

1 Introduction
Ionic liquids are solvents with high polarity, estimated between acetonitrile and methanol, and consequently capable to dissolve organic compounds, metallic salts, enzymes… [1]. In addition, these solvents contribute to ‘green chemistry’ due to their negligible vapour pressures, under usual experimental conditions, in contrast to those exhibited by common organic solvents [2]. Concerning the catalytic applications of ILs, it is noteworthy the ease to separate organic products from the ionic liquid phase by extraction, using immiscible solvents. IL phase containing the active catalyst could be readily reused without significant loss of catalytic activity [3], fact particularly interesting for asymmetric catalysis, due to the high-added value of the chiral catalysts. At present, other protocols for the separation of organic products and catalyst receive a growing attention, like supercritical carbon dioxide [4], supported ionic liquids [5] or organic solvent nanofiltration [6].
The pioneering work in this field was published by Chauvin and co-workers in 1995, concerning the use of ILs (N,N′-dialkylimidazolium salts) in biphasic catalysis for Rh-catalysed olefin hydroformylation, isomerisation and asymmetric hydrogenation, demonstrating the feasibility to recover the catalyst [7]. However, the first works using pure ILs appeared later, in 2000, when Song's team reported that N,N′-dialkylimidazolium derivatives could be appropriate solvents to perform enantioselective alkene epoxidations [8] and ring opening of epoxides [9].
Since these earliest studies, many applications of ILs have been described in asymmetric biocatalysis, organocatalysis and metal catalysis. This topic is reported in the present review which covers the literature reports published until April 2006.
For authors' convenience, the abbreviations of the ILs reported are listed in Table 1 and their structures shown in Fig. 1. In Fig. 2, chiral ionic liquids (CILs) are gathered.
Abbreviations used in this review for cations of ILs (see Fig. 1)
| Imidazolium | ||
| R1 | R2, R3 | Abbreviation |
| CnH2n−1 | H, CmH2m−1 | [CnCmim] |
| CH2CH2CH2X | CmH2m−1, CoH2o−1 | [(3′-X)C3CmCoim] |
| CnH2n−1 | CmH2m−1, CoH2o−1 | [CnCmCoim] |
| Pyridinium | ||
| R1 | R2 | Abbreviation |
| CnH2n−1 | H | [Cnpy] |
| CnH2n−1 | CmH2m−1 | [CnCmpy] |
| Ammonium | ||
| R1, R2, R3, R4 | Abbreviation | |
| CnH2n−1, CmH2m−1, CoH2o−1, CpH2p−1 | [CnCmCoCpam] | |
| Pyrrolidinium | ||
| R1, R2 | Abbreviation | |
| CnH2n−1, CmH2m−1 | [CnCmpyr] | |
| Phosphonium | ||
| R1, R2, R3, R4 | Abbreviation | |
| CnH2n−1, CmH2m−1, CoH2o−1, CpH2p−1 | [CnCmCoCpph] |
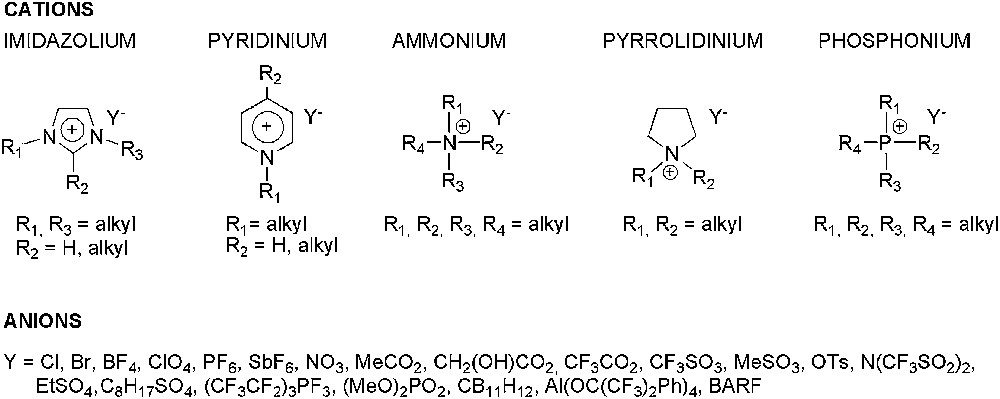
Ionic liquids commonly used in enantioselective catalytic reactions.
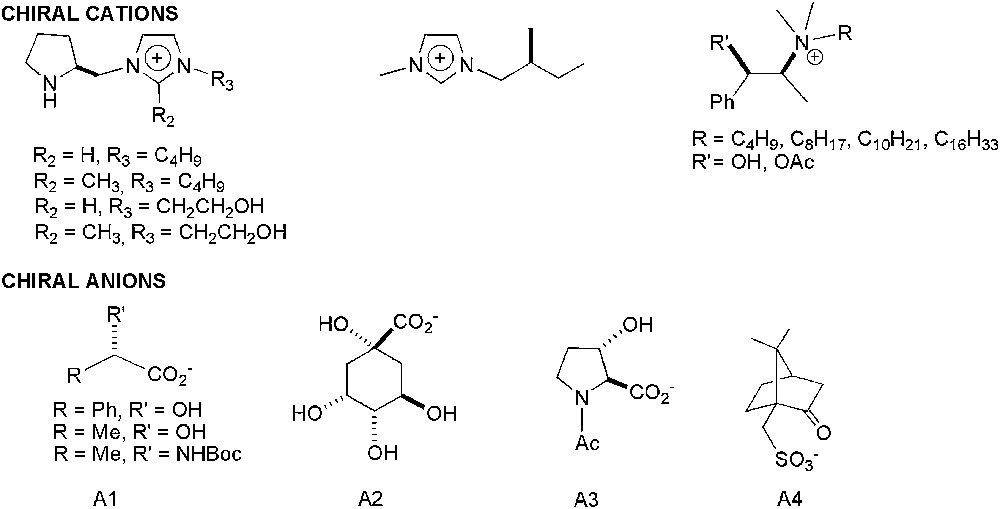
Chiral cations and anions present in CILs used in enantioselective catalytic reactions.
2 Enantioselective biocatalysis
The most frequent structure of ILs used for biocatalysis purposes [10] corresponds to imidazolium alkyl derivatives, but pyridinium, ammonium and pyrrolidinium salts have also been applied (Fig. 1).
Concerning the biocatalysts employed in IL medium, a small range of enzymes has been studied in enantioselective enzyme-catalysed processes: hydrolases such as proteases (thermolysin, α-chymotrypsin, alcalase) and lipases (Candida antarctica lipase B, CALB, Pseudomonas cepasa, PCL), oxidoreductases (Baker's yeast, peroxidases, glucose oxidase) and lyases (hydroxynitrile lyases: Prunus amygdalus and Hevea brasiliensis). With regard to the organic processes, they have been mainly focused on kinetic resolutions of alcohols or amines by acylation; but other processes like sulfoxidation, carbonyl reductions (by HCN addition, hydrogen transfer…) and Markovnikov hydration have also been investigated.
The suitable ILs for enzymatic catalysis should contain anions with low hydrogen-bond basicity to avoid interference with the internal hydrogen bonds of the enzyme; therefore, anions like BF4, NTf2 or PF6 are desirable, but not chloride, nitrate, triflate, trifluoroacetate or acetate, with high hydrogen-bond basicity [11]. These enzyme-compatible anions show low nucleophilicity and consequently low trend to modify the enzyme conformation [12]. In contrast to polar organic solvents, enzymes are active in ILs.
Erbeldinger and co-workers reported the first paper concerning enzymatic catalysis in ILs [13]. At the same time, Sheldon and co-workers published their investigations about lipase-catalysed reactions in free-water IL solvents [14]. In both cases, competitive rates in comparison with enzymatic processes in organic solvents were achieved.
2.1 Kinetic resolution processes
In 2001, the first examples related to enantioselective enzyme-catalysed processes in IL medium appeared, concerning kinetic resolution of alcohols by transesterification using lipases as catalysts (Scheme 1) [15–18].
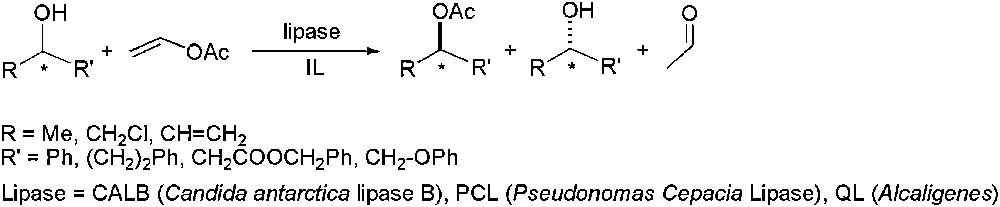
Lipase-catalysed enantioselective transesterification.
Kazlauskas and co-worker demonstrated the importance of the purity of ILs in terms of catalytic activity (Scheme 1, R = Me and R′ = Ph), obtaining comparable or even higher activities in ILs than those observed in organic solvents (10 imidazolium and pyridinium derivatives were tested comparing these with those obtained in toluene, THF, acetone, DMF, DMSO and acetonitrile) [16]. Kim and co-workers [17], using imidazolium ILs [C4C1im][BF4] and [C4C1im][PF6], obtained higher asymmetric inductions than those achieved with organic solvents (THF or toluene), in particular with hydrophobic ionic liquid [C4C1im][PF6] (Scheme 1, R = Me, R′ = (CH2)2–Ph; R = Me, R′ = CH2–COO–CH2–Ph; R = CH2–Cl, R′ = Ph; R = CH2–Cl, R′ = CH2–O–Ph); in some cases, the enantioselectivity reached a synthetically level (E > 400) [19].1 This group improved their results under heterogeneous conditions, using ionic liquid-coated enzyme ([(3′-Ph)C3C1im][PF6] with PCL) [20,21].2 Itoh and co-workers [18] also observed a similar dependence of the anion for the imidazolium derivatives in the activity and selectivity when racemic allylic alcohols were used as substrates (Scheme 1, R = CH CH2, R′ = (CH2)2–Ph), being [C4C1im][PF6] the most appropriate IL; moreover, the catalyst could be recycled up to four cycles without any loss of activity and selectivity [22]. Important enhancement in enantioselectivity has also been obtained for the enzymatic (PCL) kinetic resolution of 1,2-diols, using [C4C1im][PF6] as solvent [23].
It is important to note that immobilized enzymes (CALB) in ILs are robust under scCO2 conditions [24], and continuous biocatalytic extractive processes are feasible, as demonstrated by Iborra and co-workers for the kinetic resolution of racemic alcohols [22].
