

1 Introduction
Photovoltaic cells have gained widespread acceptance as a source of clean and renewable energy. Although the cost per peak watt of crystalline silicon solar cells has dropped significantly over the past decade, these devices are still too expensive to compete with conventional grid electricity without the benefit of government subsidies. The advent of the mesosocopic dye-sensitized solar cell (DSC) in 1991 introduced an innovative approach for low-cost alternatives to traditional inorganic photovoltaic devices [1,2]. At present, this type of inorganic-organic hybrid solar cell has attained an efficiency exceeding 11%, albeit with a volatile acetonitrile based electrolyte [3]. While the DSC exhibits excellent stability under long-term light soaking, e.g. only minor performance losses occur under light soaking for 8000 h at 2.5 times full solar the intensity, obtaining long-term stability at temperatures of 80–85 °C had remained a major challenge for over 10 years [4] and has only recently been achieved by judicious molecular engineering of the sensitizer used in conjunction with a robust and non-volatile electrolyte [5–11].
2 Operational principle of the mesoscopic dye-sensitized solar cell
A schematic presentation of the operating principles of the DSC is given in Fig. 1. At the heart of the system is a mesoscopic oxide semiconductor film, which is placed in contact with a redox electrolyte or an organic hole conductor. The material of choice has been TiO2 (anatase) although alternative wide band gap oxides such as ZnO, and Nb2O5 have also been investigated. Attached to the surface of the nanocrystalline film is a monolayer of the sensitizer. Photo-excitation of the latter results in the injection of an electron into the conduction band of the oxide. The dye is regenerated by electron donation from the electrolyte, usually an organic solvent containing a redox system, such as the iodide/triiodide couple. The regeneration of the sensitizer by iodide intercepts the recapture of the conduction band electron by the oxidized dye. The iodide is regenerated in turn by the reduction of triiodide at the counter-electrode the circuit being completed via electron migration through the external load. The voltage generated under illumination corresponds to the difference between the Fermi level of the electron in the solid and the redox potential of the electrolyte. Overall the device generates electric power from light without suffering permanent chemical transformation.
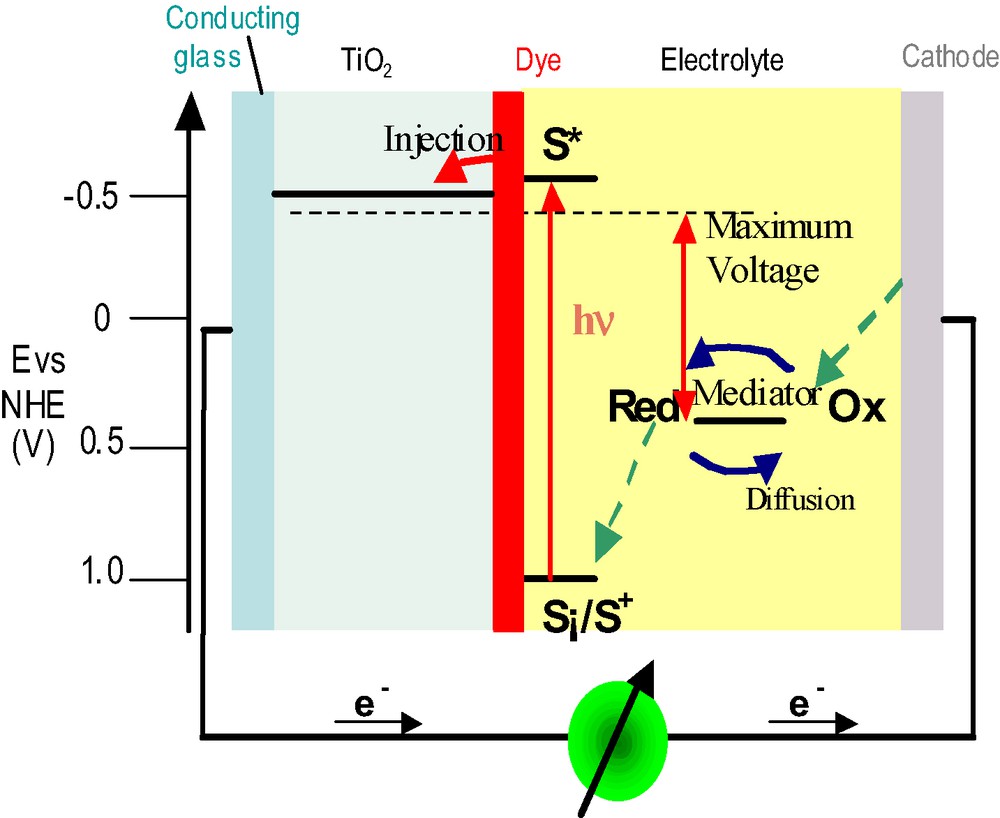
Principle of operation of the dye-sensitized nanocrystalline solar cell. Photo-excitation of the sensitizer (S) is followed by electron injection into the conduction band of an oxide semiconductor film. The dye molecule is regenerated by the redox system, which itself is regenerated at the counter-electrode by electrons passed through the load. Potentials are referred to the normal hydrogen electrode (NHE) The energy levels drawn match the redox poten tials of the standard N3 sensitizer ground state and the iodide/triiodide couple.
