



 CC-BY 4.0
CC-BY 4.0
1. Introduction
The use of mechanochemistry for the synthesis of ionic conductors is now a common method applied to compounds with different chemical hardness such as sulfides [1] or fluorides [2]. The reactivity of solid precursors under mechanical forces and local heating is controlled by crystal defects and interphases yielding final compounds with different long-range ordering, i.e., amorphous or crystallized, depending on the phase composition [3]. The reduced crystallite dimension typically found in mechano-synthesized compounds allows for the stabilization of metastable compositions, as exemplified by the solid solution BaF2–CaF2 with drastic ionic radius mismatch [4]. In addition, mechanochemistry can be further exploited to prepare thermodynamically stable phase with much lower annealing temperature and duration time compared to thermally activated solid-state chemistry [5]. Finally, ball milling treatment has also been used to investigate the effect of crystallite dimensions, microstrains [6, 7], or the presence of defects [8] on the transport properties.
In the field of fluoride ion conductors, BaSnF4 is a typical example of the beneficial use of mechanochemistry which allows it to produce a disordered structure crystallizing in the
2. Methods
2.1. Ball-milling synthesis
Powder precursors (SnF2, Sigma-Aldrich, 99%; BaF2, Sigma-Aldrich, 99.99%) were weighed and sealed in zirconia milling jars in an argon-filled glove box, with a powder to ball ratio of 1:13. The balls were 10 mm in diameter and made out of zirconia. The precursors were then milled at 400 rotations per minutes for 24 [14] or 99 cycles. Each cycle consisted of 15 min of milling and 15 min of rest to prevent overheating. The samples were labeled as c-BaSnF4-24c and c-BaSnF4-99c.
2.2. Powder X-ray diffraction
Powder X-ray diffraction was performed using a Bruker D8 Advance powder diffractometer, with a copper anode (Cu-Kα = 1.5418 Å). The powder XRD pattern was fitted using the Rietveld method as implemented in the Fullprof program, [15] with a split pseudo-Voigt function to model the peaks [16]. The instrumental X-ray line broadening was determined using the LaB6 standard sample.
2.3. Microscopy
Transmission electron microscopy (TEM) experiments, including electron diffraction (ED), high-angle annular dark-field scanning TEM (HAADF-STEM), and energy-dispersive X-ray (EDX) STEM studies, were performed on a double aberration-corrected JEOL ARM200F cold FEG microscope operated at 200 KV and equipped with a CENTURIO large-angle EDX detector, Orius CCD camera, and a Quantum GIF. The TEM samples were prepared by grinding the materials in an agate mortar in ethanol and depositing the prepared suspension on a Cu-hole carbon grid.
2.4. 119Sn Mössbauer spectroscopy
A laboratory-made Halder-type spectrometer with constant acceleration was used for the analysis. It was equipped with a Montana Instruments CryoCore cryogenic system and a 119mSn radioactive source (nominal activity 370 MBq) embedded in a CaSnO3 matrix and maintained at room temperature. All spectra were recorded at low temperature (10 K), with 50 to 70 mg of sample ([Sn] = 5 to 8 mg/cm2). The experimental data were analyzed and the 119Sn Mössbauer hyperfine parameters (δ: isomer shift, Δ: quadrupole splitting, Γ: full width at half maximum and relative absorption area) were refined using WinNormos® software (Wissenschaftliche Elektronik GmbH). Isomer shifts are given relative to CaSnO3 at room temperature (δ = 0.0 mm/s).
2.5. Impedance spectroscopy
Electrochemical Impedance spectroscopy was performed on gold-coated t-BaSnF4 pellets. Measurements were performed with a BioLogic MTZ-35 impedance analyzer with data collected in the frequency range of 3.5 × 107 Hz to 1 Hz, under an Ar atmosphere. The compactness of the pellets was approximately 90%.
