

1 Introduction
Aminoglycosides are broad-spectrum antibiotics which act by binding to the decoding site of ribosomal 16S RNA [1,2], they are used with β-lactams in polytherapies against severe infections caused by gram-negative bacteria and staphylococci, mostly of nosocomial origin. Resistance to these antibiotics arise mainly through the action of aminoglycoside-modifying enzymes which catalyze the covalent addition of acetyl, phosphate or nucleotidyl groups onto amino or hydroxyl functions [3]. Among the modifying enzymes found in clinical strains, N-6′ aminoglycoside acetyl transferases (AAC(6′)) are the most prevalent ones, accounting for 50–75% of the resistance phenotypes (their mechanism is shown in Fig. 1A). These have been classified in subfamilies (AAC(6′)-I, AAC(6′)-II, AAC(6′)-III and AAC(6′)-IV, see [2]), based on their specificity profile vs. the various clinically used aminoglycosides (Gentamicin, Amikacin, Isepamicin…). This picture has recently been getting more complicated, as very broad-spectrum variants have begun to emerge [4], which confer resistance to almost all aminoglycosides and are thus a serious threat to current therapies.
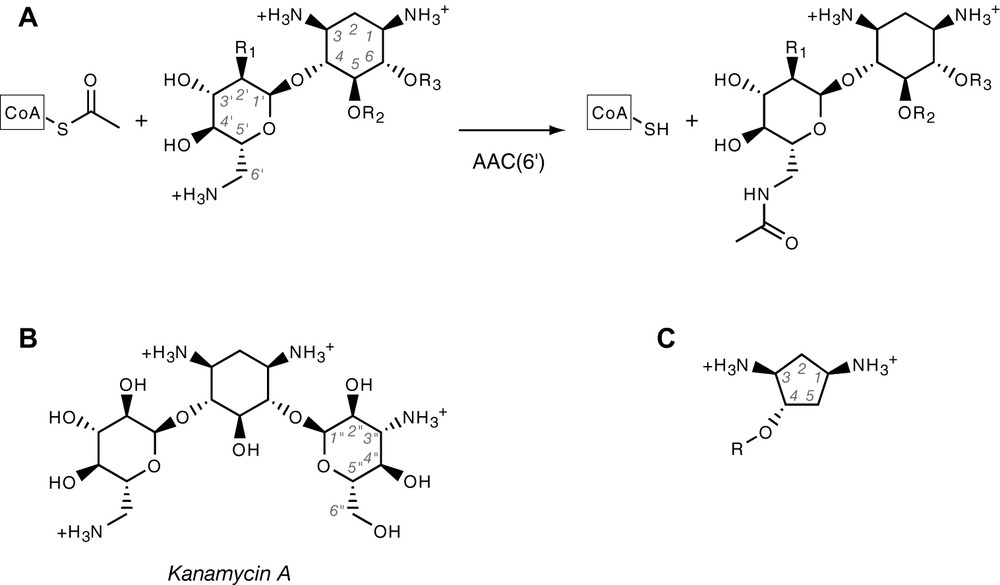
(A) Aminoglycoside acetylation reaction catalysed by AAC(6′) enzymes. Shown is the common core structure, which corresponds to neamine. Amines are shown in protonated form, as they usually are at physiological pH. CoA stands for Coenzyme A. R1 is either –OH or –NH2. Aminoglycosides are either substituted at R2 or R3, yielding the neomycin and kanamycin family, respectively. (B) Structure of kanamycin A, showing the quasi-symmetry about the median plane of deoxystreptamine ring. (C) Generic structure of the deoxystreptamine analogues used in this study.
In order to circumvent this problem, it would be desirable to either isolate new aminoglycosides, which escape the current resistance mechanisms or to design specific inhibitors of their modifying enzymes. Up to now, however, progress in these two directions has been hampered by the difficulties of aminoglycoside chemistry. These result from the combination of two factors: the large number of functional groups and chiral centers in these molecules and their intrinsic symmetry (see for instance kanamycin, Fig. 1B) and in particular that of the central ring, 2-deoxystreptamine (2-DOS), a meso compound. Synthetic routes to this compound are known but are still quite involved (reviewed in [5]) and attempts to replace it with alternate, simpler scaffolds have so far only been moderately successful [6]. There is thus a need for an efficient 2-DOS mimic, as a building block for aminoglycoside resistance enzyme inhibitors. The present work describes the identification of such a compound and of an NMR-based strategy to characterize derivatives of this molecule with improved affinities to one of the new extended-spectrum, clinical forms of AAC(6′), AA(6′)-Ib11 [4].