3 Kinetic criteria for long-term stability of the sensitizer
Fig. 2 illustrates the catalytic cycle that the sensitizer performs during cell operation. Critical for stability are any destructive side reactions that may occur from the excited state S* or the oxidized state of the dye (S+), which would compete with electron injection from the excited dye into the conduction band of the mesoscopic oxide and with the regeneration of the sensitizer. These ‘bleeding’ channels are assumed to follow first- or pseudo-first-order kinetics and are assigned the rate constants k1 and k2. Introducing the two branching ratios P1 = kinj/(k1 + kinj) and P2 = kreg/(k2 + kreg) where kinj and kreg are the first-order or pseudo-first-order rate constants for the injection and regeneration process, respectively, the fraction of the sensitizer molecules that survive one cycle is given by the product P1 × P2. After performing n cycles the remaining fraction of the sensitizer is:
| (1) |
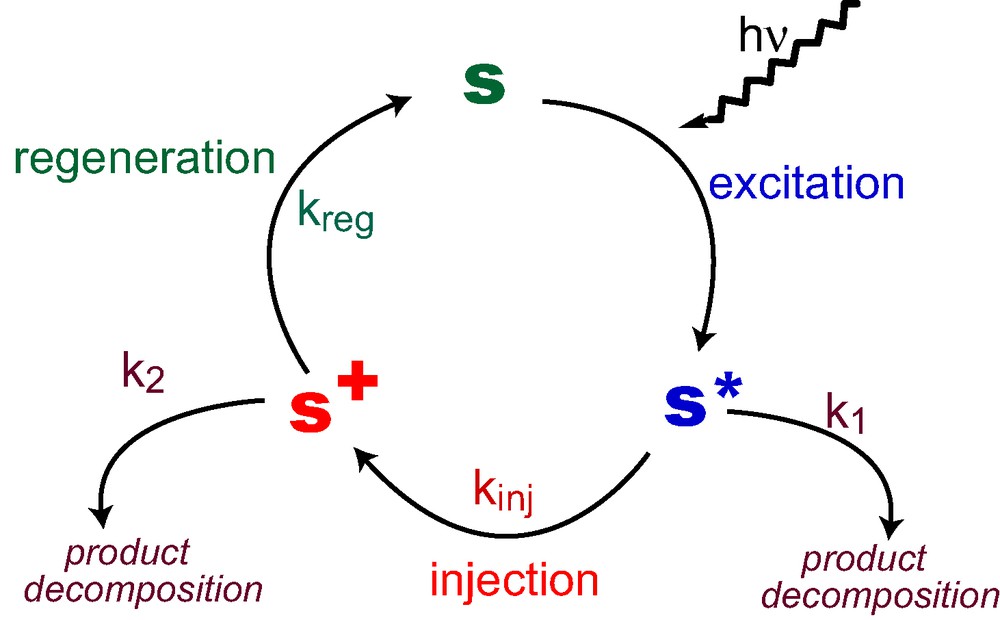
The catalytic cycle of the sensitizer molecules sustain under illumination, k1 and k2 are the rate constants (in s–1) for the destructive side reactions of the excited and oxidized forms of the sensitizer, respectively.
