

1 Introduction
Perfectly defined inorganic nanobuilding blocks such as metal–oxo–alkoxo clusters, when properly functionalised, can be assembled, without damage, into various types of structures, in order to elaborate new hybrid materials [1–3]. Hybrid organic-inorganic materials, in which organic and inorganic components are combined over a length scale ranging from a few angstroms to a few ten of nanometres, are nanocomposites that offer high potentialities for high-added-value applications. Numerous hybrid organic-inorganic materials have been developed in the past few years, mainly by taking advantage of the mild chemical conditions of the sol–gel process. Yet, the sol–gel process generally yields amorphous materials, which exhibit some polydispersity in the size and composition of the inorganic components of the hybrids. Controlling the interactions between the inorganic and the organic components is not an easy task. One of the possible solutions to overcome the polydispersity problem and achieve a better control of the interactions is to use nanobuilding blocks, less reactive than alkoxides.
Silicon, tin, or transition-metal chemistry offers a large number of metal oxo-clusters, which are potential nanobuilding blocks for hybrid materials [1–3]. The syntheses and structures of many high-valence metal or even heterometallic oxo-clusters have been described in the literature. Such species are usually prepared in solution through the controlled substoichiometric hydrolysis of the metallic alkoxides M(OR)n or the corresponding complexed alkoxides, M(OR)n–x(LZ)x, (where LZ is a complexing ligand). These clusters are isolated from the ill-defined oxo polymers concurrently formed by crystallization, frequently resulting in low yields. These species, characterized by single-crystal X-ray diffraction, present perfectly defined structures where the metallic centres usually exhibit coordination numbers larger than their valence states. The stability of these clusters is made possible by the chelating or bridging character of the organic ligands, but also by the existence of the oxo bridges.
The high ionicity of the M–C bond, where M is a transition metal, makes it very sensitive to hydrolysis or nucleophilic attacks. Therefore, the route that consists in functionalising transition-metal-based nanobricks by M–C bonds cannot be used. Different strategies have been described in the literature to overcome this problem: the functionalization through complexing group or M–O–Si–R links. By these ways, polymers reinforced by covalently bonded inorganic clusters can be obtained.
Another strategy is to exchange the alkoxo groups by appropriate nucleophilic species. The stability of titanium (IV) oxo–alkoxo clusters toward nucleophilic organic reagents (alcohols, complexing ligands) was evidenced by 13C and 17O NMR spectroscopies [4–6]. Some of the alkoxide groups are exchanged by alcohol molecules through trans-alcoholysis reactions giving rise to new TinOm(OR)z–x(OR')x species such as Ti16O16(OEt)24(OPrn)8 [5] or Ti12O13(OiPr)10(OEt)6 [4]. This stability of high-nuclearity clusters toward alcohols or diols can thus be used to establish connections between clusters [6,7]. The reactivity of titanium–oxo clusters can be also used advantageously with the aim of designing new hybrid materials with tailored textures. Thus, use of dendrimeric connectors or amphiphilic copolymers as templates has been recently reported [6,8,9].
The research work reported in this article deals with the characterisation by NMR analysis of the cluster Ti16O16(OEt)32. The structure of this cluster is described in the literature [10,11]. A main point is the simplicity of its structure, all titanium atoms being in an octahedral environment surrounded by bridging oxygen atoms. The stability of this cluster is ensured by the presence of ethoxide groups at the surface. An important difference between these ethoxide groups is the connectivity of their oxygen atom, which can be bond to one titanium atom (μ2O: ending ethoxide groups) or to two titanium atoms (μ3O: bridging ethoxide groups) (Fig. 1). So, a difference of reactivity between these two kinds of ethoxide groups is expected. The fact that only eight of them are exchanged when the cluster is treated by an excess of propyl alcohol means that the surface reactivity of Ti16O16(OEt)32 is very complex [5].
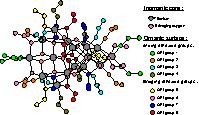
Representation of Ti16O16(OEt)32 in molecular form. Titanium atoms, in grey, are in octahedral environment with oxygen. Oxygen atoms of inorganic core, in pale red, are composed of 4 μ2-O, 8 μ3-O and 4 μ4-O. Ethoxide groups can be divided into eight packs of four ethoxide group each, in different colours.