In addition to transesterification processes, lipase-catalysed esterifications for kinetic resolution of carboxylic acids have been considered [25]. Enantioselectivity enhancements in ILs have also been stated for CALB-catalysed esterification of 2-substituted-propanoic acids with 1-butanol, compared to those obtained in organic solvents [26], trend also observed for the esterification of racemic menthol with propionic anhydride, using Candida rugosa lipase as catalyst [27]. In this context, this lipase catalyses hydrolysis of racemic esters in biphasic H2O/[C4C1im][PF6] conditions, giving the corresponding carboxylic acid optically pure (ee > 99%) with good yield (48%), the catalyst being recycled up to four runs [28].
Lipase-catalysed asymmetric ammonolysis of esters with ammonium carbamate, has also been applied for kinetic resolution purposes, using pure imidazolium derivatives ([CnC1im][Y], where n = 3–8 and Y = BF4, PF6, NO3, Br) as solvents and also tert-butanol/IL co-solvent systems, enhancing in that case, the enantioselectivity. Kinetic resolution processes involving chiral heteroatom substrates (such as amines or phosphanes) by acylation reaction, have also been investigated. Concerning amines (Scheme 2), acylation with carboxylic acids gave excellent enantioselectivities (E > 500) under reduced pressure in [C4C1im][PF6] or [C3C1im][BF4], depending on the acid nature [29]. For P-chiral substrates (Scheme 3), a strong effect on the stereoselectivity was observed depending on the anion nature of IL. Therefore, in [C4C1im][PF6] high enantiomeric excesses were obtained (ee up to 95%), while in [C4C1im][BF4] the catalytic system was not enantioselective [30].
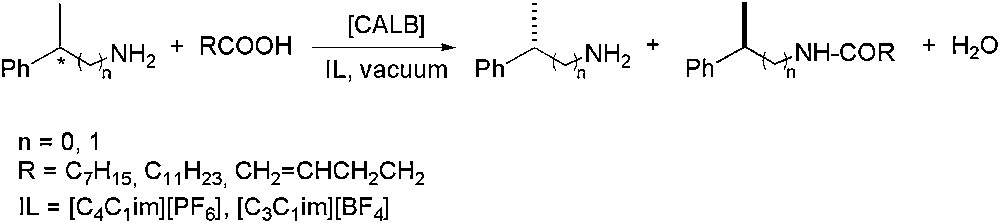
Lipase-catalysed enantioselective acylation of primary amines.
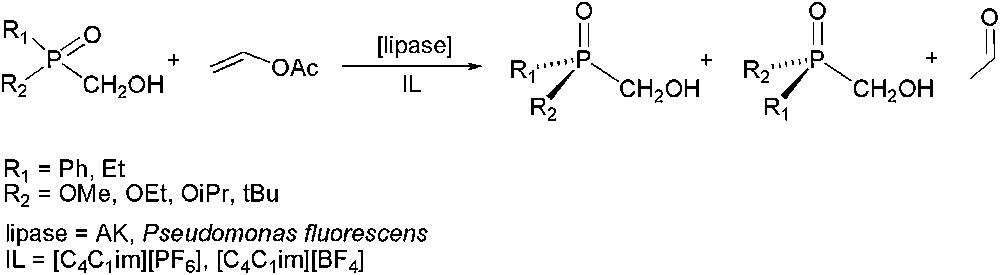
Lipase-catalysed enantioselective acylation of P-chiral substrates.
Not only lipases have been explored in enzymatic resolutions. Protease-type (alcalase) has been successfully employed in the resolution of amino acid esters (ee up to 97%) using [C2py][CF3COO] as solvent [31].
A combination of an enzymatic kinetic resolution and a metal-catalysed racemisation (chemo-enzymatic dynamic kinetic resolution) represents a useful procedure for asymmetric synthesis, becoming a convenient alternative to the classical kinetic resolution [32]. The use of ILs as solvents allows that both types of catalysts work in the same environment, being perfectly soluble. Kim and co-workers have studied lipase–ruthenium combination for the resolution of secondary alcohols (Scheme 4). They demonstrated that Ru-catalysed racemisation of secondary alcohols occurs more efficiently in [C4C1im][PF6] than in organic solvents, and lipase-catalysed acylation leads to ester formation, in high yields and excellent enantiomeric excesses [33]. Combination of two enzymes (mandelate racemase and lipase) has also been tested in the resolution of mandelic acid under biphasic conditions (water/IL); but the mandelate racemase is not compatible with the acyl-donor (vinylacetate); so, in contrast to the chemo-enzymatic dynamic resolution, both reactions could not be carried out in one-pot process [34].
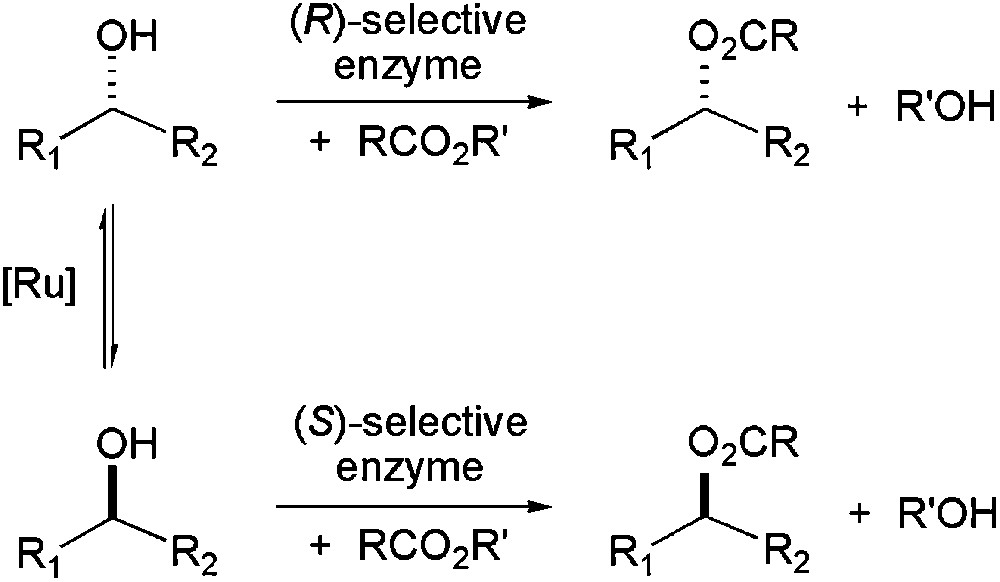
Dynamic kinetic resolution of secondary alcohols by metal–enzyme catalysis.
2.2 Other organic transformations
Besides kinetic resolution studies, other enzyme-catalysed enantioselective processes in ILs have also been investigated. Chiral sulfoxides have been obtained by enzymatic oxidation (using one peroxidase from Coprinus cinereus and one glucose oxidase from Aspergillus niger) of prochiral arylmethyl sulfides in [C4C1im][PF6], with high enantiomeric induction (ee up to 92%), similar to that reached in water, but allowing the use of water insoluble substrates or products which were soluble in IL medium, like methyl-2-naphthyl sulfoxide [35].
Hydrogen cyanide addition to aldehydes in a biphasic system IL([CnC1im][BF4], n = 2–4)/aqueous buffer, has given excellent enantiomeric excesses (up to 97%), using lyase enzymes (from P. amygdalus and H. brasiliensis) [36].
Alcohol dehydrogenase from Lactobacillus brevis, has been used as a catalyst in a biphasic system (buffer solution and [C4C1im][NTf2]), giving high enantioselectivity for the reduction of 2-octanone, substrate-coupled NADPH-regeneration with 2-propanol. In addition, this process showed high activity due to the favourable partition coefficients of 2-propanol and acetone [37].
Secondary enantiopure trimethylsilylethanol has been obtained by yeast (Saccharomyces cerevisiae)-catalysed asymmetric reduction of the corresponding ketone in biphasic IL([C4C1im][PF6])/H2O medium, with in situ coenzyme regeneration [38].
It is noteworthy that the use of ILs permits combined biological and chemical homogeneous catalysis using only a single solvent. Therefore, codeine could be converted into oxycodone, analgesic and intermediate synthon in the preparation of narcotic antagonists, using morphine dehydrogenase and bis(acetylacetonato)cobalt(II) as catalysts in [(3′-OH)C3C1im][CH2(OH)COO], in one-pot procedure [39].
After this overview on enantioselective biocatalysis in ILs, an underlying question appears: why lipases show higher enantioselectivities in ILs than in conventional organic solvents? Several factors can be involved: relative diffusion rates of substrate and product, IL water content, conformational rigidity of enzymes, hydrogen-bond basicity, polarity… But the most plausible reason could be attributed to the interaction of ILs with the charge residual found near the active catalytic centres, as suggested by some authors [23].
3 Enantioselective organocatalysis
In organocatalysed asymmetric reactions using ILs as solvents, nitrogen-containing species, mainly proline and its derivatives, have been employed as catalysts (Fig. 3).
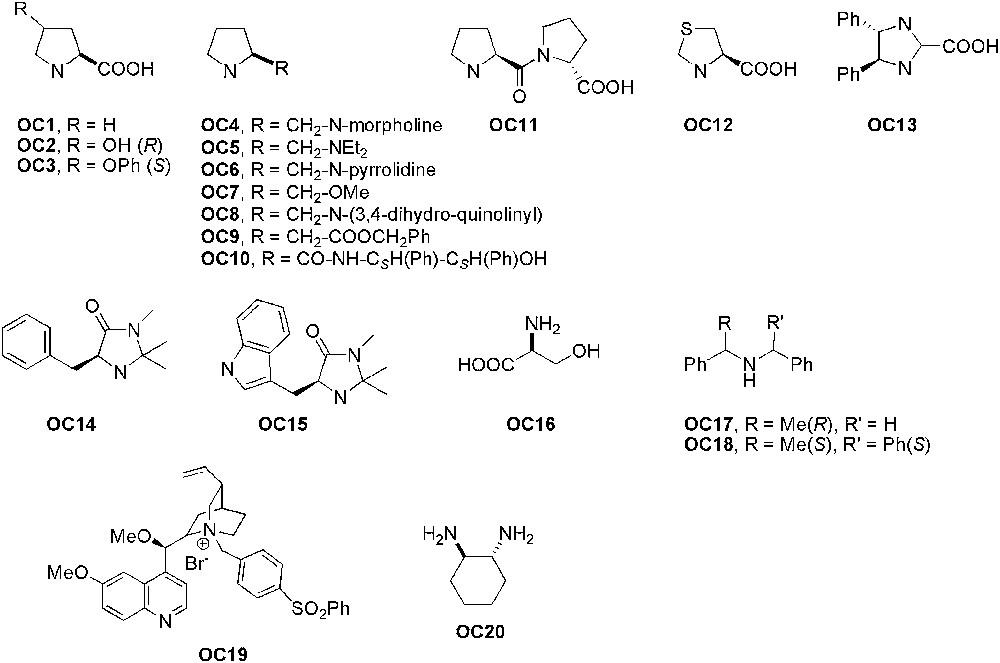
Chiral organocatalysts used for enantioselective reactions in ILs.