2 Materials and methods
2.1 Enzyme expression and purification
The enzyme used in this study is an extended-spectrum aminoglycoside 6′ N-acetyl transferase of type Ib isolated from a clinical Salmonella strain [4]. A recombinant expression system was constructed by cloning the PCR-amplified DNA coding sequence in plasmid pET101, using the pET101/D-Topo expression kit (Invitrogen). The resulting recombinant plasmid encodes the 189 aminoacid subunit of AAC(6′)-1b11 [4] under control of the T7 transcription promoter. Expression of the protein was obtained after transformation of the plasmid into strain BL21Star (Invitrogen). Cells were grown at 37 °C in LB medium supplemented with ampicillin (100 mg/l) until turbidity reached an absorbance of 1.0 at 650 nm. They were then induced by addition of 0.5 mM isopropyl-thiogalactoside (IPTG), incubation was then continued for 2 h at 30 °C. Bacteria were harvested by centrifugation, resuspended in 20 mM sodium phosphate pH 7.5 (buffer A) and lysed by sonication. Cell debris were removed by centrifugation and the crude extract was loaded on a Superdex G75 gel filtration column (2.6 × 60 cm, Amersham) equilibrated in buffer A. Fractions containing the overproduced enzyme were pooled and submitted to an ion-exchange chromatographic step: after loading on a Q-Sepaharose Hiload column (2.6 × 20 cm, Amersham) equilibrated in buffer A, the protein was eluted by applying a 0–500-mM linear NaCl gradient. The pooled protein fractions were dialyzed against 20 mM Tris–HCl pH 8.5 and submitted to a second ion-exchange chromatographic step in this buffer. Except for the increased pH, it was identical to the first ion-exchange separation. After this step, the protein was homogeneous, as judged by SDS-gel electrophoresis. It was dialyzed against 10 mM HEPES pH 7.0 and concentrated by ultrafiltration on a Centriprep cell (Amicon-Millipore). Yield was 7.5 mg of purified enzyme for a 1 l of culture.
2.2 Ligands
Kanamycin A sulfate was from Amersham-USB. Synthesis of compound 1 has been described [7], and that of 2 and 3 will be described elsewhere (Begis & Micouin, unpublished). Concentrated stock solutions (50 mM) were prepared in H2O from dry chlorhydrates (compounds 1, 2 and 3) or sulfate (Kanamycin) and the pH was adjusted to neutrality.
2.3 NMR methods
Spectra were recorded on a 600-MHz Bruker Avance spectrometer equipped with either a 5-mm standard triple resonance inverse probe, or with a 3-mm triple resonance flow-injection probe connected to a LC215 Gilson pipetting robot (Bruker BEST system). All spectra were recorded in 20-mM sodium phosphate buffer (pH 7.0) prepared in 90% H2O/10% 2H2O (by volume) and the solvent resonance was suppressed by presaturation, excitation sculpting [8] or by gradient selection for heteronuclear experiments. Resonance assignments for kanamycin were obtained using a 50-mM sample in a 5-mm tube. 2D ROESY, DQF-COSY and [1H–13C] HSQC as well as a natural abundance 3D [1H–13C] TOCSY–HSQC were recorded. The latter experiment was required to unambiguously assign the severely overlapping sugar proton massif. All experiments were recorded at 25 °C (298 K). Stereoselective assignments of the methylene protons of carbon 2 of both kanamycin and compound 1 were obtained by analyzing the relative intensity of the ROESY transfers and J-couplings with protons 1 and 3 (numbering as in Fig. 1). Assignments are indicated on Fig. 2. Ligand binding was monitored by the Saturation Transfer Difference (STD) [9] and/or the reverse NOE pumping methods [10], using, the flow-injection NMR system. Sample injection was as previously described [11], briefly, the ligand and the protein were mixed in a final volume of 180 μl of 20-mM sodium phosphate pH 7.0 and put in 96-well plates. Final concentrations were 1 mM for the ligand and 50 μM for the protein. For STD experiments, band irradiation of the protein methyl massif (0.5 ppm) was performed for 2 s at field strength of 80–100 Hz. For reverse NOE pumping, mixing times of 200 and 700 ms were used, in order to detect both strong and weak transfers.