Liquid- and solid-state NMR are appropriated tools for the selective characterization of the inorganic oxo-core (17O NMR) and the organic surface (13C, 1H NMR). In this article, NMR analysis results of Ti16O16(OEt)32 and our first results of surface reactivity towards propyl alcohol are reported.
2 Experimental section
2.1 Synthesis of the monoclinic Ti16O16(OEt)32 crystals [10,11]
A mixture of titanium tetraethoxyde, Ti(OEt)4 (purchased from Fluka), and 5% aqueous ethyl alcohol solution (1:1 v/v) was placed in a steel autoclave at 373 K during two weeks. After slow cooling (1 K h–1), colourless air-sensitive single crystals of monoclinic Ti16O16(OEt)32 were obtained. They were dried and stored in an inert atmosphere. The crystal structure was checked by the acquisition of a XRD diffraction pattern.
2.2 Substitution with propyl alcohol
1 g of Ti16O16(OEt)32 crystals was dissolved in free-water toluene (10 ml), an excess of propyl alcohol (5 ml) was added and this mixture was placed at 323 K during 24 h. After solvent evaporation, a white air-sensitive amorphous solid is obtained. Propyl alcohol was dried on molecular sieves before use.
2.3 Kinetics of reaction with propyl alcohol monitored by NMR
100 mg of Ti16O16(OEt)32 were dissolved in 4 ml of free-water toluene and then heated in a 5-mm NMR tube at 323 K. 13C and 1H blank NMR spectra were acquired before the addition of 2 ml of propyl alcohol. 13C and 1H NMR spectra, with respectively an acquisition time of 45 and 5 min, were alternatively carried out every hour during 6 h.
2.4 Liquid-state NMR experiments
For Ti16O16(OEt)32 and the substituted clusters with propyl alcohol analysis, 100 mg of compound were dissolved in 6 ml of perdeuterated toluene.
1H and 13C NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer equipped with a 5-mm z-gradient broadband inverse probe head. Kinetics of substitution with propyl alcohol were analysed in a 5-mm QNP probe at 323 K. Traces of residual solvents were used as internal standards.
2.5 Solid-state NMR experiments
13C NMR solid-state spectrum was acquired with a Bruker DSX400 spectrometer operating at 9.4 T. A 4-mm rotor spinning at the Magic Angle at 12 kHz was used.
1H–13C cross polarization was performed using Variable Amplitude Cross Polarization (VACP) [12] with a typical contact time of 1 ms. A Two-Pulse-Phase-Modulation (TPPM) [13] proton decoupling sequence was used during acquisition. Spectra were modelled using the ‘dmfit’ program [14].
3 Analysis of Ti16O16(OEt)32 by liquid and solid-state NMR
3.1 Description of the monoclinic structure
When Ti16O16(OEt)32 is in liquid state, its molecular form is studied because all of the species are separated. Nevertheless, in the solid state, it is the arrangement through Van Der Waals interactions between these neutral molecules that determine the crystal structure. Two polymorph crystal structures have been proposed in literature for Ti16O16(OEt)32, a tetragonal and a monoclinic one (depending on the water ratio used for its synthesis) [11]. These structures differ essentially by the orientation of the ethoxide groups, which leads to two different packing of Ti16O16(OEt)32 molecules. The monoclinic form, studied here, is much more stable than the tetragonal one. For simplification, Fig. 1 shows only the molecular form of the Ti16O16(OEt)32 cluster in the monoclinic crystal.
The inorganic Ti16O48 core is built up of two orthogonal blocks of eight distorted TiO6 octahedra. The sixteen titanium atoms are interconnected through four different kinds of oxygen bridges: three types of these oxygen bridges correspond to inorganic core: four bridging oxygen atoms (μ2O), eight triple bridging atoms (μ3O), four quadruple bridging atoms (μ4O). The last type of oxygen atoms corresponds to the 32 ethoxide groups, which represent the organic surface. These organic groups can be divided into two subgroups of sixteen bridging ethoxide groups and 16 ending ones. The geometry of the structure presents a 2 axis and a pseudo –4 axis so that each subgroup can be divided into four pseudo equivalent ethoxide groups. Therefore, the cluster surface can be divided into eight packs of four pseudo-equivalent ethoxide groups numbered from 1 to 8 in Fig. 1 for more clarity.