The asymmetric aldol reaction carried out in ionic liquid has been widely studied by different groups. Recently it has been reported that l-proline (OC1) catalyses the asymmetric aldol reaction in organic solvents and, among them, the most efficient one was found to be DMSO. The latter not being really suitable for industrial applications, together with the difficulties to recycle the chiral catalyst, motivated the study of ILs as reaction medium. The earliest asymmetric version of the reaction performed in ionic liquid was reported in 2002 [40]. Imidazolium salts were tested for the reaction between acetone and benzaldehyde (Scheme 5) affording ee in the range of 58–78%. The reaction in [C8C1im][Cl] was found to be faster than in any other ILs but the yield was lowered due to the formation of an important quantity of by-product resulting from the elimination reaction. Interestingly, [C4C1im][PF6] was the only one allowing a complete selectivity since no elimination product was observed in that case. It was also possible to recycle the IL containing the proline catalyst five times without significant loss of activity or enantioselectivity.
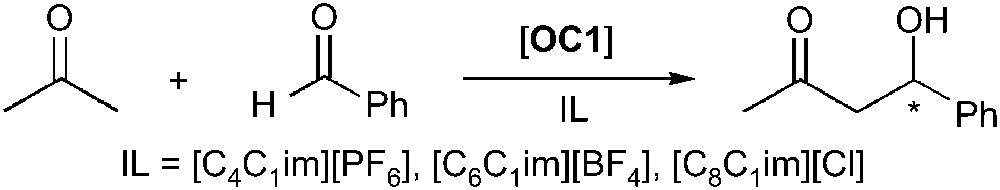
Proline-catalysed asymmetric aldol reaction.
[C4C1im][PF6] was also used to perform efficiently the aldol reaction on different kinds of benzaldehydes containing various types of substituents (Scheme 6) [41]. High yields and ee up to 82% could be achieved and two recycling runs were possible with just a slight decrease of yields and enantioselectivities. Reduction of the proline catalyst loading from 30% to 1% led to a moderate diminution of the activity but leaving the enantioselectivity unaffected.
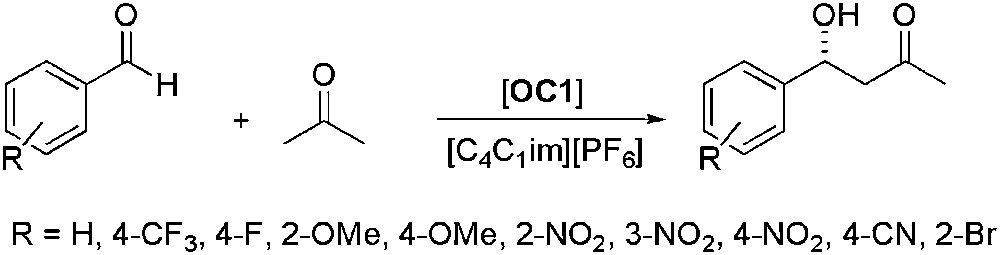
Proline-catalysed asymmetric aldol reaction of substituted benzaldehydes.
The first direct catalytic asymmetric cross-aldol reaction between two aldehydes was reported in 2004 [42]. Reaction between two molecules of propionaldehyde was reported to be faster using [C4C1im][PF6] instead of conventional organic solvents. Addition of DMF allowed preventing the oligomerisation and thus achieving higher yields. Enantioselectivities of 99% were observed, even after the fifth recycling run, using 5 mol% of catalyst. Under the same conditions, the cross-aldol reaction between two different aldehydes (Scheme 7) afforded the desired products in high yields and ee higher than 99%. The three-component version of the reaction was also tested, allowing the synthesis of pyranose derivatives, with constant yield and enantioselectivity after four runs (Scheme 8).
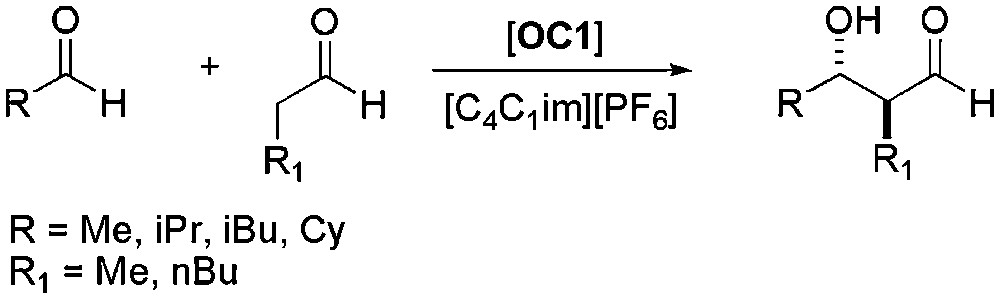
Proline-catalysed asymmetric aldol reaction between aldehydes.
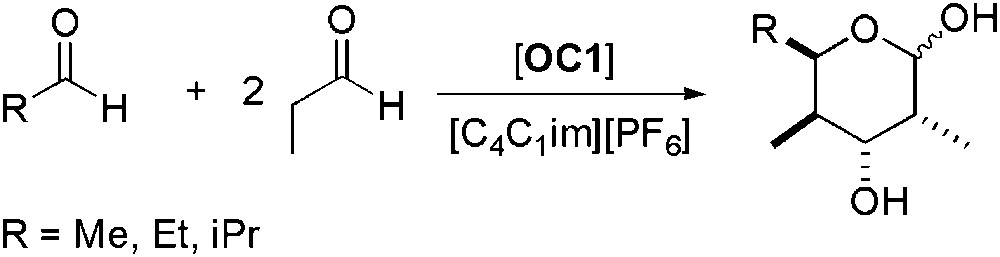
Three-component version of the aldol reaction between aldehydes.
Supported ionic liquid, covalently linked to a silica gel surface, was also used as reaction medium for the aldol reaction between acetone and benzaldehyde (Scheme 9) [43]. In all the systems tested, amounts of the undesired α,β-unsaturated product were detected. The best results were obtained using the supported derivative of [C4C1im][BF4] (ee up to 64%). When 4-nitro-benzaldehyde was used, the aldol was the only product obtained and the ee remained constant even after three recycling runs.
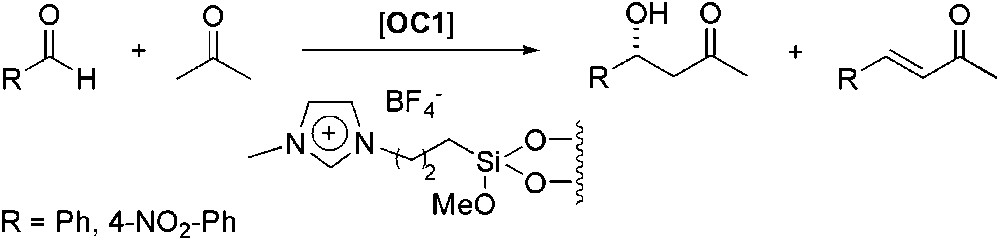
Proline-catalysed asymmetric aldol reaction in supported ionic liquid.
The higher efficiency of [C4C1im][BF4] if compared to [C4C1im][PF6] was also reported in the aldol reaction between aldehydes and ketones (Scheme 10) catalysed by a chiral l-prolinamide derived from 1,2-diphenyl-2-aminoethanol (OC10) [44]. The catalyst was claimed to be more active in ILs than in organic solvents. Various aldehydes and ketones were tested and aldol products were obtained with ee in the range of 91–99%; catalyst could be reused at least twice without any loss of its efficiency.
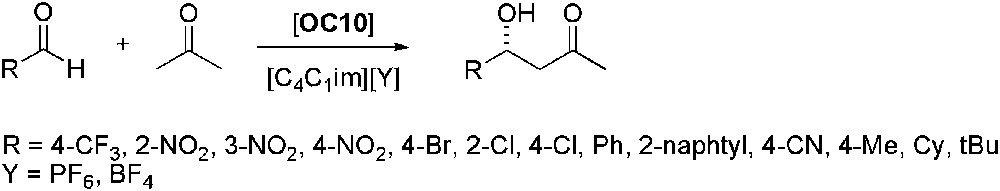
Asymmetric aldol reaction of aldehydes with acetone.
(4S)-Phenoxy-(S)-proline (OC3) was also used efficiently in the asymmetric aldol reaction between acetone and substituted benzaldehydes using [C4C1im][PF6] as solvent (Scheme 11) [45]. The latter, as in the case reported above, allowed achieving high yields and ees (up to 93% and 89%, respectively) and preventing the formation of elimination products. The catalytic system efficiency was maintained after three recycling runs. Comparison with reactions carried out in acetone evidenced also that both yields and enantioselectivities were comparable or higher when IL was used as solvent.
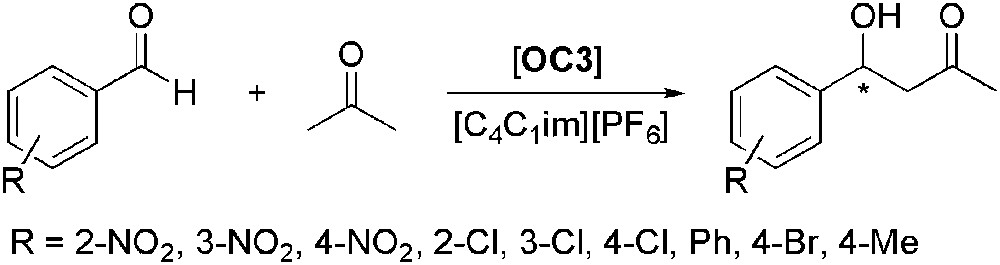
Asymmetric aldol reaction catalysed by (4S)-phenoxy-(S)-proline.
[C4C1im][PF6] was chosen as solvent for the investigation of various organocatalysts (OC1, OC4–OC8, OC17–OC18) for the asymmetric addition of methyl vinyl ketone to aldehydes (Scheme 12) [46]. The ionic liquid was found to accelerate the reaction if compared to organic solvents but the enantioselectivity remained modest (ee up to 59%).