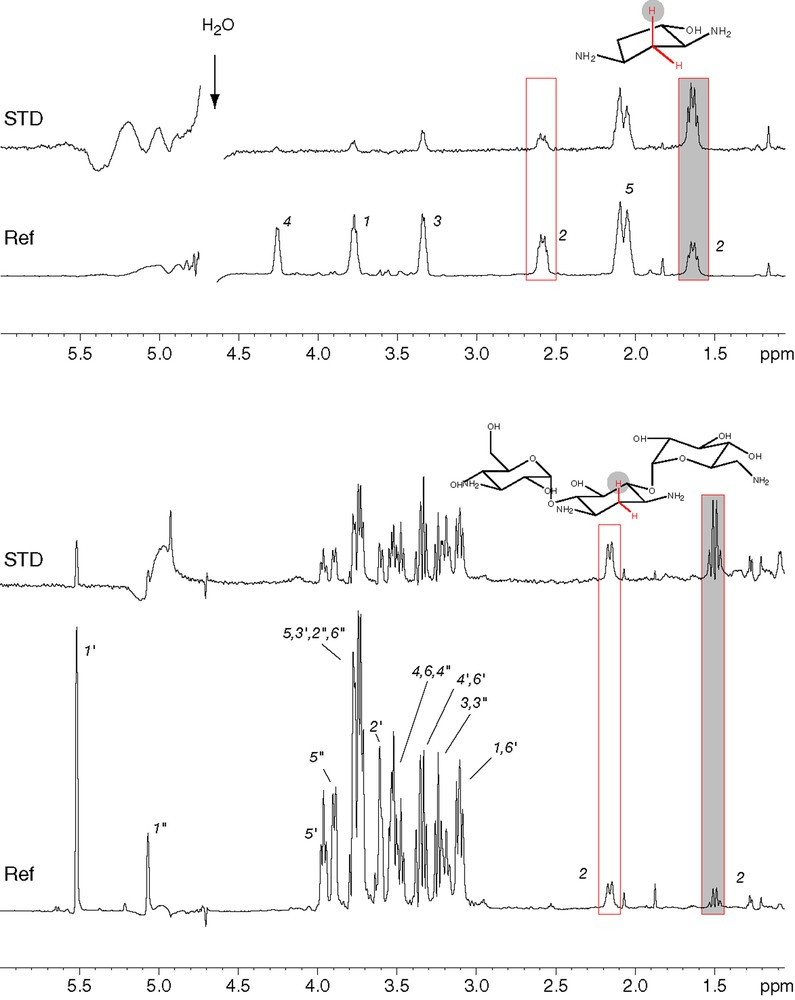
STD analysis [9] of the binding of kanamycin (bottom) and the deoxystreptamine analogue 1 (top) to AAC(6′)-Ib11. Assignments of the various protons are indicated on the reference spectra (Ref). The boxed peaks correspond to protons of the CH2 group located in between the two amino groups. In both cases, the shaded box corresponds to the axial proton of this methylene group and shows the strongest STD signal.
2.4 Biochemical affinity measurements
Fluorescence measurements were performed in a Jasco FP6200 spectrophotofluorimeter equipped with a thermostated cell. The enzyme concentration was 80 nM in 1 ml of 20 mM sodium phosphate buffer pH 7.5. Increasing amounts of the ligands were iteratively added to the protein solution, which was magnetically stirred. The intrinsic fluorescence of tryptophan residues was followed using an excitation wavelength of either 280 or 295 nm and an emission wavelength of 343 nm. All measurements were performed at 21 °C. All points were measured in duplicate and the binding parameters were derived by iterative non-linear least square fitting to a single hyperbolic function [12]. Additional binding data were also obtained by equilibrium dialysis, using disposable microcells (DispoEquilibrium Dialyzer, Harvard Biosciences, Hollington, USA). The enzyme compartment (75 μl) contained 200 μM of purified AAC(6′)-Ib and the ligand compartment (75 μl) contained an equal concentration of compound 2, either with or without 200 μM of kanamycin A. After equilibration, relative ligand concentrations in the equilibrium dialysis cell compartments were monitored by UV spectrophotometry. Dissociation constants were estimated with Mathematica (Wolfram Research).
3 Results
3.1 Design of an analogue of 2-deoxystreptamine
The common core structure recognized by N-6′ aminoglycoside acetyl transferases corresponds approximately to that of neamine (Fig. 1A), i.e. a 6-glucosamine attached to a 2-deoxystreptamine ring (2-DOS). Substituents attached to positions 5 and 6 (R1 and R2, Fig. 1A) appear not to be involved in interactions with these enzymes. This is confirmed by the crystal structure of AAC(6′)-Iy complexed with ribostamycin [13] for which the R2 group is a ribose, which lies in the solvent, making little if any contacts with the protein. This is in contrast with the 2-DOS ring, which makes electrostatic interactions with the enzyme via its two amino groups and is stacked on aromatic side chains. Thus, any scheme for designing an inhibitor of AAC(6′) must include 2-DOS or a structural analogue of it. However, 2-DOS chemistry is quite difficult, making its direct use a serious challenge and we thus chose to try and replace it by similar, more accessible scaffold. The features which we found desirable for an analogue of 2-DOS in the context of AAC(6′) inhibition were: (i) a constrained cyclic structure which would allow stacking within the enzyme active site, (ii) a pair of amino groups with a geometry close to that of 2-DOS, and (iii) an additional function for linking 6-glucosamine or a similar group in an analogous geometry. We selected derivatives of trans, trans diamino-cyclopentanol (Fig. 1C and Fig. 3) as these fulfilled the three above criteria, have no additional reactive groups apart from those strictly necessary, are no longer symmetrical and can be easily synthesized enantioselectively [7].