3.2 Correlation between 1H and 13C liquid-state NMR spectra
In the liquid state, Ti16O16(OEt)32 is in its molecular form and the eight kinds of pseudo-equivalent ethoxide groups become really equivalent, due to the Brownian motion. Effectively, the 13C spectrum presents eight resonances in the methylene region (73.7, 73.2, 72.4, 72.2, 71.6, 71.2, 70, 69.4 ppm) and the same number in the methyl region (five around 20.2 ppm and the others at 19.4, 19.3, 18.9 ppm), corresponding to the eight ethoxide groups (Fig. 2). The 1H spectrum (Fig. 2) presents eight triplets in the methyl region (1.89, 1.70, 1.61, 1.55, 1.36, 1.34, 1.33, 1.30 ppm), but is more complex in the methylene area due to the magnetic inequivalence of methylene geminal protons, which leads to overlapping of signals. Then, 13C and 1H signals corresponding to the same ethoxide groups were correlated by 1H–13C HMQC and 1H-1H COSY experiments (Fig. 3). For the sake of simplification, attribution of a letter from A to H was given for each kind of signals and their chemical shifts are summarised in Table 1.

Extracts of Ti16O16(OEt)32 liquid state 13C and 1H NMR spectra with, on the left, the methylene region and, on the right, the methyl region. Part A: 13C spectrum. Part B: 1H spectrum. * Toluene signals
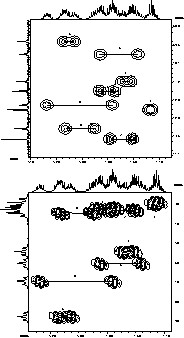
Top: extract of the methylene region of 1H–13C HMQC 2D spectrum, which shows correlation between carbon and proton of the same ethoxide groups (via 1J coupling). Bottom: extract of 1H–1H COSY 2D spectrum, which shows the correlation between the methylene geminal protons and methyl protons of the same ethoxide group (via 3J coupling).
13C and 1H chemical shifts of Ti16O16(OEt)32 ethoxide groups
| Ethoxyde groups | δCH2 (ppm) | δCH2 (ppm) | ΔδCH2 (ppm) | δCH3 (ppm) | δCH3 (ppm) | T1 of CH2 (s) |
| A | 73.7 | 4,78-4,61 | 0.17 | 20.2 | 1.34 | 0.89 |
| B | 73.2 | 5,13-4,90 | 0.23 | 20.2 | 1.36 | 0.20 |
| C | 72.4 | 4.46 | 0.00 | 19.3 | 1.30 | 0.55 |
| D | 72.2 | 5,27-4,76 | 0.51 | 20.2 | 1.70 | 0.10 |
| E | 71.6 | 4,85-4,74 | 0.11 | 20.2 | 1.33 | 0.40 |
| F | 71.2 | 4,69-4,62 | 0.07 | 19.4 | 1.55 | 0.15 |
| G | 70.0 | 4,85-4,56 | 0.29 | 18.9 | 1.61 | 0.10 |
| H | 69.4 | 5,15-5,05 | 0.10 | 20.2 | 1.89 | 0.10 |
On the one hand, it is known that the crystal structure of Ti16O16(OEt)32 presents eight kinds of ethoxide groups (numbered 1 to 8) and, on the other hand, the 13C and 1H NMR spectra present signals (names A to H) of this eight ethoxide groups. The main problem is to assign each NMR signal to each ethoxide group. In previous studies on another cluster, the Ti6(COOCH3)8(OBun)8O4, the assignment of the 13C and 1H NMR spectra were done on the basis of T1 relaxation-time measurements [15]. This cluster presents, by symmetry, three subgroups of two ending butoxide groups, and one of two bridging butoxide groups. Liquid-state 13C and 1H NMR spectra present four kinds of signals, which were correlated by 1H–13C HMQC experiment to regroup signals corresponding to the same butoxide groups. One of the four signals obtained is very different from the others, because it has the most 13C and 1H shielded signals, the difference in chemical shift between methylene geminal protons is the largest and also its T1 relaxation time is the shortest. This signal was assigned to bridging butoxide groups. The shortest T1 relaxation time and the great difference between methylene geminal proton chemical shifts were explained by the restricted rotation of these bridging groups [16], compared to ending ones.