2.6. 19F solid-state NMR
19F Magic Angle Spinning (MAS) NMR experiments were performed on Bruker Avance III spectrometers operating at B0 = 7.0 T (19F Larmor frequency of 282.4 MHz), using a 1.3 mm and a 2.5 mm CP-MAS probe head. The 19F MAS spectra were recorded in direct polarization (single 90° pulse) and using a rotor-synchronized Hahn echo sequence with an interpulse delay equal to one rotor period by default. Unless otherwise noted, the spectra shown were recorded using a Hahn echo sequence. The 90° pulse length and the recycle delay were respectively set to 1.25 μs and 300 s for c-BaSnF4-24c, 1.63 μs and 300 s for c-BaSnF4-99c, 1.25 μs and 900 s for SnF2, 2 μs and 30 s for SnF4 and 1.5 μs and 300 s for BaF2. Due to air friction heating, the sample temperature in the 1.3 mm probe head varies by up to 32 °C from 34 to 64 kHz The isotopic 207Pb chemical shift of Pb(NO3)2 was used as the NMR thermometer [17, 18]. 19F spectra are referenced to CFCl3 and they were fitted by using the DMFit software [19].
3. Results and discussion
3.1. Microstructural analysis
The mechano-synthesis of c-BaSnF4, reported in the literature results in an X-ray diffraction pattern with typically broad and asymmetric Bragg peaks [9, 14]. An example of the typical diffraction pattern is shown in Figure 1 and refers to a sample prepared by 24 cycles of high energy ball milling, i.e., c-BaSnF4-24c. Increasing the mechanical milling time up to 99 cycles, i.e., c-BaSnF4-99c, resulted in a significant increase in the broadening of the Bragg peaks (Figure 1). Both samples show close unit cell parameter values of 6.1964(2) and 6.2039(3) Å for 24 and 99 cycles, respectively. To account for the broadening and asymmetry of the Bragg peaks, we performed profile refinement using a split pseudo-Voigt function (see Methods) from which we extracted the position of each peak and the corresponding total integral width (β).

Profile matching refinements of c-BaSnF4-24c and c-BaSnF4-99c.
The origin of the X-ray peak broadening was investigated using the Williamson–Hall (W–H) plot [20] according to the following relation: 𝛽cos𝜃 = 𝜆/L + 4𝜀sin𝜃 where, 𝜆 is the wavelength, L the average crystallite size and 𝜀 the strains. Both the average crystallite size and the strains can be deduced from the intercept and slope of the W–H plot. The W–H plots obtained for both samples are summarized in Figure 2. For c-BaSnF4-24c, the plot indicates that the origin of the peak broadening is mainly dominated by strain effects. The deduced average crystallite size is 13 nm. Most noteworthy is the plot obtained for the c-BaSnF4-99c sample, which shows that the broadening of the X-ray diffraction peaks originates from a size effect in the absence of strain. It should be noted that for larger 2θ values, the shape of the peaks is more difficult to capture by the refinement, so that the peaks spread away from a straight line. However, it is clear that the broadening is solely due to a size effect, as confirmed by the average crystallite size value of 6 nm. Overall, the increasing ball-milling time results in a drastic reduction of the particle size by a factor of 2 and seems to suppress the occurrence of strains within the particles.