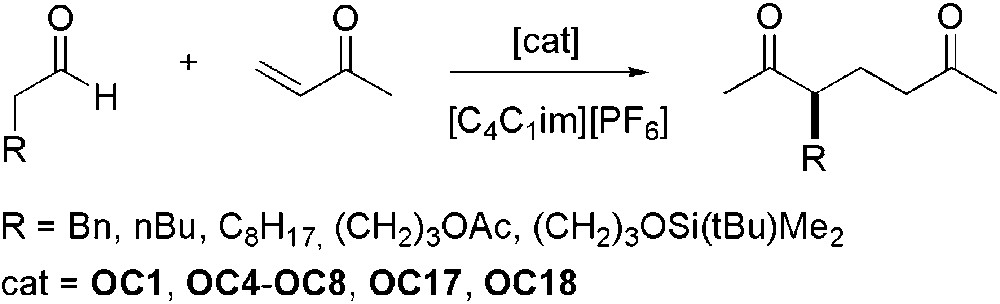
Asymmetric addition of methyl vinyl ketone to aldehydes.
l- and d-Proline were efficiently used as catalysts for the aldol reaction between aldehydes and chlorinated ketones in ILs. The corresponding condensation products, obtained with high enantioselectivities (up to 98%), could be converted, in the presence of triethylamine in [C2C1im][OTf], into optically active epoxides [47].
In addition to the aldol reaction, proline (OC1) was also used as catalyst for the asymmetric Mannich reaction carried out in [C4C1im][BF4] [48]. As preliminary tests, the reaction of the N-p-methoxyphenyl protected α-imino ethyl glyoxylate with cyclohexanone afforded the desired product with ee higher than 99% (Scheme 13). Four successive recycling runs of the proline containing ionic liquid phase afforded the same enantioselectivity and just a slight decrease in yield was observed. Similar results were observed using [C4C1im][PF6]. Replacement of cyclohexanone by different aldehydes or ketones also gave very good enantioselectivities and yields. Compared to organic solvents, ILs allow an important reduction of the catalyst loading [49]. The three-component version of the Mannich reaction was then studied and in that case again, results obtained are comparable in terms of yields and enantioselectivities to those in organic solvents, though again reaction times are shorter (Scheme 14). Some limitations of this reaction in ILs have also been reported as, for example, the use of hydroxyacetone that gave poor results in [C4C1im][BF4] if compared to DMF or DMSO.
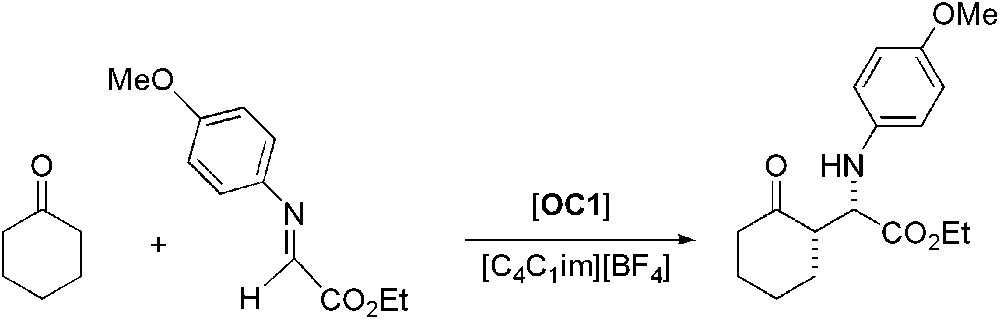
Proline-catalysed asymmetric Mannich reaction.
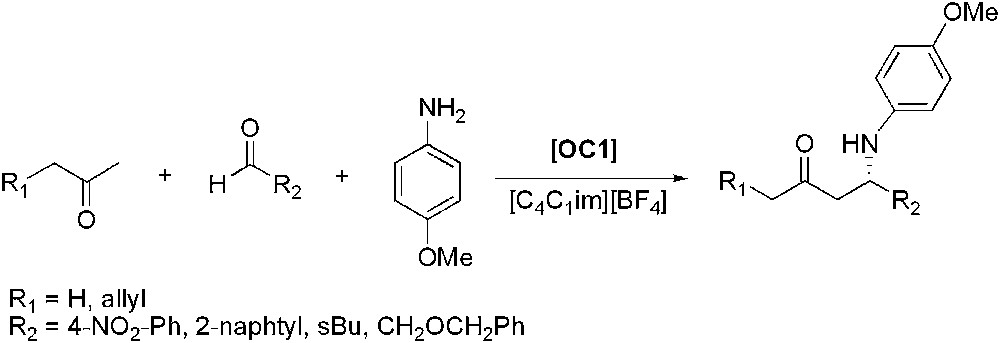
Proline-catalysed asymmetric three-component Mannich reaction.
The enantioselective Michael addition of dimethyl malonate to 1,3-diphenylprop-2-en-1-one promoted by a quaternary ammonium salt (OC19) as phase transfer catalyst has been studied in [C4C1im][PF6], [C4C1im][BF4], [C4py][BF4] (Scheme 15) [50]. Excellent yields were obtained and surprisingly enantioselectivity was reversed in the case of the imidazolium salts whereas in [C4py][BF4], it remained the same as in organic solvents; this fact has been attributed to the different nature of the ionic pair. The reaction was faster when carried out in ILs than in organic solvents, especially when [C4C1im][PF6] was used. No loss of activity was observed after three catalytic runs.
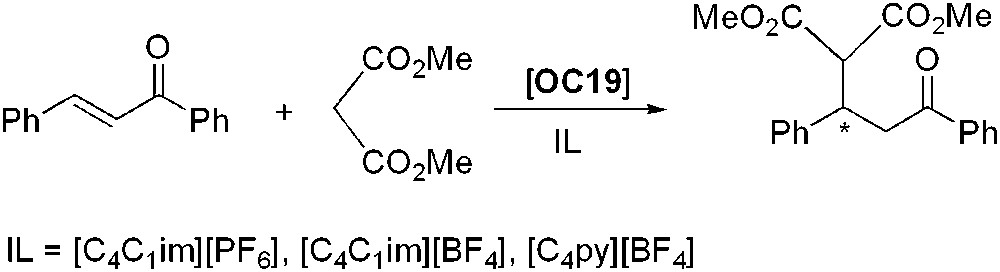
Organocatalysed asymmetric Michael reaction.
Proline was also used as catalyst for the Michael addition of cyclohexanone to nitrostyrene in imidazolium ILs (Scheme 16) [51]. Whereas [C4C1im][PF6] and [C4C1im][BF4] gave results comparable to those obtained when using organic solvents, [(1′-OMe)C2C1im][MeSO3], however, allowed an enhanced enantioselectivity, up to 75% of ee. Reusability tests of the ionic liquid phase showed that yields were not affected but that enantioselectivity dropped dramatically after the third run. The reaction was extended to acyclic ketones as substrates and showed that in that case [(1′-OMe)C2C1im][MeSO3] was not efficient and that [C4C1im][PF6] or [C6C1im][BF4] gave better catalytic results.
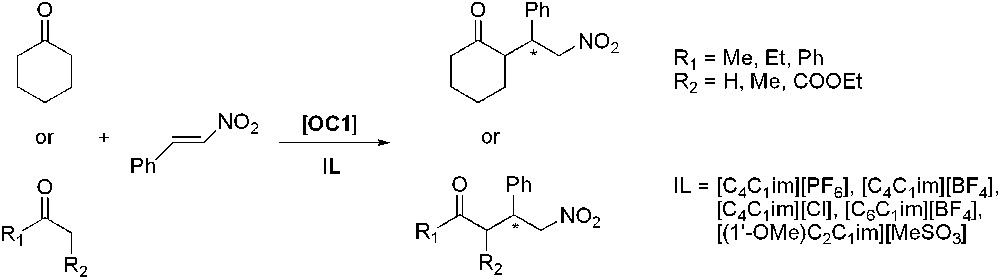
Proline-catalysed asymmetric Michael addition.
The asymmetric Michael addition of aldehydes and ketones to unsaturated nitro compounds was studied in [C4C1im][PF6] by Toma's team [52]. The reaction of nitrostyrene with isobutyraldehyde (Scheme 17) was used to test seven different amino acids or their derivatives as organocatalysts (OC1, OC2, OC4, OC12, OC14, OC14·HCl, OC15), proline being the best one. Recycling of IL/catalyst system led to a yield diminution, from 80% to 61% after the third run. Various substrates were then used as donors, also extended to 2-(β-nitrovinyl)thiophene. Aldehydes gave better results than ketones that required elevated temperatures to achieve good yields; in all cases enantiomeric excesses remained quite modest. These results were claimed to be comparable to those obtained in organic solvents but ILs allowed a diminution of catalyst loading (from 15–20% to 5%).

Asymmetric Michael addition catalysed by amino acid derivatives.
The same group has recently reported the Michael addition of thiophenol to chalcone in ILs catalysed by different chiral organocatalysts (OC1, OC9, OC9·HCl), but, unlike what was observed in CH2Cl2, the reaction carried out in IL proceeded without any stereoselectivity (Scheme 18) [53]. This fact has been attributed to a very fast addition of thiophenol to the chalcone in IL avoiding the en-iminium formation between the chalcone and the amino catalyst. Indeed it was shown that the Michael reaction proceeded smoothly even in the absence of the organocatalyst. A possible explanation could be the enhanced nucleophilicity of the thiophenol and/or its higher dissociation constant in ionic liquid. The use of other substrates showed that the catalyst-free reaction in ionic liquid also proceeded with various thiophenols and α-enones.
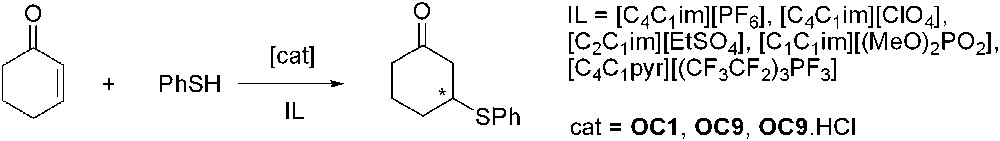
Michael addition of thiophenol to chalcone.
(R,R)-trans-1,2-Diaminocyclohexane (OC20) catalyses efficiently the asymmetric Michael addition of ethyl cyclohexanone-2-carboxylate to methyl vinyl ketone in ILs (Scheme 19) [54]. Among those tested, [C4C1im][BF4] gave the best results in terms of yields and enantioselectivities (ee up to 92%). A possible role of IL might be to favour the enamine formation, initial step of the reaction. The higher solvating ability of [C4C1im][BF4] towards water could be responsible for the observed difference in activity.