Relaxation-time T1 measurements on 13C in the methylene area were done on the Ti16O16(OEt)32 cluster (Table 1). The signals named ‘A’, ‘C’, and ‘E’ have a long T1 relaxation time (from 0.40 to 0.89 s) and could be assigned to ending groups, whereas the ‘D’, ‘G’ and ‘H’ signals have a short T1 relaxation time (about 0.10 s) and could be assigned to bridging groups. The ‘F’ and ‘B’ signals have an intermediate T1 regime (0.20 and 0.15 s) and cannot be unambiguously assigned. In contrast with the Ti6(COOCH3)8(OBun)8O4 results [15], this attribution is not confirmed by the difference between methylene geminal proton chemical shifts and the chemical shift of methylene carbons. For example, the ‘H’ signal presents a short T1 relaxation time, characteristic of bridging alkoxide groups, but the difference between chemical shifts of its methylene geminal protons is weaker than expected.
3.3 Analysis by 13C solid-state NMR
Solid-state NMR experiments can be a good tool to investigate characterization of non-soluble materials. That will be the case of cross-linking hybrids. Therefore, correlation between results obtained in liquid state NMR with solid-state NMR spectra was done.
The solid-state 13C spectrum is not well defined in the methylene region (Fig. 4A), but it presents four sharp signals (45 Hz) at 73.2, 72.3, 71.3, 70.0 ppm and two wide ones (more than 200 Hz) at 71.9 and 69.0 ppm (Fig. 4). The methyl area presents eight sharp contributions (10–20 Hz) at 21.7, 20.4, 20.1, 19.7, 19.4, 19.0, 18.8, 18.3 ppm and a wide one (about 200 Hz) at 19.4 ppm. The methylene contributions are divided into two parts: the deshielded and sharp signals between 74 ppm and 70 ppm might be assigned to ending ethoxide groups (1-4 ethoxide groups, cf. Fig. 1) because solvent residues in the crystal structure increase their mobility and also the NMR signal resolution. On the contrary, the restricted rotation of bridging ethoxide groups and the distortion of their bonds (5-8 ethoxide groups, cf. Fig. 1) causes broadening and chemical shift anisotropy of signals at 71.9 and 69.0 ppm. The better resolution of CH3 signals can be explained by the mobility of this ending carbon compared to OCH2 ones.

(A) Extracts of Ti16O16(OEt)32 13C solid-state NMR spectrum, modelled with dmfit program. (B) Comparison between solid- and liquid-state 13C NMR spectra.
Fig. 4B shows that comparison between liquid and solid-state NMR spectra is difficult in the methyl region, but is easier in the methylene region. Crystal structure of this cluster depends on the orientation of the ethoxide groups; so, relative positions and chemical shifts of these methyl groups are probably very different in liquid state compared to those in solid state. On the contrary, OCH2 range is quite similar because the carbons are close to the core and are less mobile than the methyl carbons. Modelling of the clusters’ structure in liquid state and NMR experiments at higher magnetic field may improve the resolution of 13C spectrum and allow more detailed discussion of solid and liquid NMR signatures.
4 Substitution of ethoxide groups by propyl alcohol
The trans-alcoholysis reaction occurs with the propyl alcohol used in excess in the presence of the Ti16O16(OEt)32 cluster leading to the modified cluster Ti16O16(OEt)24(OPrn)8 [5]. An extensive study of this reaction seems to be a good step towards the study of surface reactivity. The elaboration of this modified cluster can also contribute to the assignment of the NMR spectra, since the exchanges are done on selected positions in the structure.
