the ODH reaction, it was revealed that all the activity comes from the NiO phase, the papers showing insignificant levels of conversion even at the highest reaction temperature studied. Promoting NiO with cerium led to the highest conversion levels, but moderate ethylene selectivities, while using Zr as doping agent led to best ODH selectivities in the absence of CeO2 binder, but to conversion values smaller than for unpromoted catalysts. This can be partially explained by inferior oxygen transport efficiency and storage capacity of ZrO2 compared to CeO2 [127].
The binder effect among the unpromoted samples is opposed to that observed among the mixed oxides, the sample with ceria binder presenting the highest selectivity. This infers an interaction between the NiO active phase and the binder agent, as well as its high dispersion on the CeO2 coverage. However, ceria as binder has a negative effect on the selectivity of mixed oxides-containing ceramic papers, with Ni–Zr–O being the most affected.
Promoting with both Zr and Ce led to increased values for ethylene productivity compared to unpromoted catalytic systems. However, Zr-promoted catalyst with ceria binder exhibited the worst ethylene productivity, less than half of the Ni–Ce–O with zirconia binder. This may arise from different Ni/Zr and Ni/Ce ratios along with different morphology, as it is known that different catalytic performances are obtained for promoted and supported NiO catalysts [62]. A proposed explanation was partial Ce substitution by Zr, leading to a Ce–Zr solid solution capable of enhanced oxygen storage capacity, that would be negative for the ODH reaction [128, 129]. All the materials were highly stable for more than 20 h on stream, revealing no significant changes in ODH selectivity and ethane conversion.
For NiO and Ni–Ce–O, the level of dispersion is dependent on the binder used, as the ZrO2 and Y2O3-assisted ZrO2 showed spherical particle groups which were characterized by higher order and compactness, partially explaining their better catalytic performance compared to the catalysts with CeO2 binder. In comparison, Ni–Zr–O papers exhibited active phases grouped in larger aggregates of square forms.
EDX analysis on the unpromoted NiO-containing papers showed that M/Ni ratio was much higher in the case of the catalyst with CeO2 binder, a possible indication of a better Ni–Ce interaction as it exhibited the best catalytic performance in this group. Yttria-assisted ZrO2 binder proved to be superior to pure ZrO2, as Ni/ZrY papers presented a higher productivity compared to Ni/Zr ones, even if the Zr/Ni ratio was similar. This was also observed in the case of Ni–Zr–O papers. The Ni–Ce–O with Zr-containing binders showed a Ce/Ni ratio close to 0.17, value reported by the authors in a previous study to be the optimal one [130], while the catalyst with CeO2 binder presented a very high Ce/Ni ratio (1.45), thus explaining the significant difference in the catalytic properties.
The XRD analyses highlighted the reduction of NiO crystallites size when Ce was used as promoter for Zr-containing binder papers, while the CeO2 binder favored larger crystallites due to the higher Ce/Ni ratio, which leads to a higher degree of oxide segregation. A correlation was made regarding the particle dimensions and the catalytic performance as the best catalyst from the Ni–Ce–O group, NiCe/ZrY-P, presented the smallest NiO and CeO2 particles. In the case of Ni–Zr–O catalysts no diffraction lines corresponding to ZrO2 were found suggesting a very good dispersion and Ni–Zr interaction, in agreement with Wu et al. [131] showing that Zr is able to substitute Ni in NiO lattice.
Raman spectra suggested a certain degree of Ni–CeO2 interaction that leads to weaker Ni–O bonds, in accordance with the higher activity observed. On the other hand, the promoted catalysts that did not contain CeO2 as binding agent showed the main band shifted to significantly higher wavenumbers, indicating the incorporation of the promoters into the lattice with the formation of new active species for ethane ODH as their catalytic performance significantly improved.