Introducing P1 × P2 = 1 – ɛ and rewriting Eq. (1) in logarithmic form gives:
| (2) |
For any sensitizer showing reasonable stability, the product P1 × P2 must be very close to 1 and hence ɛ << 1. This allows using the first term of a Taylor expansion for the logarithm, yielding ln (1 – ɛ) = – ɛ. Hence:
| (3) |
Expressing P1 and P2 by the rate constants and taking into account that k1 << kinj and k2 << kreg, one obtains:
| (4) |
Finally, as we are interested in the lifetime τ of the dye, we note that at the time t = τ, ln ([S]/[S]t = 0) = –1. Therefore:
| (5) |
Thus the simple result of this consideration is when the outside exposure time of the cell reaches the sensitizer lifetime, the sum of the branching ratios for the two decomposition channels becomes equal to the reciprocal value of the turnover number of the dye.
Using Eq. (5), the branching ratios that correspond to a sensitizer lifetime of 20 years under outdoor operation of the solar cell can now be calculated. The surface concentration of the currently employed ruthenium complexes is about 2 × 10–7 mol cm–2 and the photocurrent delivered in full sun is between 15 and 20 mA cm–2. Assuming for the photocurrent density, averaged over day and night and over the different seasons a value of 3 mA cm–2, the turnover frequency of the dye is derived to be 0.155 s–1. Hence each dye molecule adsorbed at the surface of the mesoscopic oxide film performs the cycle presented in Fig. 2 on the average about once in 7 s. Twenty years correspond to 631 113 472 s, yielding a turnover number of 9.78 × 107, i.e. about 100 million. The upper limit for the sum of the two branching ratios is therefore 1 × 10–8.
4 Recent kinetic measurements
For most of the currently employed sensitizers the rate constant for electron injection from the excited state in the conduction band of the mesocopic TiO2 particles is in the femto-second range. Taking for kinj a value of 1 × 1013 s–1, a destructive side reaction with k1 < 105 s–1 could be tolerated. Ruthenium sensitizers of the N3 type readily satisfy this condition. They can undergo photo-induced loss or exchange of the thiocyanate ligand, which however occurs at a much lower rate than the 105 s–1 limit. It is also debatable whether this pathway is destructive as the product formed, often an iodo or solvato complex, still acts as a charge transfer sensitizer. In ethanolic solution prolonged photolysis of N3 dye leads to sulfur loss and formation of the cyanato-ruthenium complex probably via photoxidation by oxygen [12]. However this reaction is not observed when the dye is adsorbed on the surface of titanium dioxide surfaces [12] where the extremely rapid deactivation of the excited state by interfacial electron injection in the conduction band precludes the degradation reaction.
Precise kinetic information has also been gathered for the second destructive channel involving the oxidized state of the sensitizer, the key parameter being the ratio k2/kreg of the rate constants for the degradation of the oxidized form of the sensitizer and its regeneration. The S+ state of the sensitizer can be readily produced by chemical or electrochemical oxidation and its lifetime determined independently by absorption spectroscopy. Data from a recent study [13] of the monosodium salt of the amphiphilic dye Ru(4-carboxylic acid-4′-carboxylate)(4,4′-dinonyl-2,2′-bipyridine)(NCS)2, coded as Z-907 are shown in Fig. 3. Upon addition of the NOBF4 one-electron oxidant, the Z-907 peaks shift to the red. The insert in Fig. 3 shows the spectrum of the oxidized sensitizer exhibiting two broad LMCT peaks with maxima at 594 and 768 nm. By monitoring the absorption change at 770 nm, the formation of the oxidized form of Z-907 is seen to occur over the first 8–10 min after the addition of the oxidant. The subsequent decay occurs with a lifetime of 75 min corresponding to k2 = 2.2 × 10–4 s–1. The regeneration rate constant for this sensitizer and a typical iodide/triiodide redox electrolyte [13] is at least 2 × 105 s–1. Hence the branching ratio is about 109 that is well above the limit of 108 required to achieve the 100 million turnovers and a 20-year lifetime for the sensitizer.