4.1 Kinetics of substitution by propyl alcohol monitored by NMR
Treatment in solution of Ti16O16(OEt)32 with an excess of propyl alcohol leads to the formation of the Ti16O16(OEt)24(OPrn)8 cluster. The stability of the inorganic core was demonstrated [5] and the substitution reactions lead to an exchange of only eight ethoxide groups with propyl alcohol. The 13C spectrum obtained after a one-day reaction is in agreement with this result. No interpretation in terms of assignment with the crystallographic structure has already been done. Fig. 5 shows the two spectra obtained for the both clusters, Ti16O16(OEt)32 and Ti16O16(OEt)24(OPrn)8. The ‘B’, ‘D’, and ‘E’ 13C signals are not present on the spectrum after substitution, but three new signals appear at 79.7, 78.2 and 71.9 ppm. Two of them are more deshielded and correspond to the propoxide groups, while the third one corresponds to a shift of an ethoxide group’s signal. If there are only two signals for the propoxide groups, that may signify that only two kinds of ethoxide groups have been exchanged. On the 1H spectrum, only the ‘B’ and ‘E’ signals have disappeared, not the ‘D’ one. It means that ‘E’ and ‘B’ signals correspond to ethoxide groups that have been exchanged with propyl alcohol and that the ‘D’ signal has been shifted in the 13C spectrum.
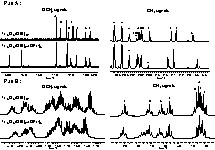
Comparison between Ti16O16(OEt)32 and Ti16O16(OEt)24(OPrn)8 liquid-state NMR spectra. (A) Details of 13C NMR spectra. (B) Details of 1H NMR spectra.
Fig. 6 (limited to the methylene part of 13C spectrum and the methyl part of 1H spectra) shows the evolution of the trans-alcoholysis reactions at 323 K during 6 h (delay not long enough to substitute the eight ethoxide groups). The large excess of propyl alcohol modifies the magnetic susceptibility and leads to a shielding of all signals compared to the first spectrum presented (Fig. 6). After 5 min, the intensity of the ‘B’ signal decreases significantly and a new 13C signal emerges at about 79.5 ppm, corresponding to the first propoxide group incorporated in the cluster. At the same time, the ‘E’ signal has been slightly deshielded. After 1 h, the ‘B’ signal has totally disappeared. The ‘E’ signal starts vanishing, whereas a second deshielded 13C signal emerges at about 78 ppm, corresponding to a second type of propoxide groups. At the same time, the ‘D’ and ‘H’ signals have been significantly deshielded. Disappearance of the ‘E’ signal is quite effective after 6 h.
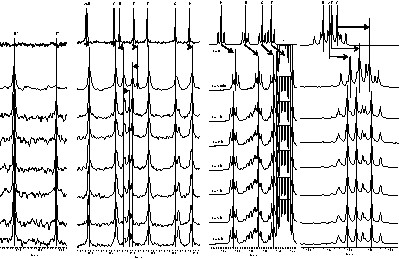
On the left, methylene region of 13C spectrum. On the right, methyl region of 1H spectrum. t is the time spent after introduction of propyl alcohol. * Toluene signal.
The last 13C spectrum was recorded at ambient temperature and presents the signatures of a mixture of Ti16O16(OEt)24(OPrn)8 and Ti16O16(OEt)32 clusters.
So, time is needed to achieve the nucleophilic substitution and this time will probably increase with longer carbon chain alcohols (work in progress). Also, only two kinds of ethoxide groups are reactive towards propyl alcohol, corresponding to ‘B’ and ‘E’ NMR signals. One of these two groups is quickly exchanged (‘B’ signal) compared to the other (‘E’ signal). If we look at the steric hindrance, ending ethoxide groups 1 and 3 (Fig. 1) are the most accessible, and they can be assigned to the ‘E’ and ‘B’ signals, respectively.
5 Conclusion
A better understanding and control of the local and semi-local structure of organic-inorganic hybrid materials is an important issue, especially if tailored properties are thought. To achieve the control of the material structure, the assembling of well-defined nanobuilding blocks is an interesting approach. The feasibility of the trans-alcoholysis reactions on the cluster Ti16O16(OEt)32 allows us to envisage functionalisation of this cluster with reactive groups such as polymerisable functions, in order to cross-link by covalent bonds a polymer matrix. Nevertheless, the reactivity of the surface of this cluster is not understood yet. This study shows the difficulties encountered to characterize its surface. A better control of its surface reactivity should lead to a better control of the material structure. The use of NMR methods in the liquid and the solid state allows good understanding of the organic ligands. We reported in this paper our first results dealing with this characterization. We demonstrated that chemically equivalent ethoxide groups do not have the same reactivity toward transalcoholysis by propyl alcohol. This could open new perspectives for the cross-linking on selected positions in hybrid materials.