

1 Introduction
Perylene dyes have found widespread applications ranging from industrial pigments [1] to components of molecular photonic and electronic devices. In recent years, there has been growing interest in developing organic chemistry, photochemical and photophysical properties of perylene-3,4:9,10-bis(dicarboximide) (PDI) derivatives because of their exceptional chemical and photochemical stability [2]. These molecules, classified as n-type semiconductors [3], have been used as building blocks for molecular switches, wires, logic gates [4], organic field-effect transistors [5], organic light-emitting diodes [6] and solar cells [7]. Their possibility to undergo photoinduced energy or electron transfer process justified also their attractivity for the artificial mimicking of photosynthesis and light-harvesting applications [8]. In these purposes, molecular systems that contain electron or energy donor–acceptor pairs are of particular interest with regard to the possibility to control this energy or electron transfer. Efforts are especially stimulated to create artificial donor–acceptor architectures in an attempt to mimic the conversion of light into chemical or electrical energy. Some examples of based PDI-dyads have been reported very recently in the literature, those concerning the use of oligo(p-phenylenevinylene) unit [9], pyrene or ferrocene [10] as the donor. In this work, we have been interested in the synthesis, electrochemical and photophysical properties of PDI-based dyads 1 and 2 in which this highly fluorescent acceptor perylene-3,4:9,10-bis(dicarboximide) is covalently attached to the non fluorescent tetrathiafulvalene (TTF)1 or the high fluorescent perylene-3,4-mono(carboximide) (PMI) (Scheme 1)2.
2 Results and discussion
2.1 Synthesis
The most appropriate retrosynthetic analysis to reach dyads 1 and 2 consists in an esterification reaction involving carboxylic acids 3 and 4 in the TTF and PMI series, respectively, and the unique PDI derivative 5 functionalized by the alcohol group (Scheme 2).

The synthesis of TTF monoacid 3 started by the trimethylphosphite-mediated cross-coupling of the two corresponding 2-(thi)oxo-1,3-dithiole moieties affording 2,3-bis(methyloxycarbonyl)TTF 6 in 59% yield. [13] Further monodecarboxymethoxylation of the diester 6 was cleanly achieved with LiBr in DMF under reflux (95% yield) and subsequent ester hydrolysis of compound 7a afforded TTF monoacid 3 in 90% yield according to reported procedures (Scheme 3) [14].
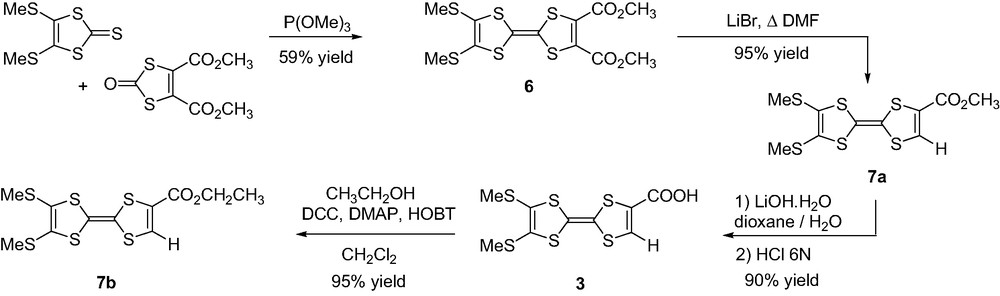
A chief challenge in working with PMI or PDI dyes is to overcome their low solubility. A widespread approach consists in incorporating 2,6-diisopropylphenyl groups located at the N-imide positions. Thus N-(2,6-diisopropylphenyl)perylene-3,4-dicarboximide was used as the starting material for preparing PMI derivative 4. Bromination using bromine in chlorobenzene [15] afforded a mixture of mono and tribrominated compounds 8a and 8b, respectively, which were separated by silica gel column chromatography (CH2Cl2 as the eluent). The electron-donating character of the PMI derivative was enhanced with substitution of the bromine atom by the pyrrolidinyl group [12] leading to compound 9 in 68% yield. In order to introduce the carboxylic acid functionality, the 2,6-diisopropylphenyl group was first removed to afford 9-(N-pyrrolidinyl)perylene-3,4-dicarboximide 10 in 25% yield. Required carboxylic acid 4 was further obtained in quantitative yield by reaction with glycine in alkaline medium (Scheme 4).