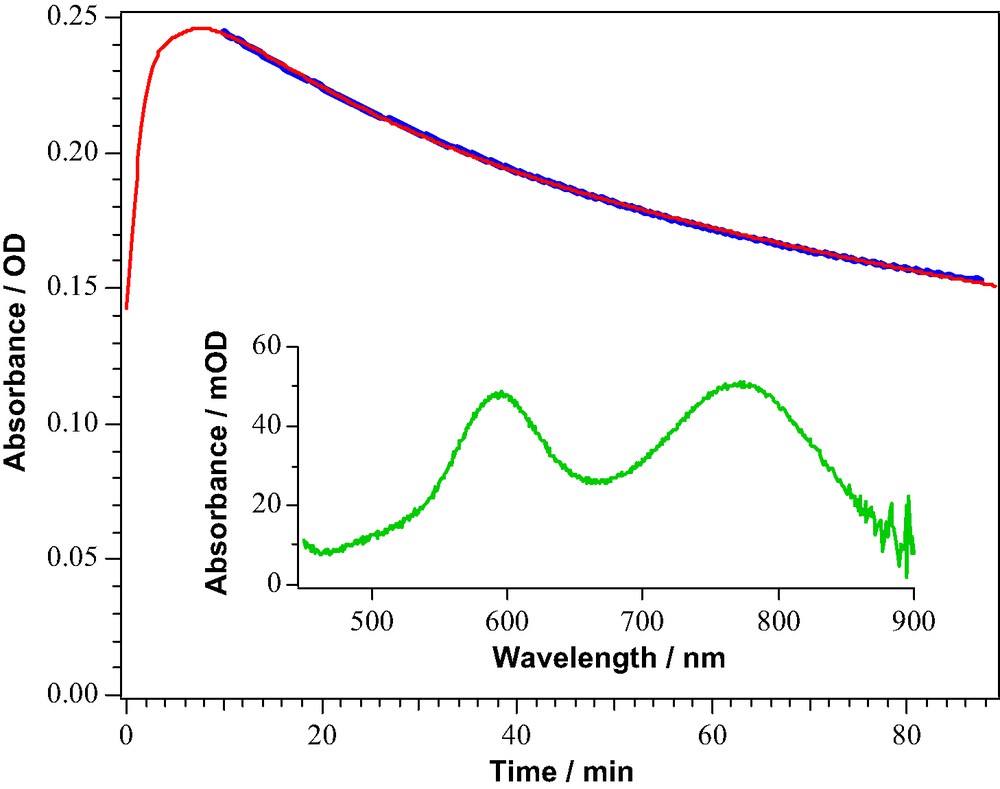
The time dependence for the evolution and decay of the oxidized form of Z-907 in solution. The insert shows the spectrum of the oxidized form derived from the subtraction of the starting material at t ≈ 0.
5 Recent experimental results
Many long-term tests have been performed with the N3-type ruthenium complexes confirming the extraordinary stability of these charge transfer sensitizers. For example, a European consortium financed under the Joule program has confirmed cell photocurrent stability during 10,000 h of light soaking at 2.5 suns corresponding to ca. 56 million turnovers of the dye without any significant degradation [14]. These results corroborate the projections from the kinetic considerations made above. A more difficult task has been to reach stability under prolonged stress at higher temperatures, i.e. 80–85 °C. Recent stabilization of the interface by using self- assembly of sensitizers in conjunction with amphiphilic co-adsorbents has been particularly rewarding [5–11] in this respect allowing for the DSC to meet for the first time the specifications for outdoor applications of silicon photovoltaic cells.
For example we consider the new amphiphlic sensitizer [9] cis-Ru(4,4′-dicarboxylic acid-2,2′-bipyridine)(4,4′-bis(p-hexyloxystyryl)-2,2′-bipyridine)(NCS)2, coded as K-19, which exhibits an increased extinction coefficients due to extension of the π conjugation of the hydrophobic bipyridyl and the presence of electron donating alkoxy groups [10]. The structure of the new dye is shown below. Taking advantage of the enhanced optical absorption of this new sensitizer and using it in conjunction with decylphosphonic acid (DPA) as coadsorbent and a novel electrolyte formulation a ≥ 8% efficiency DSC has been realized showing strikingly stable performance under both prolonged thermal stress and light soaking [11].
The electrolyte contained 0.8 M 1-propyl-3-methylimidazolium iodide, 0.15 M I2, 0.1 M guanidinium thiocyanate, and 0.5 M N-methylbenzimidazole in 3-methoxypropionitrile. At 30 °C, this novel electrolyte formulation has a conductivity of 14.2 mS cm–1. The conductivity–temperature data for the electrolyte are well fitted to the Vogel–Fulcher–Tammann (VFT) equation.