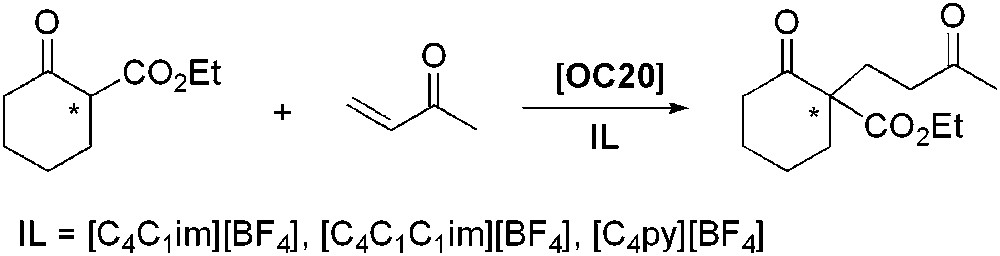
Asymmetric Michael addition catalysed by (R,R)-trans-1,2-diaminocyclohexane.
The enantioselective addition of aldehydes to diethyl azodicarboxylate (Scheme 20) in ILs in the presence of different chiral organocatalysts (OC1, OC2, OC4, OC11–OC13, OC16) has been investigated and quantitative yields (up to 99% of isolated product) and enantioselectivities were obtained (up to 94% ee) [55]. The best results were obtained in [C4C1im][PF6] and [C6C1im][BF4], where the catalyst was more active than in organic solvents. The catalysts performance can be compared to what is observed in conventional organic solvents, the reaction being, however, apparently faster in ILs. Recycling attempts showed that enantioselectivity remained high but the yield dropped probably due to a partial organocatalyst loss during the extractions.
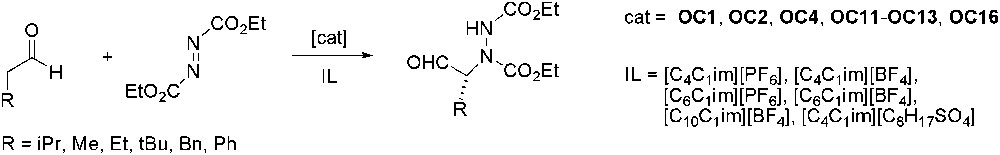
Asymmetric addition of aldehydes to diethyl azodicarboxylate.
Various imidazolium ILs were used for the asymmetric Diels–Alder reaction between cyclohexadiene and acroleine catalysed by OC14·HCl (Scheme 21) [56]. Good yields (up to 85%) and enantioselectivities (up to 93% ee) were obtained for the reaction carried out in [C4C1im][PF6] and [C4C1im][SbF6]. However, reactions in [C4C1im][BF4] or [C4C1im][OTf] gave racemic products in low yields. This observation was attributed to the hydrophilic character of ILs. Kinetic measurements evidenced that the reaction was faster in [C4C1im][PF6] than in CH3CN. In the former case, the ionic liquid phase containing the catalyst could be recycled three times without significant loss of yield or enantioselectivity. The reaction was extended to cyclopentadiene and cinnamaldehyde (Scheme 21), but in that case better results were obtained in CH3CN than in ILs and after the fourth recycling run in IL, enantioselectivity and yield dropped dramatically.
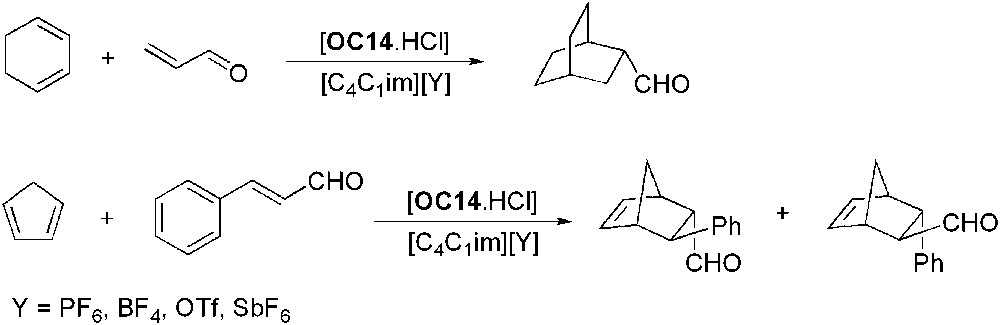
Organocatalysed asymmetric Diels–Alder reaction.
The use of chiral solvents in asymmetric synthesis has already been reported in the literature [57], but low enantioselectivities were obtained. Moreover, the high cost of chiral solvents and their difficult preparation have limited their progress. Different kinds of chiral ionic liquids (CILs) have been developed for various applications including chiral stationary phases for chromatography, shift reagents for NMR, chiral liquid crystals, optical resolution of racemates or solvents for stereoselective radical polymerisation of alkenes [58]. Their facile synthesis associated to the possibility of their recycling made them much more attractive than conventional organic solvents as reaction medium for asymmetric organocatalysis.
In 2004, Baylis–Hillman reaction of benzaldehyde with methylacrylate was carried out using CILs and DABCO (1,4-diazabicyclo[2.2.2]octane) as catalyst (Scheme 22) [59]. The use of CILs afforded high yields and moderate enantiomeric excesses (up to 44%), in contrast to good enantioselectivities (up to 84%) obtained in organic solvents using Lewis acid catalysts containing chiral ligands [60]. The modification of different parameters like the nature of the anion of the IL or the use of substituted aldehydes as substrates did not allow any improvement in terms of enantioselectivity. The presence of the hydroxy group on the backbone of the cation was claimed to be essential for the transfer of chirality, due to the formation of hydrogen bonds between the solvent and the carbonyl groups of substrates.
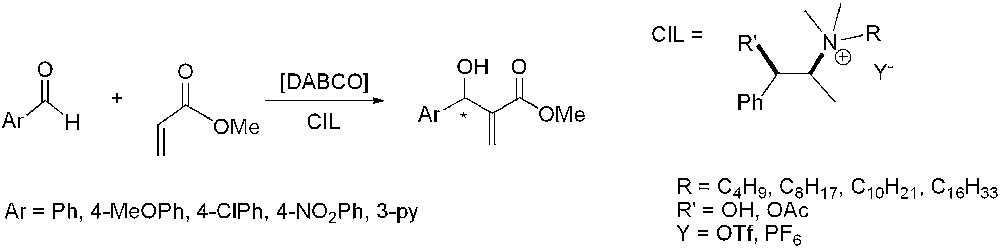
Asymmetric Baylis–Hillman reaction in chiral ionic liquids.
Based on the observation that chiral pyrrolidines and imidazolines can act as highly enantioselective organocatalysts, new pyrrolidine-based CILs were prepared and efficiently applied to the Michael addition of cyclohexanone to nitroolefins (Scheme 23) [61]. In that case, CILs act as both solvent and chiral catalyst. The catalytic reactions were performed in the presence of trifluoroacetic acid as co-catalyst, but no co-solvent was added. The pyrrolidine–imidazolium bromide and tetrafluoroborate demonstrated the best performance with almost quantitative yields and enantioselectivities up to 99%. Introduction of a methyl substituent on the imidazolium ring or of the OH group on the side chain of the cation led to a decrease in both catalytic activity and enantioselectivity. Recycling experiments evidenced that high selectivities were maintained but that a loss of activity appeared for the third and fourth runs, therefore, an increase of the reaction time from 8 to 48 h was required to achieve similar yields. The reaction was also efficiently extended to other Michael donors such as cyclopentanone, acetone or aldehydes but with moderate to good asymmetric inductions.
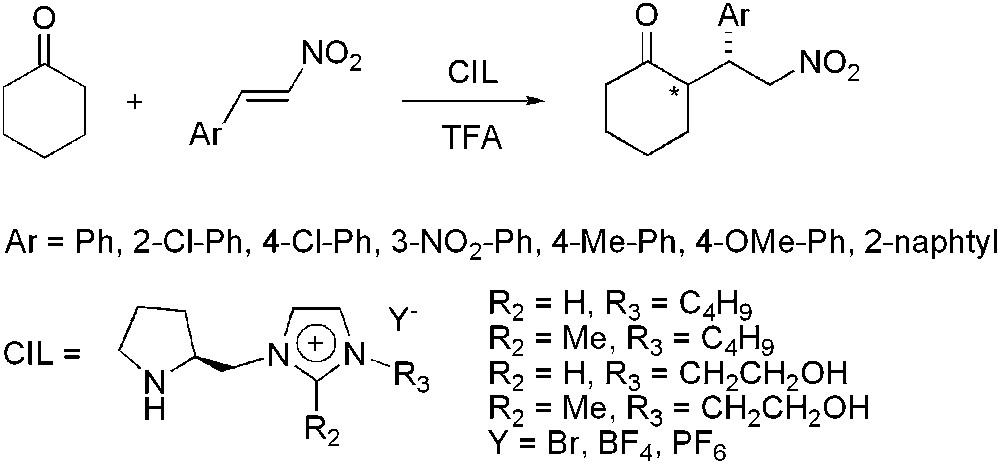
Michael addition reaction catalysed by chiral ionic liquids.
4 Enantioselective metal catalysis
In transition-metal-catalysed asymmetric reactions using ILs as solvents, P- and N-donor ligands have been mainly employed as chiral source (Figs. 4 and 5).
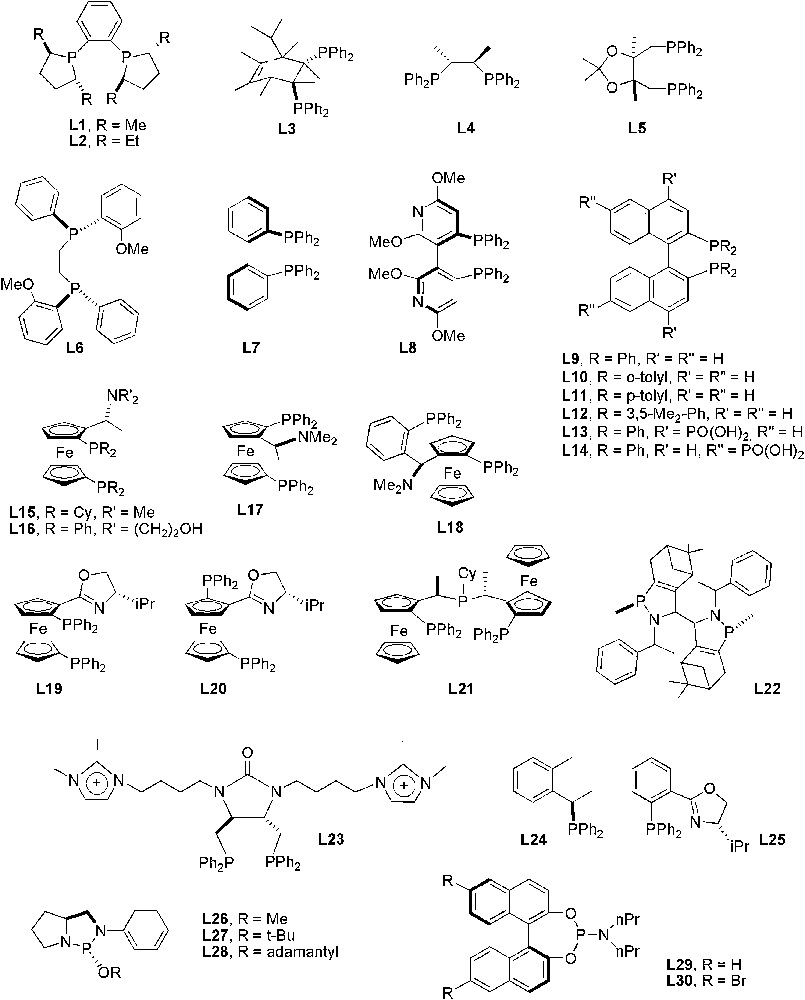
P-donor chiral ligands used in asymmetric catalytic processes in ILs.