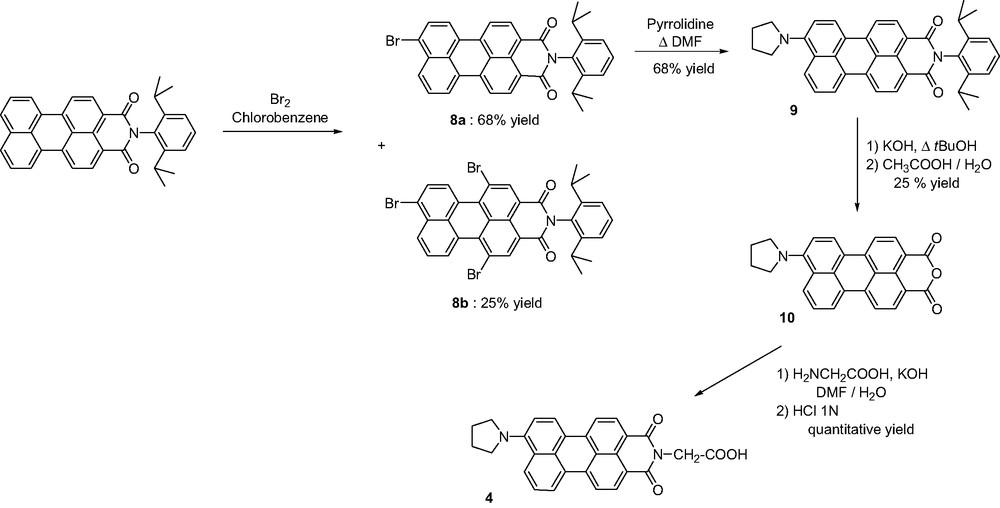
The other strategy commonly used to easier manipulate the quite insoluble PMI and PDI derivatives requires the introduction of substituents at the bay region, such as chlorine atoms. Their presences undergo a twist of the perylene core, therefore restricting π-stacking interactions. For the synthesis of compound 5, we have chosen to develop a new methodology to reach dissymmetrical tetrasubstituted perylenediimide at the bay region. Instead of applying methods reported to reach 3,4:9,10-perylenetetracarboxylic monoanhydride monoimide derivative [16], we have realized the direct condensation of 1-pentylamine and 2-ethanolamine in stoichiometric ratio on 1,6,7,12-tetrachloroperylenetetracarboxylic dianhydride as the starting material. After precipitation of symmetrical perylenediimide dialcool 11, both symmetrical and dissymmetrical compounds 12 and 5, respectively, were separated by column chromatography on silica gel (CH2Cl2 followed by CH2Cl2/EtOAc 1:4 as the mixture of eluents) (Scheme 5).

Esterification between TTF monoacid 3 and perylenediimide monoalcool 5 in the presence of DCC as an activating reagent with both DMAP and HOBT afforded dyad TTF–PDI 1 in 94% yield. TTF derivative 7b was thus prepared in 95% yield to act as a reference compound for further electrochemical and photophysical studies. Using the same reaction, dyad PMI–PDI 2 was synthesized in 34% yield by replacing TTF monoacid 3 by PMI monoacid 43.
2.2 Electrochemical and optical properties
The cyclic voltammogram (Fig. 1a and Table 1) of TTF–PDI 1 showed two one-electron reversible oxidation waves at E1/2 ox1 = + 0.65 V and E1/2 ox2 = + 1.05 V (vs. AgCl/Ag) corresponding to the successive generation of the cation radical (TTF+•–PDI) and dication (TTF2+–PDI) of the TTF moiety. The oxidation of the PDI moiety was not observed as it should occur above 1.95 V, which is the limit potential in our experimental conditions. As chlorine atoms at the bay region of the perylenediimide core present electron-withdrawing effects, the oxidation potential of 12 must be shifted to a more positive value by comparison with the non-substituted perylenediimide [17]. Two one-electron reversible reduction processes were shown for the PDI moiety at E1/2 red1 = –0.32 V and E1/2 red2 = –0.52 V which were ascribed to the successive formation of the anion radical (TTF–PDI–•) and dianion (TTF–PDI2–) of the perylene-3,4:9,10-bis(dicarboximide) unit.


(a) Cyclic voltammogram of TTF–PDI (c = 10–3 M) in n-Bu4NPF6 0.1 M in CH2Cl2, platinum wire as working electrode and AgCl/Ag as reference electrode. (b) Cyclic voltammogram of PMI–PDI (c = 2.5 10–4 M) in n-Bu4NPF6 0.1 M in CH2Cl2, platinum wire as working electrode and AgCl/Ag as reference electrode.
Electrochemical data for 1, 2 and reference compoundsa
| Compound | E1/2 red4 | E1/2 red3 | E/2 red2 | E1/2 red1 | E1/2 ox1 | E1/2 ox2 |
| 1 | – | – | –0.52 V | –0.32 V | 0.65 V | 1.05 V |
| 12 | – | – | –0.53 V | –0.34 V | –b | –b |
| 7b | – | – | – | – | 0.63 V | 1.05 V |
| 2 | –1.51 V | –1.00 V | –0.53 V | –0.33 V | 0.69 V | 1.40 V |
| 8a | – | – | –1.34 V | –0.89 V | 1.50 V | – |
| 9 | – | – | –1.54 V | –1.07 V | 0.67 V | 1.37 V |
a CV measurements in dichloromethane solution using n-Bu4NPF6 (0.1 M) as supporting electrolyte, platinum wire as working electrode and AgCl/Ag as reference electrode.
b Not measurable.