Importantly, photoanodes based on TiO2 nanocrystals derivatized by K-19 and DPA were found to maintain a strikingly stable performance under thermal stress and long-term light soaking. Hermetically sealed cells were used for long-term thermal stress test of cells stored in the oven at 80 °C. As shown in Fig. 3, the VOC of such a device drops only by 25 mV during 1000 h aging at 80 °C while there is a ~70 mV decline (not shown) for device B in the case of film stained with the K-19 sensitizer alone. The stabilizing effect of the DPA is attributed to the formation of a robust and compact molecular monolayer at the mesoscopic TiO2 surface, reducing the amount of adsorbed water and other interfering impurities. This stabilization of the VOC allows device A to sustain the high conversion efficiency during extended heat exposure. Fig. 4 shows that this device maintained over 98% of its initial conversion efficiency after 1000 h aging at 80 °C. While no change was observed for the fill factor, after 1000 h aging, the measured JSC of 15.38 mA cm–2 was even higher than its initial value of 15.16 mA cm–2. The opposite change of Jsc and Voc reflects probably a small positive-shift of flat-band potential of the mesoporous titania film under the thermal stress, which can result in a net enhancement of photo-induced charge separation efficiency in DSC.
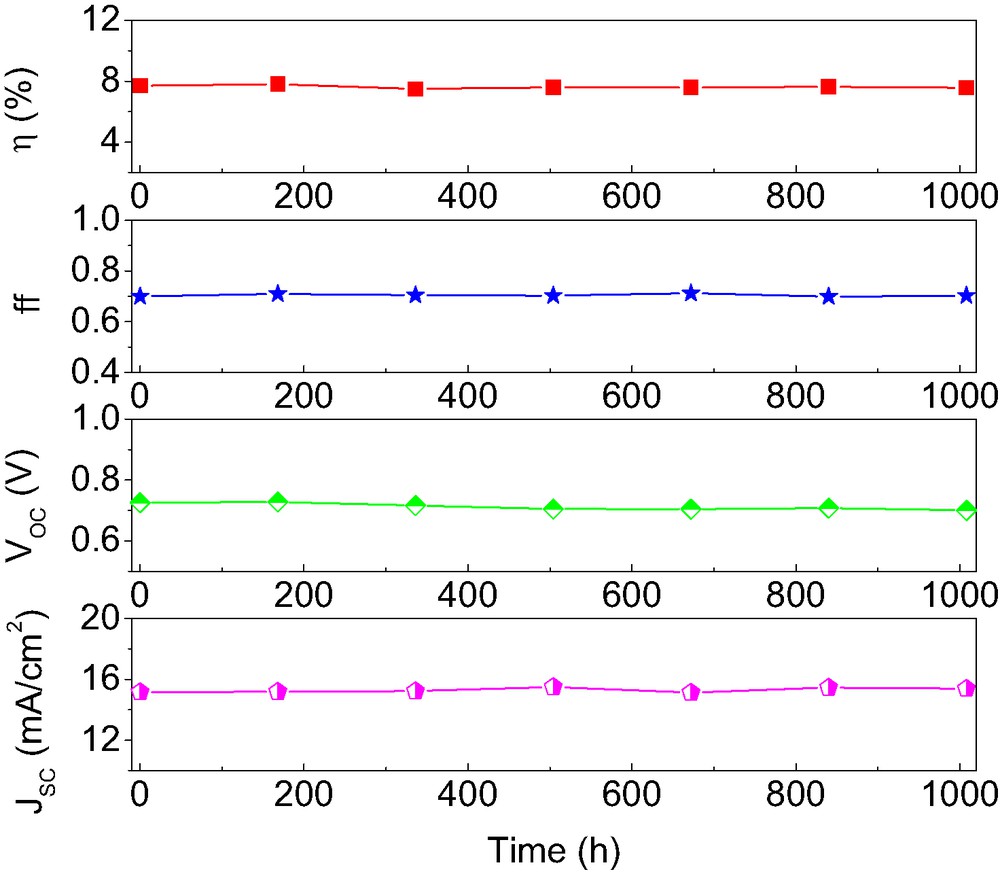
Temporal evolution of photovoltaic parameters (AM 1.5 full sunlight) of device A during continued thermal aging at 80 °C in the dark.
Cells covered with a 50-μm-thick polyester film (Preservation Equipment Ltd, UK) as a UV cut-off filter (up to 400 nm) were irradiated at open circuit and 60 °C in a Suntest CPS plus lamp (ATLAS GmbH, 100 mW cm–2). As shown in Fig. 5, device A also showed an excellent stability under the dual stress of heating and visible light soaking, retaining 97.7% of its initial power conversion efficiency. Impressively, the measured JSC of 15.53 mA cm–2 after 1000 h aging was still higher than the initial value of 15.15 mA cm–2 while a 16 mV drop in VOC and less than 3% decrease in fill factor were observed. This confirms again the photochemical inertness of the styryl unit attached to the bipyridly ligand to prolonged visible light soaking [10].