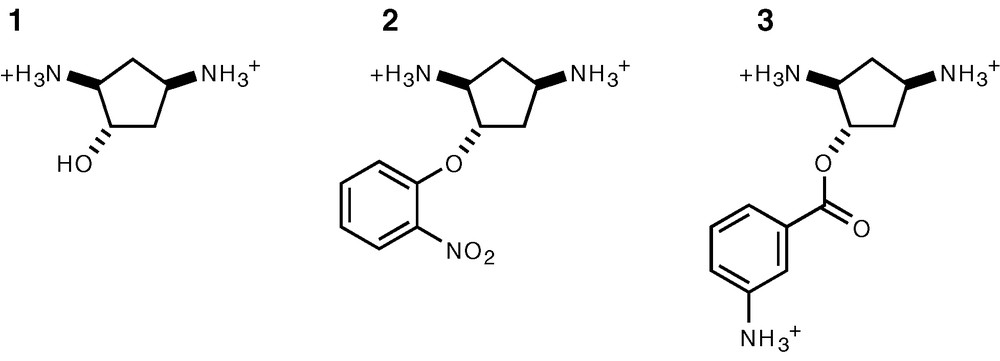
Structure of the AAC(6′) ligands used in this study. These were racemic mixtures of the two possible enantiomers.
3.2 diamino-cyclopentanol binding to AAC(6′)-Ib
The binding of diamino-cyclopentanol (compound 1, Fig. 3) to AAC(6′)-Ib was analyzed and compared to that of kanamycin A, a natural substrate of the enzyme. As it is a very simple pharmacophore, one could expect it to have a significantly reduced affinity. We thus used NMR interaction screening, as this technique is able to detect weak interactions and provides information on the binding mode of the ligand. Both STD and reverse NOE pumping experiments were used to evidence magnetization transfer between the protein and specific protons from the ligand. Similar results were obtained with both techniques and a typical STD experiment is shown in Fig. 2. Two pieces of information can be derived from these approaches: observation of transferred magnetization demonstrates that compound 1 binds efficiently to the enzyme. For both STD (Fig. 2) and NOE pumping (not shown), a significantly stronger signal is observed for the axial proton located on carbon 2 (Fig. 2, numbering as in Fig. 1). This indicates that kanamycin binds the enzyme with the amino face of 2-DOS in close contact with AAC(6′)-Ib, a result which is in keeping with the crystal structure of the distantly related AAC(6′)-Iy [13], and that compound 1 binds in a similar fashion, also with the two amino groups facing the enzyme surface. Equilibrium dialysis experiments confirmed that diamino-cyclopentanol derivatives such as compound 2 do bind to AAC(6′)-Ib, with a Kd in the 10−5 M range, and that this interaction is efficiently antagonized by kanamycin (not shown) and thus that binding is competitive. This experiment was performed with compound 2, as its concentration can be easily monitored by UV absorption. Overall, this strongly suggests that binding of diamino-cyclopentanol and its derivatives occurs at the same site and in the same orientation as that of 2-DOS in kanamycin.