Cordierite monoliths present several advantages over fixed-bed reactors, including a high geometric surface area, minimization of hot spots runaways and a lower pressure drop, and, hence, they were used to disperse Ni/Al2O3 [56]. Three types of catalysts were, thus, prepared, each with 15 wt% Ni: Ni/Al2O3(C)-Cor—colloidal alumina deposited onto cordierite monolith followed by deposition of nickel nitrate and subsequent calcination, Ni/Al2O3(S)-Cor—mixture of powder and colloidal alumina deposited onto cordierite monolith followed by deposition of nickel nitrate and subsequent calcination, and Ni/Al2O3(P)-Cor—powder Ni/Al2O3 deposited onto cordierite monolith. Among them Ni/Al2O3(S)-Cor exhibited the best catalytic performances, amassing a selectivity higher than 70% even at 30% conversion. Ni/Al2O3(C)-Cor presented similar conversion values, while Ni/Al2O3(P)-Cor was the least active with a conversion lower than 10% at 500 °C. The conversion increases at the expense of the selectivity for all the catalysts, indicating that ethylene overoxidation proceeds to some extent. Nevertheless, their behavior is different: by varying the contact time, Ni/Al2O3(P)-Cor showed the steepest decrease, while the other two catalysts presented a similar, flatter selectivity drop.
The differences in catalytic activity were explained by the different accessibility of reactants to nickel active sites. For the most active catalyst, Ni/Al2O3(S)-Cor, NiO species were well dispersed on the Al2O3 support and showed strong Ni-support interactions, as SEM-EDX, XPS and Raman measurements showed. Nickel sites were accessible to the reactants, therefore good conversion and selectivity values were obtained. For Ni/Al2O3(C)-Cor, due to Al2O3 support particles entering the macropores of the cordierite monolith, some of the Ni species were not accessible to the reactants, therefore the ODH performance was diminished compared to Ni/Al2O3(S)-Cor. For the Ni/Al2O3(P)-Cor sample, some of the Al2O3 support particles were placed during the synthesis on top of the catalytic coating containing NiO, therefore not allowing the reactants to reach the active sites and decreasing the activity. The covering of the surface catalytic coating also leads to an increase in Brønsted and Lewis acidity, favoring ethylene decomposition.
In an attempt to further improve the catalytic performance, the authors modified the Ni/Al2O3(S)-Cor system with La during impregnation, in order to create four Ni–La–O mixed oxides (noted xLaNi-M, with x being the theoretical La/Ni molar ratios ranging from 0.09 to 0.25) [42]. The actual ratio, however, was determined by EDX analysis to vary from 0.01 to 0.11. Lanthanum incorporation led to an increase in conversion at the expense of ODH selectivity. The best catalytic system in terms of ethylene yield appeared to be the 0.08LaNi-M, showing almost three times increase compared to the unpromoted catalyst—13.4% versus 4.9%.
Nevertheless, the conversion does not follow a linear trend with the amount of lanthanum, the highest conversion at 400 °C being attained by the same solid, 0.08LaNi-M (ca. 25%). The ethylene selectivity decreases with increasing the La concentration, as well as with temperature. Interestingly, for all catalysts, except 0.01LaNi-M, the selectivity remained almost constant at temperatures exceeding 350 °C. Since 0.01LaNi-M presented a negative slope in the selectivity versus temperature plot, indicating that overoxidation plays an important part, as was the case for the unpromoted catalytic system, it can be concluded that by increasing the La concentration the catalysts become more active towards ethane than ethylene. One possible explanation for lanthanum addition reducing the selectivity was proposed to be the weakening of the Ni-support interactions by the added promoter.
From the Raman, XPS, FTIR and H2-TPR measurements the authors suggested a weakening of the nickel-alumina interactions possibly due to the contribution of Ni–La and La–Al2O3 interactions. Since no bands attributed to La2O3 were observed, it was suggested that La freely disperse in 𝛾-Al2O3 at La/Al ratios between 0.037 and 0.19, in agreement with Haack et al. [132], with the investigated systems having La/Al ratios smaller than 0.04. The formation of new, more reducible nickel species was evidenced by H2-TPR, and correlated well with the increased activities of the La-modified catalysts. It was concluded that increased conversion at the expense of ethylene selectivity is partially due to La easing the NiO reducibility by weakening its interaction with alumina and facilitating the presence of unselective oxygen species.