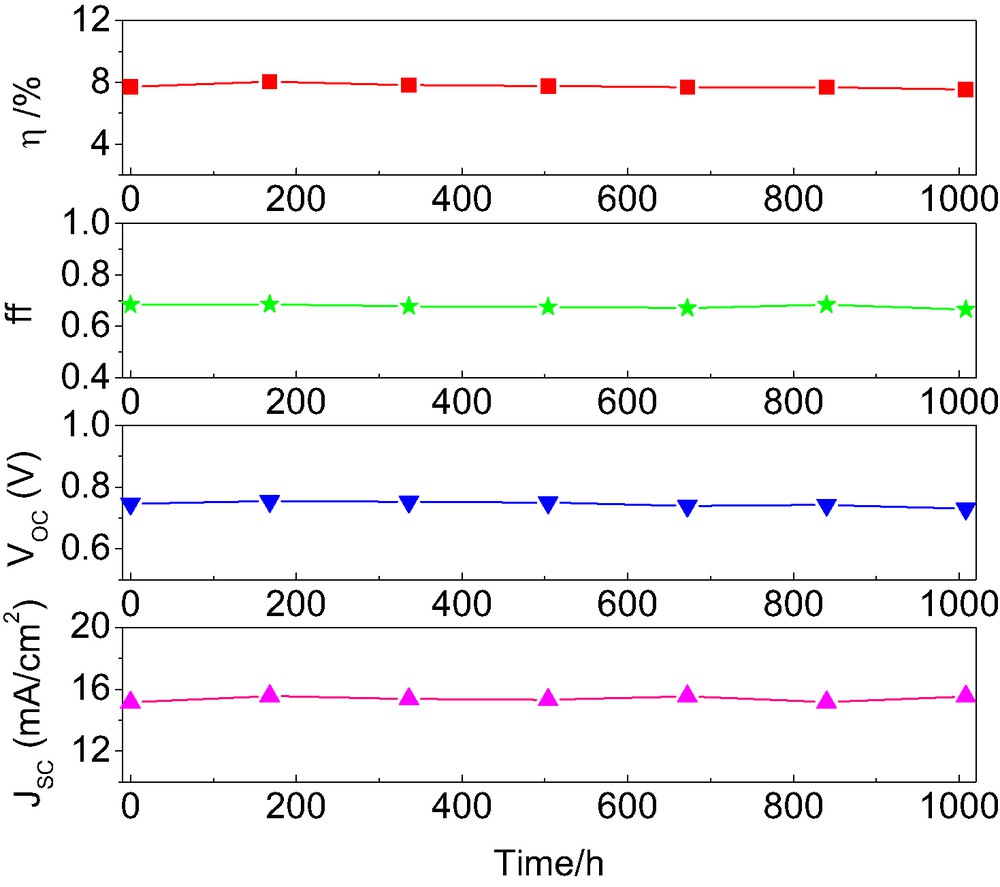
Temporal evolution of photovoltaic parameters (AM 1.5 full sunlight) of device A during continued one sun visible light soaking at 60 °C.