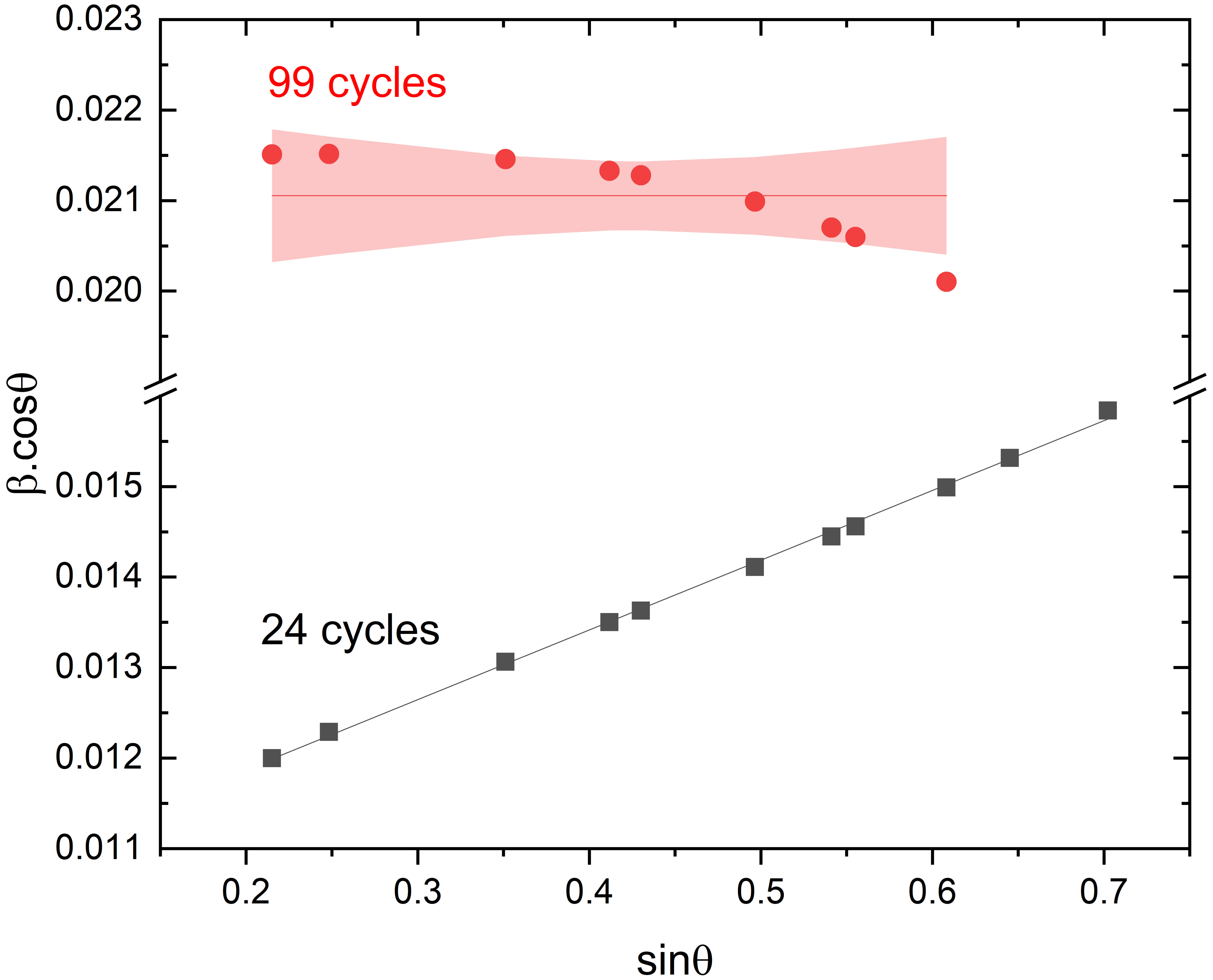
Williamson–Hall plots for c-BaSnF4-24c and c-BaSnF4-99c samples. The pink area refers to a 95% confidence interval.
The presence of strain in c-BaSnF4-24c has been previously observed using HRTEM data, where local strain was detected by visible changes in inter-reticular spacing and the appearance of defects [14]. Further investigations using HAADF-STEM, first confirm that c-BaSnF4-24c has a crystallite size of about ten nanometers and that other sources of strain are visible through the presence of twin defects (Figure 3a). Interestingly, c-BaSnF4-99c did not show the presence of these twin defects while the crystallite size distribution appears to be narrower and less than ten nanometers (Figure 3b) with well-crystallized aspects. This shows that under mechanical stress, twin defects can act as cleavage planes, reducing both crystallite size and microstrains without implying any recrystallization [21]. In addition, we did not observe any particular changes in the elemental atomic distribution at the particle level (Figure S1).

HAADF-STEM images from c-BaSnF4-24c (a) and c-BaSnF4-99c (b).
3.2. Sn environments and electron lone pairs
119Sn Mössbauer spectroscopy, which probes Sn properties such as the oxidation state, the coordination number and lone pair electrons, was used to investigate how the ball-milling treatment affects these physical properties. The spectra of both samples were reconstructed to extract the hyperfine parameters (Figure 4 and Table 1). The spectrum of c-BaSnF4-24c is consistent with previous reports, with two symmetrical quadrupole doublets characteristic of covalently bound Sn(II) and a large quadrupole splitting parameter (Δ> 1.5 mm⋅s−1) indicating that Sn has a stereochemically active lone pair [22]. These two components (quadrupole doublets) have previously been attributed to different local environments (coordination geometry and bond lengths) for Sn(II) atoms [14]. Prolongation of the ball milling treatment did not affect these environments with similar isomer shift, quadrupole splitting and relative intensity. However, a stronger contribution of Sn(IV) was detected with an isomer shift of −0.14 mm/s. Such a value, intermediate between those of Sn(IV)F4 and Sn(IV)O2 (Figure 4, Table 1) combined with a large quadrupole splitting value (0.72 mm/s), suggests mixed oxide–fluoride and distorted Sn(IV) environments.