Recently, the same group studied the effect of the geometry and nature of the ceramic structure on the catalytic performance of NiO/Al2O3-coated structured catalysts [57]. Thus, three different ceramic structures, i.e., a cordierite monolith, an alumina foam and an alumina-silica paper, were used as ceramic substrates for the deposition of the NiO–Al2O3coating to obtain the NiO–M, NiO–AF and NiO–CP structured catalysts, respectively. For comparison purposes, a powder NiO/Al2O3 catalyst, noted NiO-P, with similar NiO amount, was prepared by the wet impregnation with a nickel nitrate solution. It has been shown that the nature of the ceramic substrate strongly influences the distribution of the catalytic coating and the properties of the NiO active phase. Thus, better distribution and stronger NiO–Al2O3interactions were observed for the NiO–M and NiO–AF systems, which gave high ethylene selectivities (85 and 79%, respectively) with similar conversion levels (6.1 and 6.9%, respectively) at 400 °C. Notably, the ethylene productivity was higher for NiO–AF than for NiO–M catalyst, i.e., ca. 232 versus 150 g/kgNi∕h. These performances were superior compared to NiO-P system, which showed 4% conversion, 77% ethylene selectivity and 115 g∕kgNi∕h productivity, clearly demonstrating the superiority of these structured catalysts. On the other hand, a heterogeneous distribution of the NiO active phase with NiO agglomerations along the fibers, and poor NiO-support interaction were noticed for the NiO–CP system, which displayed the highest conversion level (9.9%) and the lowest ethylene selectivity (ca. 27%) and productivity (113 g∕kgNi∕h) in the same reaction conditions. It is worth noting that the addition of Ce to the NiO–M and NiO–AF systems resulted in a significant increase of conversion but with an ethylene selectivity drop. However, a significant increase of the ethylene productivity for both NiCeO–M and NiCeO–AF systems was noticed. Notably, the effect of cerium addition on the ethylene selectivity in the supported systems is opposite compared to that observed in bulk NiO.
The different supported NiO catalysts hereby described are summarized in Table 4 together with their preparation methods, reaction conditions and catalytic performance in ethane ODH into ethylene.
NiO catalysts deposited on different supports for ethane ODH
| Catalyst | Preparation method | Reaction conditions | Best performance | Ref. |
|---|---|---|---|---|
| NiO/MgO (1, 2, 3, 5, 10 and 15% Ni) | Impregnation of MgO with Ni(acac)2 | C2H6∕O2 = 1∕2 – 1/1 (molar); 30 mL⋅min−1; T = 450–600 °C; 60 mg catalyst | Smax = 54.3% for NiO/HS-MgO calcined at 600 °C, at 32% C2H6 conversion | [55] |
| Ni/Al2O3 (0.12 and 0.24 g NiO/100 m2 support) | Impregnation of Al2O3a with Ni(NO3)2 | 10% C2H6, 10% O2, 80% N2 (molar); GHSV = 338 mL C2H6 g−1⋅h−1; T = 350–450 °C; 0.5 g catalyst | Smax = 65.3% for 0.24NiO/S2, at 450 °C and 59% C2H6 conversion | [54] |
| Ni/Al2O3 ( ∼11, 16 and 20 wt% Ni) | Impregnation of Al2O3 with either Ni acetate or nitrate; Mechanical mixing of NiO and Al2O3 | 8% C2H6, 3% O2, 89% He (molar); 100 mL⋅min−1; T = 500 °C; 0.05–045 g catalyst | Smax = 86% for Ni–Al–NO3-19.7, at 500 °C and 10% C2H6 conversion | [41] |