6 Overall efficiency improvements, a new record cell
The overall conversion efficiency of the dye-sensitized cell is determined by the photocurrent density measured at short circuit (Isc), the open circuit photo-voltage (Voc), the fill factor of the cell (ff) and the intensity of the incident light (Is)
ηglobal = iph × Voc × ff/Is (6)
Under full sunlight (air mass 1.5 global intensity Is = 1000 W cm–2) short circuit photo-currents ranging from 16–22 mA cm–2 are reached with state of the art ruthenium sensitizers, while Voc is 0.7–0.80 V and the fill factor values 0. 65–0.75. A certified overall power conversion efficiency of 10.4% was attained [15] in 2001. A new record efficiency of 11% was achieved recently and Fig. 6 shows current voltage curves obtained with this cell. The main improvement is in the open circuit cell voltage (Voc), which was increased to 0.86 V by the addition of guanidinium thiocyanate (GuSCN) to the electrolyte without sacrificing short circuit photocurrent. The GuSCN appears to block the dark current, i.e. the reduction of triiodide by conduction band electrons leading to the observed augmentation in the Voc value. Increasing the injection and lowering the recombination rates is critical for maximizing the open circuit voltage of the cell as shown by the equation
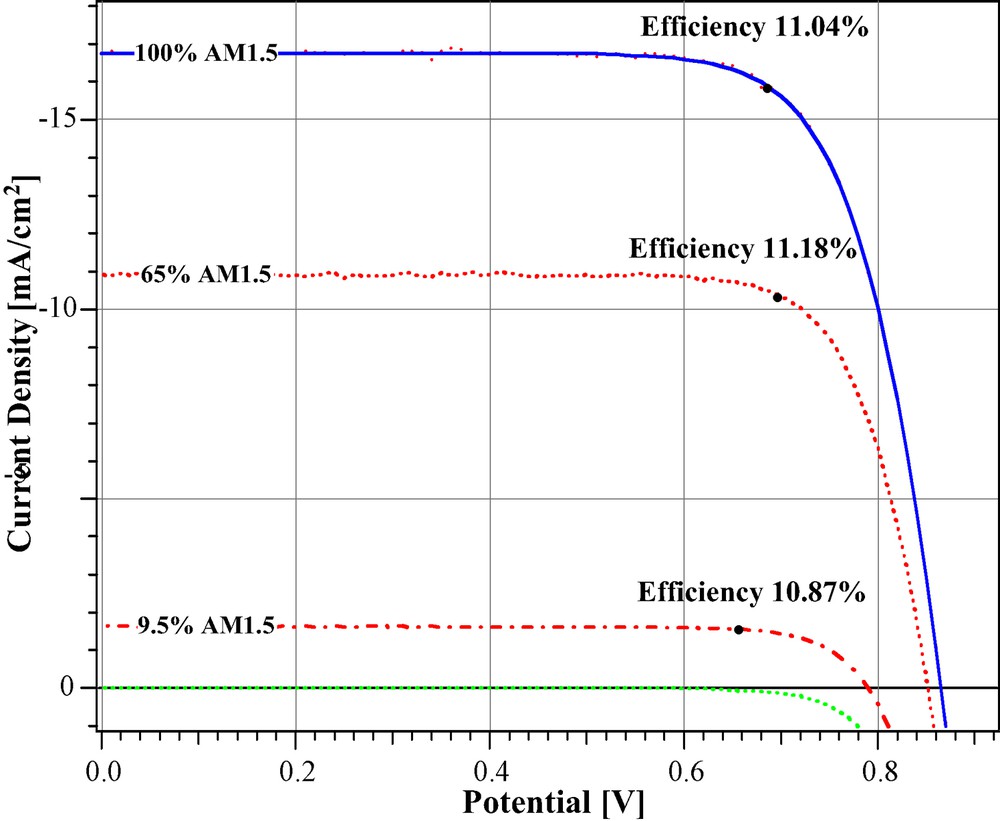
Photocurrent–voltage curve of a DSC at different light intensities. The conversion efficiency in full AM 1.5 sunlight was 11.04%. It increased to 11.18% at 65% full sunlight.
Voc = (n R T/F) ln (K Φ/(kr [S+] + kd [D+]) (6)
where KΦ is the charge carrier photo-generation rate, n is the ideality factor of the junction, kr is the rate constants for recombination of the conduction band electrons with oxidized sensitizer molecules and kd is the rate constant for the dark reduction of triiodide to iodide by the conduction band electrons.
7 Conclusions
Research on both the stability and efficiency of dye-sensitized solar cells has progressed remarkably over the last two years since the IPS-14 conference was held in Sapporo Japan, Apart from providing new physical insight in the operation of these cells these findings will foster practical DSC applications.
Acknowledgements
The authors would like to thank T. Koyanagi (CCIC, Japan) for a free sample of the 400-nm-sized light scattering anatase particles and P. Comte, R. Charvet, and S. Ito for some help on experiments. Thanks are also due to Dr. Karlheinz Hausmann (DuPont Packaging and Industrial Polymers, PO Box 50, CH 1218 Le Grand Saconnex, Switzerland) for providing samples of the Bynel films. Financial support by the Swiss Science Foundation under National Program No. 46, the Swiss Federal Office for Energy (OFEN), the European Joule FP 6 NANOMAX project, and the European Office of the U.S. Air Force under Contract No. F61775-00-C0003 is gratefully acknowledged.