Low-temperature (10 K) 119Sn Mössbauer spectra of c-BaSnF4-24c and c-BaSnF4-99-c and reference SnO2 and SnF4.
Low-temperature (10 K) 119Sn Mössbauer hyperfine parameters for c-BaSnF4-24c and c-BaSnF4-99c and reference compounds SnF4 and SnO2
| Sample/components | δ (mm/s) | Δ (mm/s) | Γ (mm/s) | Relative area (%) |
|---|---|---|---|---|
| c-BaSnF4-24c | ||||
| Sn(II)-A | 3.18(1) | 1.81(1) | 0.91(1) | 55(3) |
| Sn(II)-B | 3.50(1) | 1.74(1) | 0.93(2) | 37(3) |
| Sn(IV) | −0.14(2) | 0.48(6) | 0.90(3) | 8(3) |
| c-BaSnF4-99c | ||||
| Sn(II)-A | 3.17(1) | 1.78(1) | 0.98(3) | 46(3) |
| Sn(II)-B | 3.51(2) | 1.68(2) | 0.96(3) | 31(3) |
| Sn(IV) | −0.11(1) | 0.72(2) | 0.95(3) | 23(3) |
| SnF4 | −0.24(1) | 0.46(3) | 1.07(3) | 100 |
| SnO2 | 0.09(1) | 0.53(3) | 1.06(3) | 100 |
3.3. Fluoride ion mobility
The influence of the milling time on the ionic conductivity and activation energies was investigated by temperature-dependent electrochemical impedance spectroscopy. The temperature-dependent ionic conductivity shown in Figure 5, exhibits a linear Arrhenius behavior. The activation energy extracted from the Arrhenius plots equals to 0.57 eV in both samples. This shows that increasing the ball milling time does not affect the energy barrier for fluoride ion jumps. In addition, the ionic conductivity decreases from 4.60 × 10−6 to 1.58 × 10−6 S⋅cm−1 at 30 °C when the ball-milling time is extended.

Arrhenius plot of the ionic conductivity for c-BaSnF4 prepared with 24 and 99 cycles.
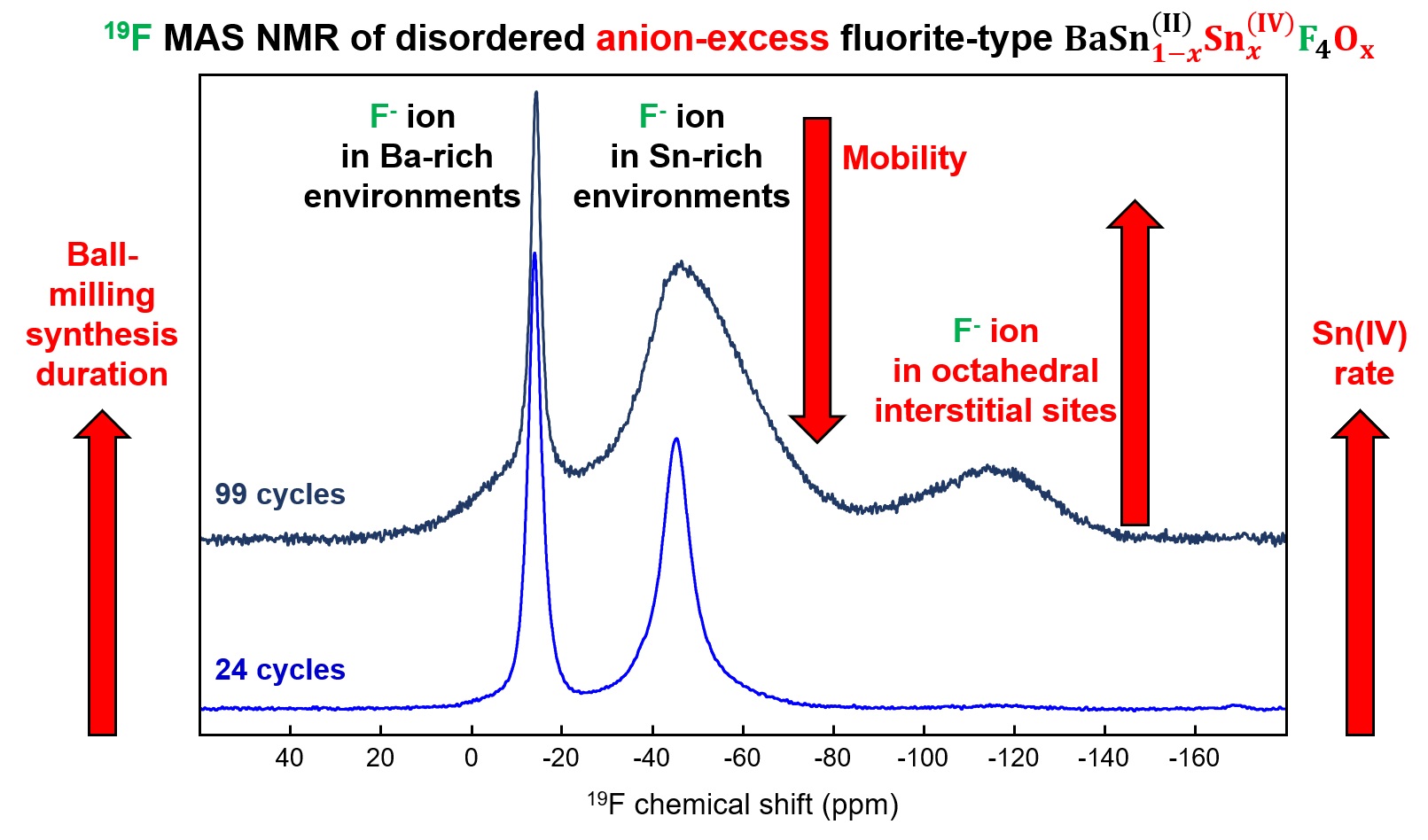