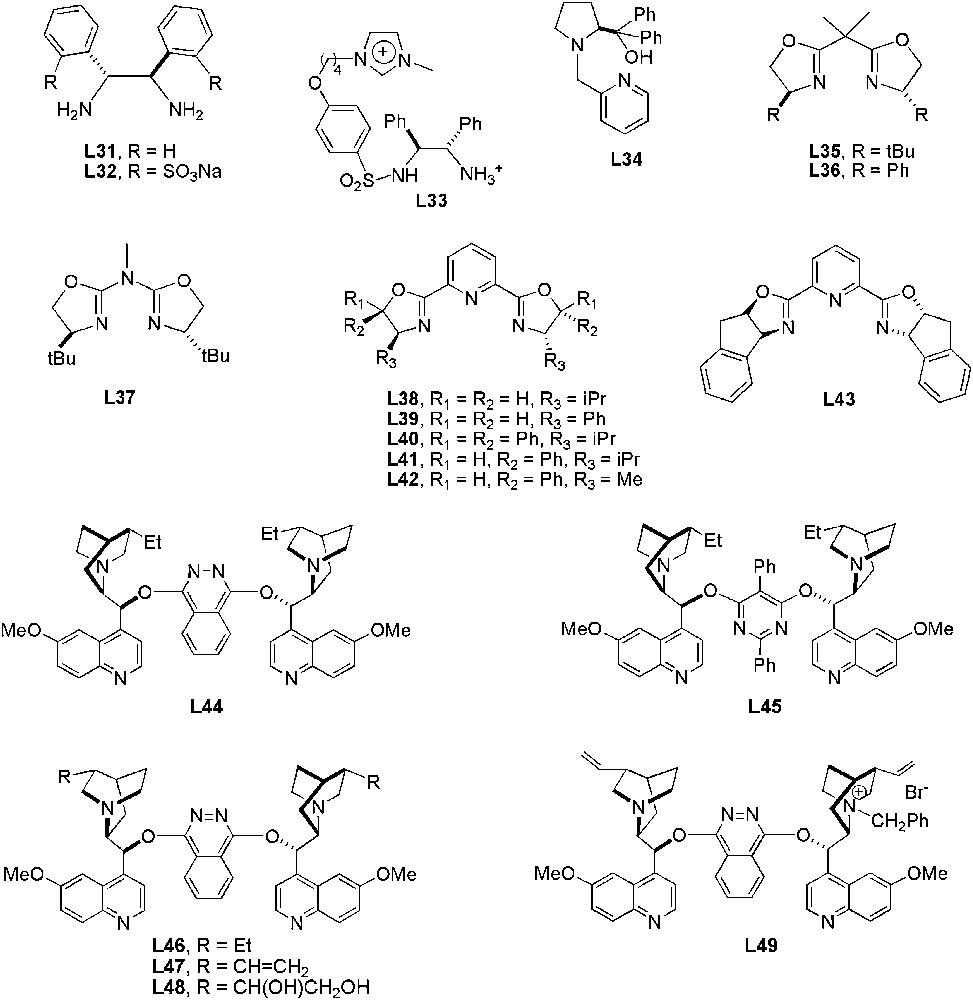
N-donor chiral ligands used in asymmetric catalytic processes in ILs.
4.1 Hydrogenation
The catalytic reduction of unsaturated bonds in the presence of a hydrogen source, molecular hydrogen or hydrogen donors, is one of the most powerful chemical way for the industrial production processes of fine and bulk hydrogenated chemicals. Transition-metal-catalysed asymmetric hydrogenation has become one of the most important tools for the preparation of optically active compounds and high activity and enantioselectivity are observed using Ru, Rh and Ir complexes [62].
For hydrogenation, the solubility of molecular hydrogen in the reaction medium is a crucial factor to be considered. The solubility of hydrogen and the corresponding Henry coefficients for several ILs have been determined at 100 bar H2 pressure [63]. The concentration of hydrogen in these solvents is very low, lower than in molecular organic solvents, and close to that found in water (Table 2).
Hydrogen concentration at 298 K and atmospheric pressure in some organic solvents and ionic liquids
| Organic solvent | H2 (mM) | Ionic liquid | H2 (mM) |
| Water | 0.81 | [C4C1im][BF4] | 0.86 |
| Methanol | 3.75 | [C4C1im][PF6] | 0.73 |
| Ethanol | 2.98 | [C4C1im][SbF6] | 0.93 |
| Benzene | 4.47 | [C4C1im][OTf] | 0.97 |
| Toluene | 3.50 | [C4C1im][NTf2] | 0.77 |
| Cyclohexane | 3.63 | [C4C1im][CF3CO2] | 0.98 |
| [C6C1im][BF4] | 0.79 | ||
| [C8C1im][BF4] | 0.62 |
Classical homogeneous Rh(I)- and Ru(II)-catalysed asymmetric hydrogenations can be transposed to liquid–liquid two-phase systems using imidazolium-based ILs as the mobile phase. The important kinetic parameter to be considered when comparing studies performed under different gas–liquid mass transfer conditions is the hydrogen concentration in the ionic phase rather than the hydrogen pressure in the gas phase. This is particularly evident for [C4C1im]-ionic liquids, where the hydrogen solubility at 50 atmospheres is almost four times higher in the hydrophilic tetrafluoroborate [C4C1im][BF4] than in the hydrophobic hexafluorophosphate [C4C1im][PF6] [64]. Jessop et al. demonstrated that the selectivity in hydrogenation processes depends on the nature of IL employed: for tiglic acid (CH3CHC(CO2H)CH3), higher ee (93%) was obtained in [C4C1im][PF6] than in [C4C1im][BF4] (88%), while for atropic acid (CH2C(CO2H)Ph), [C4C1im][BF4] gave better enantioselectivity than [C4C1im][PF6], 37% versus 22% [65,66]. In addition, by using mixtures of neoteric solvents like IL and scCO2, the hydrogen concentration could be significantly increased [67].
For hydrogenation reactions, imidazolium-based ILs, mostly BF4 and PF6 salts, are commonly employed, and anions like triflate or acetate can deactivate the catalyst due to their coordinating properties [68]. Asymmetric hydrogenation reactions could be also performed with classical Ru- and Rh-complexes dissolved in IL and the substrates, in an appropriate organic solvent. In 1995 Chauvin et al. proposed the first enantioselective metal-catalysed reaction in ILs [7]. They obtained (S)-phenylalanine (64% ee) by hydrogenation of α-acetamidocinnamic acid dissolved in 2-propanol, using [Rh(cod){(−)-DIOP}](PF6) complex ((−)-DIOP = L5) as catalyst dissolved in [C4C1im][SbF6]. The product could be separated quantitatively and the ionic liquid phase could be reused. Similarly, the asymmetric hydrogenation of 2-phenylacrylic acid and 6-methoxy-2-naphthylacrylic acid was catalysed by [RuCl2{(S)-BINAP}]2·NEt3 precursor ((S)-BINAP = (S)-L9, Fig. 4) immobilized in [C4C1im][BF4] [69]. The ees obtained were similar to those obtained in homogeneous media, but the important advantage was that the catalytic ionic solution could be reused several times without any loss of activity or enantioselectivity [3,69].
Concerning the asymmetric hydrogenation of enamides, [C4C1im][PF6] was found to provide an extra stability to the air-sensitive chiral rhodium complex [Rh(cod)(L1)](CF3SO3) and the ees obtained were in the range of 95% (Scheme 24) [70]. Catalyst recycling was possible without loss of enantioselectivity, and the products were separated by decantation of 2-propanol phase. Recently, Sheldon and co-workers have reported the recovery of N-acetyl-(S)-phenylalanine methyl ester (APAM), by using supercritical carbon dioxide as a co-solvent in extractions or as an anti-solvent in precipitations [4]. The extraction of APAM from IL phase was easy due to its high solubility in scCO2 whereas [C4C1im][BF4] and substrate showed a negligible solubility in that co-solvent. On the other hand, scCO2 could be used as an anti-solvent to precipitate APAM out of the ionic liquid phase because its solubility in IL/scCO2 mixture is low compared to the solubility in pure IL.
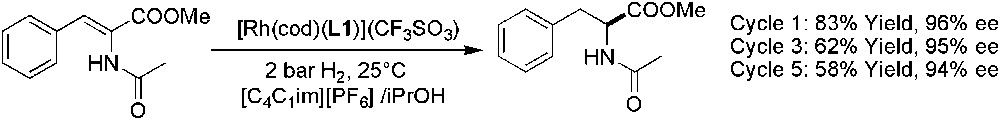
Rh-catalysed asymmetric reduction of methyl-(Z)-α-acetamidocinnamate.
In addition, in enamide hydrogenation, the catalytic activity of [Rh(cod)(L1)](CF3SO3) was largely decreased after the first run, certainly due to the leaching or instability of the catalyst [70]. It has been demonstrated that attachment of imidazolium salts to achiral Rh- and Ru-catalysts increased the preferential solubility of the catalyst in IL, and the catalysts were reused without significant loss of catalytic efficiency [71,72]. This ionic tag strategy was applied to Rh-catalysed asymmetric hydrogenation in ILs [73]. Chiral Rh-complex involving ligand L23 (Fig. 4) which contains two 1,2-dimethylimidazolium cationic groups, demonstrated its catalytic efficiency and reusability in asymmetric hydrogenation of an enamide (Scheme 25), without catalyst leaching.