3.3 NMR provides qualitative information on AAC(6′) ligand affinity
The above results show that compound 1 is indeed a simple scaffold that can mimic 2-deoxystreptamine in the design of competitive inhibitors of AAC(6′) enzymes. Because of its small size, its binding affinity, is however, limited and it failed to significantly inhibit the enzyme in standard acetylation assays (not shown). Nevertheless, it provides a specific anchor point on the enzyme, from which more complex molecules can be constructed, by linking additional pharmacophores to improve the affinity. Several derivatives of compound 1 were thus synthesized by substituting the hydroxyl group with different types of linkages. Two such compounds with aryl groups replacing the 6-glucosamine groups are shown in Fig. 3. Because of the lack of an efficient enzymatic test at this early stage of inhibitor design, we wanted to investigate whether NMR interaction screening techniques such as reverse NOE pumping could provide qualitative information on the binding mode and affinity of these bipartite ligands of AAC(6′). We chose compounds 2 and 3 (Fig. 3) since their NMR spectra show completely separate signals for the cyclopentane moiety and the aromatic ring, on either side of the spectral window (Fig. 4). It was expected that for all such ligands, the constant cyclopentane ring would bind in most cases, whereas, depending on the nature of the linkage and of the substituent, the other moiety would either fit into the active site or be rejected outside the protein. In the former case, NOE transfer is expected to occur for both parts of the molecule, whereas in the latter, transfer would only occur for the cylcopentane moiety, and not for the substituent. The result of such an experiment for compounds 2 and 3 is shown in Fig. 4. Compound 2 does show a contact between an aromatic proton and the protein (emphasized by a black circle on Fig. 4), whereas compound 3 does not show any significant transfer. This suggests that the aromatic ring of compound 2 slips at least in part into the active site, whereas that of compound 3 cannot, possibly because of the different linkage. In order to see whether these observations could be correlated with the affinity of these two ligands for the enzyme, fluorescence titration were performed. We indeed observed that ligand binding induces a large quenching of the intrinsic fluorescent signal of tryptophan residues (50–70% quenching). This was not unexpected as AAC(6′)-I enzymes share conserved tryptophan residues within their active sites, on which the 2-DOS ring of the substrate was observed to stack [13]. The resulting fluorescence data were compared to that obtained with the parent compound 1, in order to see whether the substituents provided either an improvement or a loss of affinity, and this is shown on Fig. 5. The three compounds indeed showed differential affinities, with calculated dissociation constants of 41 ± 3 μM for compound 1, 23 ± 2 μM for compound 2 and 57 ± 6 μM for compound 3. This correlates very well with the NMR results, as the additional contact zone observed with 2 indeed results in a twofold improvement of the affinity, whereas the small loss observed with 3 can probably be ascribed to a steric hindrance of the substituent, which apparently does not fit into the protein.

Interaction of ligands 2 (top) and 3 (bottom) with AAC(6′)–Ib11. Shown are reference proton spectra (Ref) immediately above the corresponding reverse NOE pumping experiments (NOE), which show, the intermolecular NOE transfer between the ligands and the protein [10]. Aliphatic resonance (1.5–5.5 ppm) belongs to the cyclopentane ring, whereas the leftmost protons (6.5–8.0 ppm) correspond to the aromatic ring substituent.

Fluorescence titration of the binding of the various ligands to AAC(6′)-Ib. The fluorescence data were normalized, so that the initial signal was set to 100% and the fully quenched signal extrapolated at infinite ligand concentration corresponded to 0%. Individual data points are shown on top of the fitted curves. The titration of the reference compound 1 is shown with solid circles and a thick line. Data corresponding to compounds 2 and 3 are indicated by diamonds and a thin line, or by triangles and a dashed line, respectively.
4 Conclusion
This work describes an iterative strategy for designing ligands of AAC(6′), which can be used for isolating inhibitors of these antibiotic resistance enzymes. It is based on the characterization of a primary pharmacophore showing specific binding, the affinity of which is subsequently improved by ‘decorating’ it with additional functional groups. Both stages of this approach rely on the use of NMR to detect and characterize ligand interactions. In the first step, NMR was used to reveal weak but specific interactions of simple compounds, which cannot be reliably detected by enzymatic tests. It also allowed controlling that they were likely to bind in the correct orientation. In a latter stage, the present study shows that the NMR approach allows a qualitative ranking of the various substituents of the anchoring pharmacophore, diamino cyclopentanol. This can provide a rationale for the early guidance of medicinal chemists and thus could significantly speed up the discovery process. This is particularly interesting for those molecules, which cannot be easily tested by fluorimetric techniques. A number of the derivatives of compound 1, which were originally synthesized indeed either showed a significant fluorescent background or exhibited a significant UV absorbance at the typical tryptophan excitation wavelengths, which prevented their direct analysis by fluorescence methods. Those molecules can, however, be productively tested by the NMR approach. Using the above strategy, ligands with affinities in the micromolar range have presently been isolated, and all of these derive from an original and easily accessible scaffold. This is already significant and further application of this strategy should in principle allow for the identification of inhibitors with higher affinities. Its combination with robotized flow-injection NMR makes it attractive for a medium scale screening approach of aminoglycoside resistance enzyme inhibitors.
Acknowledgments
The authors gratefully acknowledge Drs. E. Collatz and O. Pajot for gift of strains and plasmids. F. Maurice is recipient of a studentship from the French ‘Ministère de la Recherche et de la Technologie’. Supported in part by an ACI ‘Jeune Chercheur’ to L. M.