19F NMR was used to record the effect of ball-milling duration on the environments and mobility of fluorine. The spectrum of c-BaSnF4-24c shows two distinct contributions at −14 ppm and −45 ppm, which are assigned to broad Ba-rich and Sn-rich fluorine environments, based on their proximity to the resonance of BaF2 (𝛿iso = −14 ppm) and SnF2 (−44 and −51 ppm), respectively (Figure 6) [14]. In fact, due to fluorine exchange between different sites within the host framework, the rich variety of fluoride ion environments is not observed in the experimental 19F MAS NMR spectrum. Fluorine exchange on the NMR time scale has been assumed to occur at different rates, between Ba-rich sites, on the one hand, and between Sn-rich sites, on the other hand, each giving rise to a single observed resonance, have been assumed [14].

19F MAS NMR spectra of SnF4 (30 kHz), SnF2 (64 kHz), BaF2 (60 kHz), c-BaSnF4-24c (64 kHz) and c-BaSnF4-99c (54 kHz). Fit of the spectra of c-BaSnF4-24c (64 kHz) and c-BaSnF4-99c (54 kHz) and characteristics of the lines used for these fits are given as Supplementary Materials (Figures S2 and S3, Tables S1 and S2).
In c-BaSnF4-24c, a third type of fluorine exchange between Ba-rich and Sn-rich environments has been demonstrated using variable-temperature 19F MAS NMR spectroscopy. As the temperature increases, the relative intensity of the peak assigned to fluoride ions in Sn-rich environments also increases, at the expense of the peak assigned to fluoride ions in Ba-rich environments, confirming some degree of fluoride-ion exchange between Ba-rich and Sn-rich environments [14]. This can also be observed by comparing the 19F MAS NMR spectra of c-BaSnF4-24c in Figures 6 and 7, recorded at 64 kHz (at 64 °C due to frictional heating) and 44 kHz (40 °C).