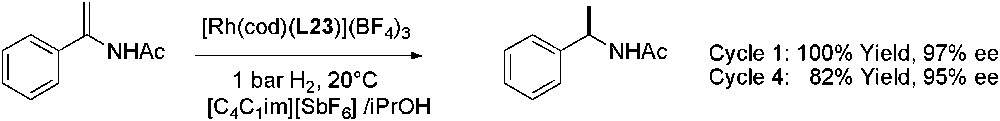
Rh-catalysed asymmetric reduction of N-acetylphenylethenamine.
Recently, the [Rh(cod)(L6)](BF4) complex was chosen as catalyst for the asymmetric hydrogenation of enamides using different ILs as solvent and 2-propanol as co-solvent. The best enantioselectivity was 87% in [C4C1im][BF4] [74]. Interestingly, the use of ‘wet ILs’ (mixtures of ILs and water) gave better ees than pure ILs or 2-propanol/ILs mixtures in Ru-catalysed hydrogenation of tiglic acid in [C4C1im][PF6] [65]. In the asymmetric reduction of acetamidocinnamate, addition of water to [C8C1im][BF4] and [C4C1im][NTf2] led to higher ees and reaction rates compared to common organic solvents when Rh-ferrocenyl-diphosphine catalyst was used [75]. When ‘wet ILs’ are used, the choice of ILs is very important to prevent a possible leaching of IL into the water phase. [C8C1im][BF4] and [C4C1im][NTf2] were generally selected for their poor solubility in water. Ferrocene-based diphosphines (L19 and L20 [75]) and L2 [76,77], were efficiently used in asymmetric hydrogenation of enamides. The association of a second solvent with ILs in the case of L2 ligand increased the efficiency of the catalytic cycle [77].
Noyori et al. reported good enantioselectivities for the reduction of β-ketoesters using Ru/(R)-BINAP ((R)-BINAP = (R)-L9) system as catalyst [78]. For β-aryl ketoester substrates, low ee values were obtained, but they were improved modulating L9 at 4 and 4′-positions, obtaining high enantiomeric excesses (>99%) for a wide range of β-aryl ketoesters [79]. The effect of bulky or electrodonating substituents was very marked on the ee values obtained relative to (R)-L9 [80,81]. The best ee (>99%) was achieved when two phosphonic acid substituents were introduced at 4 and 4′ positions, ligand (R)-L13 (Scheme 26) [80].
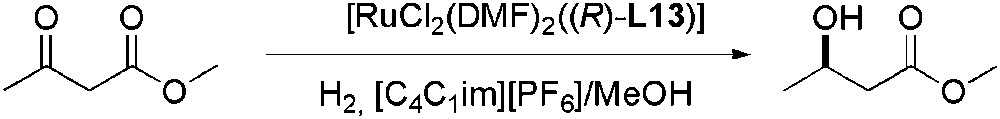
Ru-catalysed asymmetric hydrogenation of β-ketoesters.
In the asymmetric hydrogenation of methyl acetoacetate, other ligands like L8 led to higher conversion and enantioselectivity than when (R)-L9 was used [82].
[RuCl2(L31)(L)] precatalysts containing polar biphosphonic BINAP derivatives (L), (R)-L13 and (R)-L14, and the chiral diamine L31, were immobilized in room temperature ionic liquids for asymmetric hydrogenation of aromatic ketones, giving ee values up to 98% (Scheme 27). The performance of these catalysts was found dependent on the nature of imidazolium ILs. Low conversions were observed in [C4C1im][BF4] and [C4C1im][PF6] [83]. In contrast, [C4C1C1im][BF4] or [C3C1C1im][NTf2] permitted total conversions for all the substrates tested under the same conditions. The low conversions were certainly due to catalyst deactivation via generation of carbenes under catalytic conditions when ILs with an acidic proton were used. Other chiral diamines, such as L32, were used in the same asymmetric reaction and the synergy between the diamine and KOH allowed the reaction acceleration and enhanced the enantioselectivity [84].
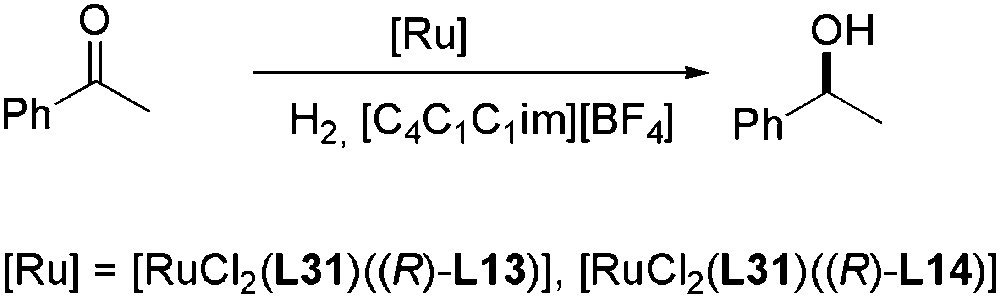
Asymmetric hydrogenation of aromatic ketones catalysed by Ru-complexes.
A rhodacarborane catalyst with the chelating ligand (R)-L9 was tested in this reduction reaction using the classical [C4C1im][PF6] and [C8C1im][BF4] ILs, and also IL involving 1-carbadodecaborate as anion, [C4py][CB11H12] [85]. The most efficient IL was [C4py][CB11H12], whose B–H bonds may be probably involved in the reaction.
Recently, Xiao and co-worker demonstrated that pyridinium-based ILs, [C2py][CF3CO2] and [C2py][BF4], gave quantitative yield and ee up to 71% in the asymmetric reduction of aromatic ketones using chiral ligands L29 and L30 in the presence of lithium aluminium hydride [86]. The best results were found in [C2py][CF3CO2] using L30 as ligand, but the catalyst recycling was difficult, in contrast to when [C2py][BF4] was used as solvent. This different behaviour could be explained by the complex formation between LiAlH4 and CF3CO2−.
Iridium complexes bearing bidentate phosphine or phosphino-oxazoline ligands were tested in the enantioselective hydrogenation of imines [67,87]. More particularly, cationic iridium complexes with chiral phosphino-oxazolines were used in the asymmetric hydrogenation of N-(1-phenylethylidene)aniline in the biphasic system IL/CO2 [87]. [C2C1im][NTf2] was selected to assess the influence of CO2 on the hydrogenation process using Ir/L25 as catalyst (Scheme 28). In the absence of CO2, significant conversion required hydrogen pressures as high as 100 bar whereas quantitative formation of the hydrogenated product was observed at a hydrogen partial pressure of 30 bar in the presence of CO2. With other ILs the effect of the CO2 pressure was reflected at shorter reaction times. This positive effect could be attributed to a higher hydrogen solubility and lower viscosity in IL/CO2 mixture. No influence of carbon dioxide on the enantiomeric excess was detected.
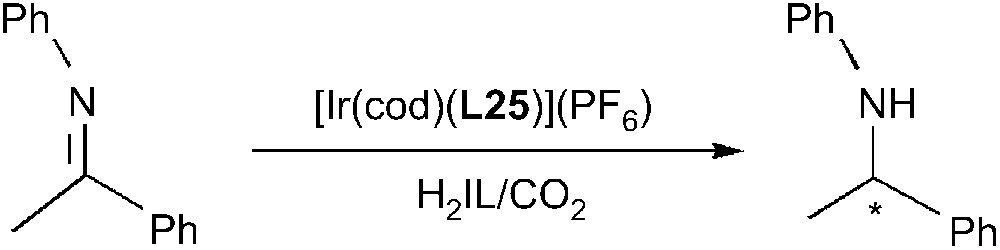
Enantioselective hydrogenation of N-(1-phenylethylidene)aniline.
Lately, the application of organic solvent nanofiltration (OSN) for the simultaneous recycling of asymmetric hydrogenation catalysts and ILs has been reported [6]. In the reaction of asymmetric hydrogenation of dimethyl itaconate using Ru/(R)-BINAP catalytic system, a surprising and significant increase in the enantiomeric excess has been observed in methanol/[(C6)3(C14)ph][Cl] medium. OSN has been proven to offer an effective and simple alternative for the separation of ILs and the catalyst from the product in a single-phase mixture.
A new recyclable heterogeneous system has been described for transition-metal-catalysed hydrogenation in IL. The preparation of supported ionic liquid phase (SILP) is made by a mixing of the metal and the ionic liquid with poly(diallyldimethylammonium chloride) [5]. This polymeric phase simultaneously heterogenises the transition-metal complex and the IL. In this asymmetric reduction of methyl acetoacetate using [RuCl(η6-p-cymene)((S)-L9)]Cl, the catalyst reuse was possible without loss of activity in the second cycle. However, mass transfer became critical for the polymeric system and reaction rates were lower than those obtained under homogeneous conditions.
An interesting alternative to hydrogenation using molecular hydrogen is the transfer hydrogenation reaction. The hydrogen donors most commonly used for ketones are 2-propanol (generally used with a base) and formic acid/triethylamine azeotropic mixture. Schotten et al. described microwave-assisted reduction in ILs with a palladium on charcoal catalyst and formate salts [88]. Rhodium-catalysed transfer hydrogenation in phosphonium tosylates with 2-propanol as proton source, has been reported [89]. The reduction of acetophenone by [Rh2(OAc)4]/(−)-DIOP ((−)-DIOP = L5, Fig. 4) immobilized in [(C8)3C2ph][OTs] in the presence of KOH/iPrOH, afforded 1-phenethylethanol in 50% yield and 92% ee (Scheme 29). These tosylate salts are liquid at the reaction temperature and solid at room temperature, thereby facilitating the catalyst recovery.
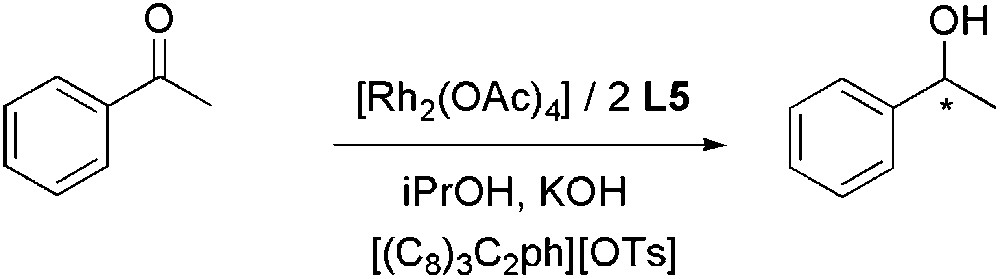
Rh-catalysed asymmetric transfer hydrogenation of acetophenone.