| NiO/Al2O3-5% SiO2 (8% and 16% NiO) | Impregnation of silica-doped alumina (SDA) with Ni(NO3)2; support calcined at 700 and 1100 °C; catalysts calcined at 400 and 500 °C | 10% C2H6, 10% O2, 80% N2 (molar); 10 mL⋅min−1; W/F = 0.6 g⋅s⋅mL−1; T = 300–500 °C; 0.5 g catalyst | Smax = 80% at 5% C2H6 conversion and 400 °C for 16%NiO/SDA700 calcined at 400 °C | [63] |
| NiO/PCH (pillared clay heterostructures with SiO2) and NiO/PCH-Ti (with TiO2); 5; 10; 17; 50; 80% NiO); NiO/SiO2 | Evaporation of a solution of Ni(NO3)2 in ethanol, with oxalic acid and containing the support | C2H6∕O2∕He = 3∕1∕26 or 3/3/24 (molar); 50 mL⋅min−1; T = 300–500 °C; 100 mg catalyst | Smax = 67.3% for 17NiPCH-Ti at 450 °C and 51.5% C2H6 conversion | [58] |
| NiO/PCH-Ti (10 wt% NiO) NiO/Ti-anatase (20 wt% NiO) NiO/Ti-P25 (20 wt% NiO) NiO/SiO2 (10 wt% NiO) | Impregnation | C2H6∕O2∕He = 3∕1∕26 (molar); 50 mL⋅min−1; T = 450 °C; 100 mg catalyst | Smax = 89% for NiO/Ti-P25 at 17% C2H6 conversion | [62] |
| 15% Ni/Al2O3/cordierite monolith | ∙ Colloidal (C) or suspension (S) of Al2O3, deposited on cordierite monolith, followed by impregnation with Ni(NO3)3; | 6% C2H6, 6% O2, 88% He (molar); W/F = 0.48 g⋅s⋅mL−1; T = 350–500 °C | Smax = 70% for Ni/Al2O3(S)-Cor at 500 °C and 30% C2H6 conversion | [56] |
| ∙ Powder Ni/Al2O3 prepared by impregnation of Ni(NO3)3, then a suspension of the powder deposited on cordierite monolith | ||||
| Ni–La/Al2O3-cordierite (NiLa(X)-M) with Ni/Al = 0.2; X = La/Ni = 0.09, 0.13, 0.17, 0.25 | Al2O3 deposited on cordierite monolith, Ni and La from nitrate precursors introduced by immersion of the coated monolith in the solution | 6% C2H6, 6% O2, 88% He (molar); W/F = 0.2–1.3 g⋅s⋅mL−1; T = 300–450 °C; 0.4 g catalytic coating per sample | Ymax = 13.4% for NiLa(0.17)-M at 400 °C | [42] |
| NiO/TiO2 (1–80% NiO) | Evaporation of Ni(NO3)2 solution in ethanol, with oxalic acid/Ni = 3, in the presence of TiO2b | C2H6∕O2∕He = 3∕1∕26 or 10/10/80 (molar); T = 300–500 °C; W/F = 0.2–12 g⋅s⋅mL−1 | Smax = 73.9% for 50Ni-B at 450 °C and 55.2% C2H6 conversion | [59] |
| NiO/TiP2O7 (Ni(x)TiP, with x = 10, 15, 18, 22, 26 wt% Ni) | Wet impregnation of TiP2O7 with Ni(NO3)2 solution in ethanol, with oxalic acid/Ni(NO3)2 mol ratio 1:1 | C2H6∕O2 = 0.5–3 (molar); T = 350–450 °C; W/F = 0.18–1.09 g⋅s⋅mL−1; 0.7 g catalyst | Smax = 70% for Ni(26)TiP at 425 °C and 36% C2H6 conversion | [60] |
| NiO/SiO2–Al2O3 fibers (ceramic papers) with CeO2, ZrO2 or Y2O3-stabilized ZrO2 binders | Impregnation of ceramic papers with Ni, Ce, Zr nitrates solutions | C2H6∕O2∕He = 6∕6∕88 (molar); T = 280–440 °C; W/F = 0.08 g⋅s⋅mL−1; 65 mg catalyst | Smax = 70% for NiZr-PZr at 360 °C and 26% C2H6 conversion | [64] |
| NiO/SiO2–Al2O3 fibers (ceramic papers)—NiO-CP NiO/alumina foam—NiO–AF NiO/cordierite monolith—NiO-M | Impregnation of the substrates with Ni nitrate solution | C2H6∕O2∕He = 6∕6∕88 (molar); T = 350–450 °C; W/F = 0.48 g⋅s⋅mL−1 for alumina foam and monolith, 0.08 g⋅s⋅mL−1 for ceramic papers; 400 mg catalyst for alumina foam and monolith, 60 mg catalyst for ceramic papers | Smax = 85% for NiO-M at 400 °C and 6.1% C2H6 conversion | [57] |
a Al2O3 obtained by six methods: from high purity aluminum (S1), precipitation of Al2(SO4)3 with (NH4)2CO3 (S2), precipitation of Al2(SO4)3 with NH3 (S3), hydrolysis of aluminum isopropoxide (S4), thermal decomposition of Al(NO3)3 (S5), precipitation of Al(NO3)3 with urea (S6). b Pure anatase—A; Degussa P25—B; nanocrystalline anatase—C.