The cyclic voltammogram of PMI–PDI 2 (Fig. 1b and Table 1) showed two one-electron reversible oxidation wave at E1/2 ox1 = + 0.69 V and E1/2 ox2 = + 1.40 V (vs. AgCl/Ag) corresponding to the successive generation of the pyrrolidinium cation then the oxidation of the PMI unit of 2 [18]. Four one-electron reversible reduction processes were shown at E1/2 red1 = –0.33 V, E1/2 red2 = –0.53 V, E1/2 red3 = –1.00 V, and E1/2 red4 = –1.51 V. These were assigned to the successive generation of the anion radical (PMI–PDI–•), then the dianion (PMI–PDI2–), the trianion radical (PMI–•–PDI2–) and finally the tetraanion species (PMI2––PDI2–).
The UV–vis spectrum of dyad 1 in CH2Cl2 showed a wide absorption in the visible range between 400 and 550 nm with the absorption maximum centered at λ = 517 nm. This absorption feature could be attributed to the PDI moiety by comparison with absorption spectra of both references 7b and 12. This UV–vis absorption spectrum matches the profile obtained by summation of the spectra of both donor and acceptor units, indicating the absence of significant interaction in the ground state between TTF and PDI for dyad 1, this being confirmed by electrochemical measurements. The UV–vis spectrum of dyad 2 in CH2Cl2 was characterized by a supplementary broad absorption band with the maximum at λ = 620 nm corresponding to the absorption of the PMI part, since compound 9 exhibited the same characteristic features at this wavelength (Fig. 2).
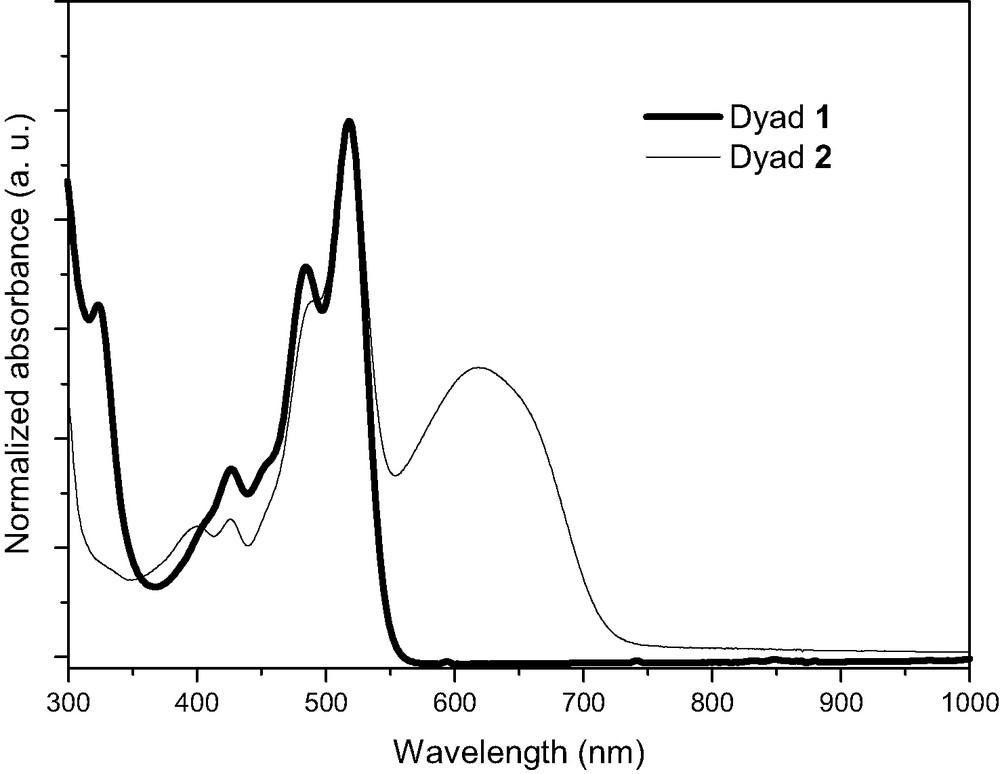
Normalized absorption spectra of dyad 1 and dyad 2 in dichloromethane.
We were first interested in studying the TTF–PDI dyad 1 since one of the most interesting properties of TTF is that it can be oxidized successively and reversibly to the cation radical and dication species within a very accessible potential window.