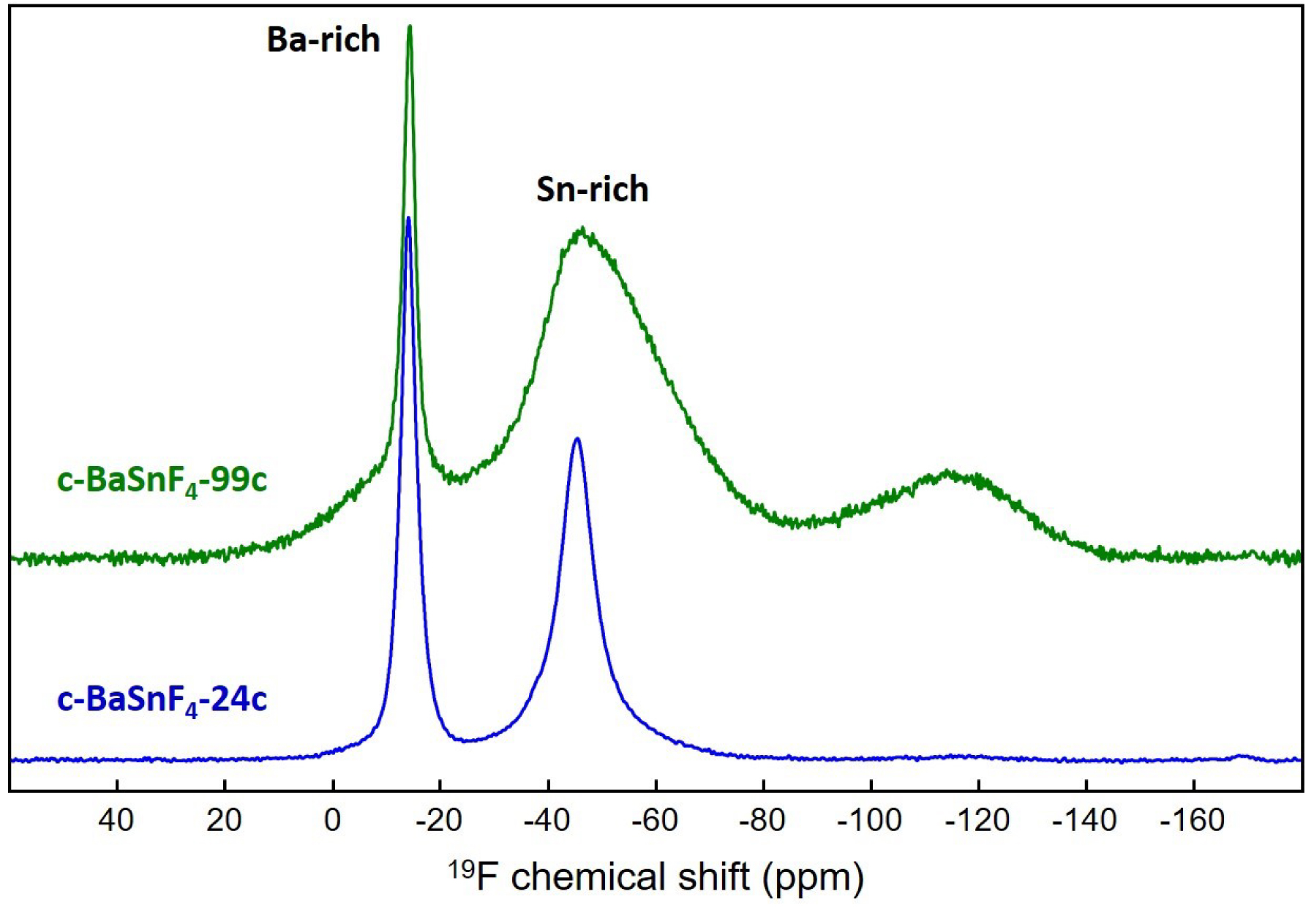
19F MAS (44 kHz, 40 °C) NMR spectra of c-BaSnF4-24c (blue) and c-BaSnF4-99c (green). Fits of these spectra and characteristics of the lines used for these fits are given as Supplementary Materials (Figures S4 and S5, Tables S3 and S4).