4. The reaction mechanism
Several studies by our group [26, 49, 93] and other research groups [27, 51, 52, 82, 91], including density functional calculations [133, 134], were conducted in order to understand the ethane ODH reaction mechanism over NiO-based catalysts and have been discussed in detail. Based on these data, the ethane ODH reaction mechanism first proposed by Schuurman et al. [23] has been, in principle, confirmed. Thus, NiO, which is a p-type semiconductor with positive holes (h+) as the main charge carriers, contains an oxygen excess that is associated to the surface lattice O− species which are considered the catalytic active sites. In fact, the latter represent the chemical sites of the positive holes, according to the following exchange reaction:
- (1) The attack of the first C–H bond in ethane by surface lattice O− species (also noted O∙−) leading to the homolytic scission of this bond with formation of an ethyl radical chemisorbed on the surface:
Notably, to date, it is not clear if the ethyl radical formed in this reaction step is chemisorbed on a nickel or an oxygen surface site.
- (2) The 𝛽-hydrogen abstraction from the surface-bonded ethyl radical leading to ethylene:
As different oxygen species were shown to exist on the surface of the NiO-based oxides, it is worth noting that, according to some authors [27], the active O− species are also the selective sites provided that they are less numerous and, hence, more isolated on the surface. When their concentration on the catalyst surface is high, they lead to total oxidation. According to other authors [52], O− species represent the selective sites, while responsible for the total oxidation are species. Finally, there are authors [51] considering that the 𝛽-hydrogen abstraction step involves surface lattice O2− species, which are believed to be the selective sites, rather than O− species.
- (3) The elimination of water from two adjacent HO− groups with formation of a surface oxygen vacancy (□O) in the oxide lattice:
- (4) The replenishing of the oxygen vacancy with oxygen coming from the gas phase:
Overall, this corresponds to a Mars–van Krevelen mechanism, for which the redox cycle involves the Ni3+–Ni2+ transition, as suggested by the “breathing” redox behavior of the NiO-based catalysts under successive reductive and oxidative gaseous atmospheres [26, 49, 93].
5. Conclusions
NiO-based catalysts proved to have a huge potential in the ethane oxidative dehydrogenation reaction, their catalytic performance being strongly determined by their non-stoichiometric oxygen content which can be controlled by the preparation procedure and the calcination temperature of the oxide, the addition of modifiers or the dispersion of the active phase on an appropriate support. The crystallite size, the extent of isolation of non-stoichiometric oxygen, the low p-type semiconducting character and the acid-base properties of the catalyst were also shown to influence their catalytic behavior.
In general, both promoted and supported NiO-based catalytic systems showed remarkable performance in ethane ODH, with improved conversion and ethylene selectivity compared to bare NiO. Notably, Ti and Nb dopants managed to continuously deliver top-notch catalytic performances. Nevertheless, Nb-doped NiO catalysts exhibit a much greater ethane conversion for roughly the same ethylene selectivities compared to their Ti-containing counterparts. Another distinctive feature for Nb-containing catalysts is the lower reaction temperature required to achieve similar conversion levels. Therefore, Nb-doped NiO, specifically Ni0.85Nb0.15O, as this formulation was repeatedly demonstrated to be the best in terms of ethane ODH performance, is very close to industrial applications requirements. Unfortunately, its Achilles’ heel is the slow, but steady deactivation in time. One of the main ways used to overcome this drawback was to employ different synthesis methods, which was, up to now and to the best of our knowledge, unsuccessful. Another way, less exploited but which seems to give interesting results, is the addition of low amounts of a third metal cation M resulting in a M–Ni–Nb synergistic interaction with positive consequences on the catalytic performance, including the stability of the ternary system. As there are many metals that could prove synergism with both Nb and Ni, much work should be done in this direction in the future. Also, even though the overall ethane ODH reaction mechanism over NiO-based catalysts was shown to be a heterogeneous redox mechanism (Mars–van Krevelen), both experimental studies and theoretical calculations should be done to clarify in great detail and to understand the chemistry of the different individual reaction steps involved.
All in all, the correlations preparation procedure—physicochemical characteristics—catalytic properties evidenced in the different studies discussed here might allow us to further improve the existing NiO-based catalysts or to design new effective catalytic systems for ethane ODH reaction.
Conflicts of interest
Authors have no conflict of interest to declare.