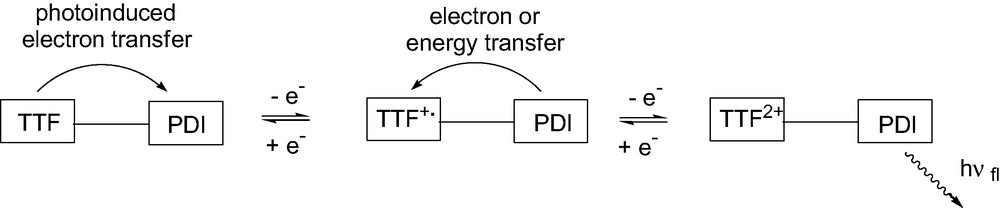
The steady-state fluorescence emission in CH2Cl2 of PDI was quasi quantitatively quenched in dyad 1 (99.5% compared to fluorescence emission of 12 in same conditions). This could be explained by an intramolecular photoinduced electron transfer (PET) from the donor TTF to the acceptor PDI. Thus it was expected that the chemical or electrochemical oxidation of TTF would hinder the fluorescence quenching. Indeed, intramolecular PET would not be possible anymore as TTF loses its donating character upon oxidation. Experiments of emission fluorescence measurements coupled to electrochemistry were carried out at controlled potentials [19]. After applying an oxidation potential of +0.85 V where the TTF+•–PDI species must be predominant, surprisingly the fluorescence emission of the PDI part was not recovered. This behavior could result from a new quenching process such as an energy transfer from PDI to TTF+•. This process is allowed according to the energy position of the lowest singlet excited states of both PDI and TTF+• units, which can be estimated at 2.32 and 1.61 eV, respectively (the energy position of the lowest singlet excited state of TTF unit was estimated at 2.85 eV, which hindered such energy transfer process)4. A reverse electron transfer from PDI to TTF+• species could be also envisaged. Finally, when the potential was fixed at +1.25 V, where TTF2+–PDI must be the main species present, the fluorescence intensity increased slowly to reach a stable limit value after more than 5 min. Energy transfer from PDI to TTF2+ could exist as the energy position of the lowest singlet excited state of TTF2+ unit, was estimated at 1.90 eV. Chemical oxidation experiments were performed in order to confirm the behavior of this TTF2+–PDI species. Thus, dyad 1 in CH2Cl2 solution was treated by an excess of (diacetoxyiodo)benzene in the presence of triflic acid (PhI(OAc)2/CF3SO3H) used as oxidizing reagent [20]. The same phenomenon was observed since the fluorescence emission intensity increased to a limit value corresponding to around 16% of the initial fluorescence emission of the PDI reference compound 12.
To study the reversibility of the process, zinc powder was added in excess to this solution in order to reduce TTF2+–PDI into neutral TTF–PDI. The initial fluorescence spectrum of the PDI in the dyad was completely recovered.
3 Conclusion
We have demonstrated that the modulation of the fluorescence emission intensity (quenching by electron and/or energy transfer or partial restoration) in dyad 1 can be reversibly controlled depending on the oxidation state of the TTF unit (Fig. 3). Such a unique behavior can be attributed to peculiar properties of the TTF linked to the PDI acceptor which fluoresces intrinsically and consequently this dyad 1 can be considered as a new redox-fluorescence molecular switch.
Photophysical studies of dyad 2 are currently underway and this dyad should present a different behavior because an energy transfer process could be allowed from PDI to PMI, according to preliminary UV–vis and fluorescence emission spectroscopic investigations.
Acknowledgments
The authors acknowledge the ‘Région Pays de la Loire’, the CNRS, TotalFinaElf and the ‘Conseil général du Maine-et-Loire’ (France) for the grants of L. Perrin and J. Baffreau, respectively, ADEME and CEA through the CSPVP ‘Cellules solaires photovoltaïques plastiques’ research program, BASF-AG (Ludwigshafen) for providing starting materials (1,6,7,12-tetrachloroperylenetetracarboxylic dianhydride and N-(2,6-diisopropylphenyl)perylene-3,4-dicarboximide).
1 To our knowledge, only a triad TTF–PDI–TTF has been recently reported and partial quenching of fluorescence was ascribed to a photoinduced electron transfer interaction. In our work, we have been interested in the system TTF–PDI 1 because results obtained from a dyad with the 1:1 stoichiometry between donor and acceptor are expected to be unambiguous compared to those resulting from a triad in which the role of the partner in excess is usually not really defined [11].
2 To our knowledge, only a perylene-3,4-mono(carboximide) unit linked to a naphthalene-1,8:4,5-bis(dicarboximide) (PMI-NI) dyad has been recently reported to produce new materials for photorefractive liquid crystals composites in the near-infrared region [12].

3 All new compounds gave satisfactory analytical and spectral data.
4 Values corresponding to the peak of highest energy featured in absorption spectra or UV–vis spectroelectrochemical spectra recorded in dichloromethane.