The 19F MAS NMR spectra of c-BaSnF4-99c also show these two resonances, with the one assigned to Sn-rich sites being much broader. Moreover, in contrast to c-BaSnF4-24c, the relative intensity of this contribution does not increase and its width does not decrease with increasing temperature (Figure 8). Then, for c-BaSnF4-99c, unlike c-BaSnF4-24c, motional narrowing of the Sn-rich peak with increasing temperature is not observed, indicating that the so-called fast-exchange regime is not reached and there is no evidence of exchange between Sn-rich and Ba-rich F sites. This results in a lower ionic conductivity.

19F NMR spectra of c-BaSnF4-99c at various MAS frequencies and temperatures. Fits of these spectra and characteristics of the lines used for the fits are given as Supplementary Materials (Figures S3, S5 and S6 and Tables S2, S4 and S5).
3.4. Environment of F− ions from the third contribution of the 19F NMR spectra
In addition to these two main resonances, the spectra of c-BaSnF4-99c (Figure 6) show at ∼−116 ppm a third broad and less intense resonance. A closer look at the spectra of c-BaSnF4-24c also shows the presence of this contribution (see Figure 7), at a chemical shift close to −115 ppm, which we did not mention before [14] because of its very low intensity (∼1%).
The width of this third contribution indicates a disorder due to the very different environments of the corresponding F-ion sites. In addition, this peak remains almost unchanged with increasing temperature (Figure 8), indicating that these fluoride ions have low mobility.
We have tried to consider the various hypotheses that could explain the origin of this contribution. First, such an additional contribution whose average chemical shift is significantly lower than that of the two main resonances, could indicate overcoordinated F-ions (the higher the F-ion coordination number, the lower its 19F chemical shift [23, 24]), i.e., coordination number greater than 4. Indeed, ab initio molecular dynamics simulations have revealed, in c-BaSnF4, a complex local structure with a high degree of intrinsic fluoride ion disorder, where 1/3 of the fluoride ions occupy octahedral “interstitial” sites [14]. The detection of resonances characteristic of fluoride ions occupying within these interstitial sites can be explained by the slower exchange in c-BaSnF4-99c than in c-BaSnF4-24c (for which the contribution of these sites does not (or almost does not) appear in the spectra). This hypothesis is supported by the observation in the 19F NMR spectra of the tetragonal phase of BaSnF4 of a contribution at a near 𝛿iso value (−121 ppm on average with a maximum at −118 ppm) assigned to F− ions partially occupying the octahedral interstitial sites [25]. The occurrence of F-ions occupying octahedral sites is consistent with the accommodation of Sn(IV) and O2− oxide ions in the structure and then with mixed oxide-fluoride and distorted Sn(IV) environments evidenced by Mössbauer. Using the relative area of the signals assigned to Sn(IV) in Table 1, we evaluated the corresponding formulas, which yielded
The second hypothesis concerning the origin of the third contribution in the 19F NMR spectra is the formation of a Sn(IV) oxide fluoride impurity and Sn(II) depletion in the fluorite. Since it is not detected in the X-ray diffraction patterns (Figure 1), the impurity must be amorphous. Assuming that all Sn(IV) atoms and all oxide ions are involved in the formation of the impurity, its formula would be Sn(IV)OF2 [26]
We can now check whether the fraction of 19F nuclei involved (=2x/4) is equal to half the fraction of Sn(IV) atoms (=x). This is the case for c-BaSnF4-99c (x = 0.23 (Table 1) and 2x/4 = 0.122 (Table S6)), but not at all for BaSnF4-24c (x = 0.08 (Table 1) and 2x/4 = 0.009 (Table S7)). Thus, in BaSnF4-24c, whether most of the Sn(IV) is not present in the SnOF2 impurity, or the impurity has a lower F content and therefore a formula of the type SnO2−y/2Fy, which also implies that a fraction, albeit smaller, of the Sn(IV) remains in the fluorite.
Knowledge of the 19F isotropic chemical shift values of these compounds is required to support or exclude the above hypotheses. Unfortunately, 19F NMR studies of Sn(IV) oxyfluorides are scarce. In addition to SnOF2, which is obtained in amorphous form from SnF2(ONO2)2 and is expected to adopt the ReO3 type structure after crystallization [26], there is Sn4O2F10, a mixed-valence tin oxyfluoride, whose structure has been reported [27], but neither of these compounds appears to have been studied by solid-state NMR. In contrast, more work has been done on fluorine-doped tin oxide (SnO2:F, FTO), in particular an extensive solid-state NMR study, with a 19F MAS NMR spectrum showing evidence of a single, relatively broad resonance at −113.9 ppm, which, on the basis of experiments and first-principles quantum-chemical calculations, has been assigned to F atoms occupying O vacancies in SnO2:F (at 1.2 mol%), i.e., F atoms triple-coordinated to Sn(IV) [28]. Even if the doping rate of the supposed impurity SnO2−y/2Fy is significantly higher, the hypothesis is still convincing. However, if there is an impurity in c-BaSnF4-99c, its formula is SnOF2, in which the F atoms are doubly coordinated to Sn(IV), while the chemical shifts of 19F of the third contributions are very close in both samples. Similar 19F chemical shift values for double and triple coordinated F atoms should exclude the presence of impurities in both samples. However, we lack experimental data on Sn(IV) fluorides and oxyfluorides to completely rule out this hypothesis. In addition, we have observed that the influence of the coordination number of the fluorine through Sn(IV) on its chemical shift is minimal or rare. In fact, in SnF4, half of the F atoms are terminal and half are bridging [29], and their chemical shift values, close to −150 ppm, differ by only a few ppm ([[30]], Figure 6). In contrast, in NbOF3 and TaOF3 which adopt the SnF4-like structure, the chemical shift values of the terminal and bridging F atoms differ by 200 ppm and 170 ppm, respectively [31].
After outlining various hypotheses, the most plausible is the stabilization of an anion-excess fluorite phase rather than the formation of an impurity. This is supported by microscopic analysis, which shows that the particles are well crystallized and have a homogenous elemental distribution, while X-ray diffraction does not reveal any impurity. The stabilization of an anion-excess fluorite phase is further supported by Mössbauer spectroscopy and the associated Sn(IV) in mixed oxide–fluoride environments. It is also consistent with the additional resonance observed by 19F NMR and assigned to fluoride ions located in interstitial octahedral sites. They exhibit low mobility, which may be due to the formation of Sn(IV)–F bonds.
Overall, this work confirms that fluorite is an adaptable framework that can accommodate different cations and anions with deviations from stoichiometry. To determine the stabilization of an anion-excess fluorite phase, the intentional introduction of oxide anions and/or Sn(IV) can be considered. In addition, further characterizations, such as 19F 2D homonuclear correlation spectroscopy, could show that all the fluorine atoms are in the same phase. Finally, a comprehensive study of Sn-based fluoride oxides using 19F NMR and Mossbauer spectroscopy would be invaluable for a better understanding of the mechanochemistry applied of these compounds.
4. Conclusions
Mechanosynthesis of BaF2 and SnF2 yields cubic BaSnF4. While short milling times yield the desired product, they also introduce microscopic defects such as strain and twinning regions within the roughly 13 nm crystallites. Interestingly, these defects are suppressed by longer milling times, which also reduce the particle size to 6 nm, suggesting that these twin defects may act as cleavage planes under mechanical stress. The resulting particles are well crystallized and the absence of a second phase has been confirmed by XRD and EDX analysis. However, Mössbauer spectroscopy revealed the presence of Sn(IV) in a disordered mixed oxide–fluoride environment. In addition, 19F NMR indicated the presence of a peak assigned to fluoride ions located in octahedral sites. We hypothesize that an anion-excess fluorite phase was formed with the general chemical formula
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
BM, CM, and DD thank the French National Research Agency (STORE-EX Labex Project ANR-10-LABX-76-01) for financial support. DD wishes to thank for support a French government grant managed by the Agence Nationale de la Recherche under the France 2030 program, reference 23-PEBA-0002.
Supplementary data
HAADF-STEM images and corresponding EDX elemental mapping of c-BaSnF4-99c, experimental and fitted 19F MAS NMR spectra of c-BaSnF4-24c and -99c and features of the NMR lines used in the fits.
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.361 or from the author.