Recently, the asymmetric transfer hydrogenation of acetophenone in IL using a modified arene–Ru(II) complex containing chiral 1,2-diphenylethylenediamine derivatives with catalyst recycling, has been described [90,91]. With 2-propanol/KOH, good results in terms of catalyst retention and recycling were achieved, and with formic acid, conventional catalysts afforded better results due to a necessary extraction step with water [91]. A novel task-specific chiral ionic ligand, with an attached imidazolium group, L33 (Fig. 5), has been recently used in asymmetric transfer hydrogenation of ketones by HCOOH–Et3N azeotrope in ILs [92]. In this study, the ionic catalyst Ru/L33 showed excellent conversion (99%) and ee (93%) up to the fourth catalyst reuse.
4.2 Olefin oxidation
4.2.1 Epoxidation
Optically pure epoxides represent a useful preparative method in organic synthesis, chiral Jacobsen and Katsuki manganese catalysts being the most effective systems [93]. Attempts to immobilize these types of catalysts on solid supports have led to less efficient systems [94]. However, successful procedures have been published by Song and Smith groups concerning the immobilization of salen–manganese(III) Jacobsen and Katsuki catalysts in ILs (Fig. 6).
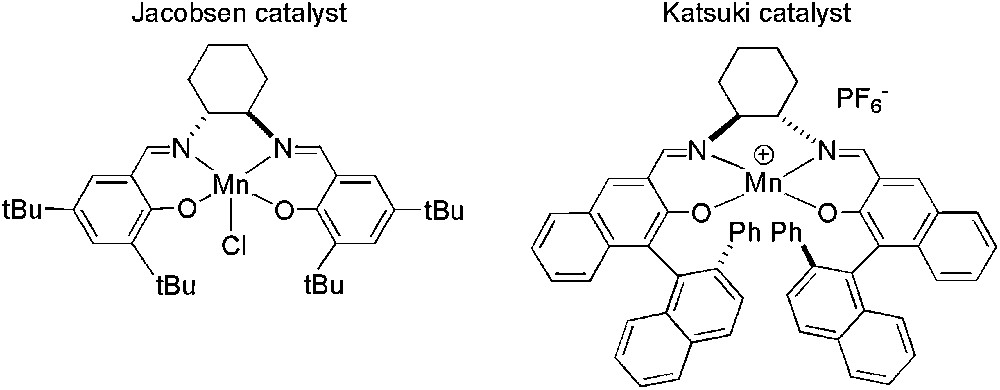
Jacobsen and Katsuki chiral catalysts used in asymmetric epoxidation.
Song's work demonstrated that Jacobsen chiral catalyst gave better activities in [C4C1im][PF6] than in organic solvent obtaining comparable enantioselectivities, for the epoxidation of several alkenes using aqueous buffered sodium hypochlorite solution as oxidant. In addition, the catalyst was easily recovered and recycled [8]. Similar results were obtained by Smith group for the asymmetric epoxidation of 1,2-dihydronaphthalene when Katsuki catalyst was used in the presence of 4-phenylpyridine N-oxide (4-PPNO) as a co-ligand, without any loss of activity and enantioselectivity up to four catalyst reuses [94] (Scheme 30).
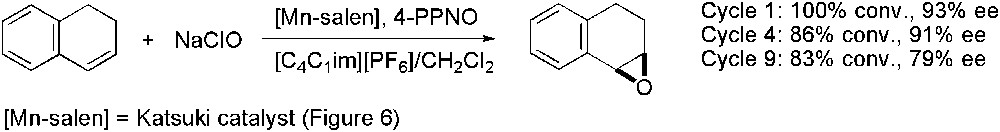
Mn-catalysed asymmetric epoxidation.
Chiral salen–cobalt catalysts have been successfully employed for hydrolytic kinetic resolution of epoxides in THF–IL medium (IL = [C4C1im][Y], Y = PF6 or NTf2) (Scheme 31) [95]. They led to the separation of optically pure (S)-epoxide (when the substrate used was a racemic mixture of epichlorohydrin, R = CH2Cl) and the formation of the corresponding diol with an ee up to 92%. In contrast to those results obtained in pure THF, the cobalt species recovered after hydrolytic kinetic resolution showed +3 oxidation state, without formation of inactive Co(II) complexes, as observed from XPS analysis.
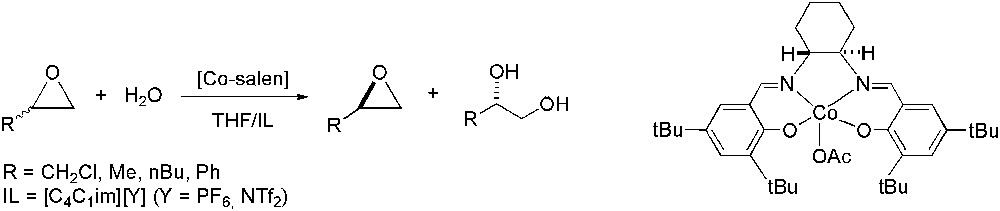
Co-catalysed hydrolytic kinetic resolution of epoxides.
The same group has also investigated the asymmetric ring opening reactions of epoxides with TMSN3 in [C4C1im][Y] (Y = PF6, SbF6, BF4, OTf), obtaining enantiomeric excesses up to 94% using chiral Cr–salen catalysts, without any activity and selectivity changes after five recycling runs [9].
4.2.2 Dihydroxylation
The Sharpless asymmetric dihydroxylation (AD) constitutes a versatile methodology for the synthesis of a wide range of chiral 1,2-diols from the appropriate olefins [96]. However, its industrial viability suffers from several disadvantages, mainly the high cost of osmium and chiral ligands, and the metal toxicity which can contaminate the chiral organic product. Catalyst immobilization in IL seems to offer an interesting way to plan large-scale production.
In 2002 for the first time, Afonso and Song teams reported independently olefin AD in ILs [97,98]. Afonso and co-workers [97] demonstrated for the dihydroxylation of mono- and disubstituted olefins, using K2[OsO2(OH)4]/K3[Fe(CN)6]/L∗ (where L∗ is a bis(cinchona) alkaloid: L44 and L45, Fig. 5) as catalyst that biphasic system IL/H2O or IL/H2O/tert-butanol instead of common H2O/tert-butanol medium allowed recovering the catalyst up to 10 runs, without any loss of activity and selectivity, due to the higher catalyst affinity for IL phase (Scheme 32).
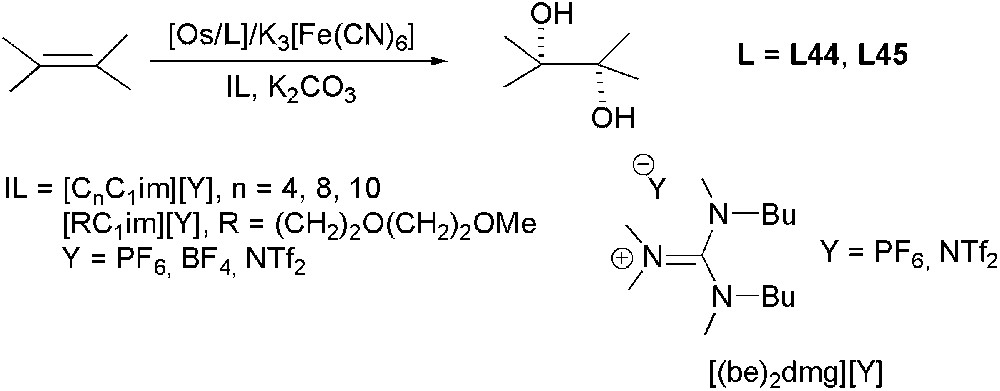
Os-catalysed asymmetric dihydroxylation.
For these kinds of processes, using N-methylmorpholine (NMO) as co-oxidant instead of K3[Fe(CN)6], combination of pure ILs ([CnC1im][Y], n = 4, 8; [C1C1im][Y]; Y = PF6, BF4, NTf2) and scCO2 allowed an efficient separation of organic products from IL medium due to the high solubility of scCO2 in IL and also the easy catalyst recovery (osmium content in organic extracts is less than 0.4%) [99]. Recently, Afonso group has developed the synthesis of new chiral ILs proving their asymmetric induction ability [100]. These CILs are constituted by tetra-n-hexyl-dimethylguanidinium cation ([(di-H)2dmg]) and natural chiral anions (A1–A4 in Fig. 2). The AD of 1-butene using [(di-H)2dmg][A2] as solvent and in absence of bis(cinchona) alkaloid chiral ligand, gave the corresponding diol with an ee = 85% (Scheme 33), similar to that obtained in tert-butanol/water [97b] in the presence of L44 or L45.
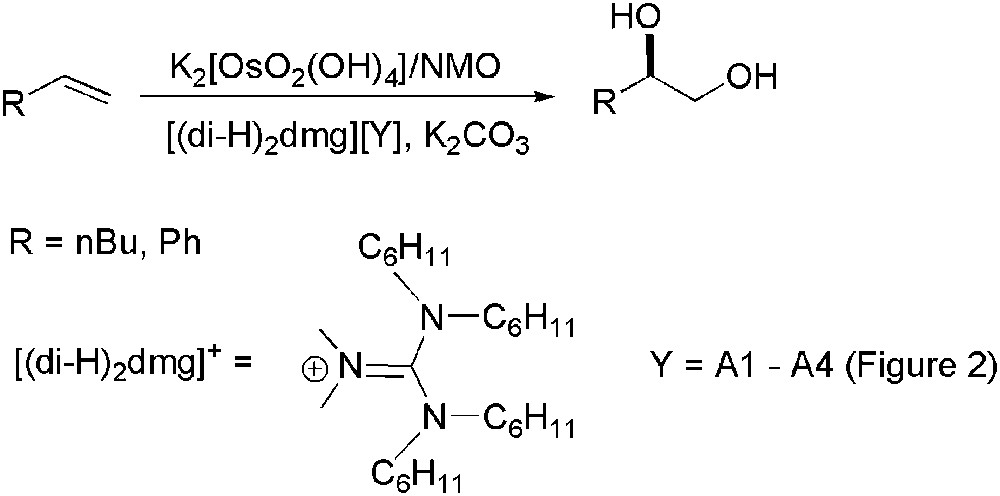
Os-catalysed asymmetric hydroxylation using CILs.
Song and co-workers [98] demonstrated that when AD catalysed by OsO4 with L47 (Fig. 5) and NMO, for several disubstituted olefins, was carried out in IL/acetone/water solvent (IL = [C4C1im][Y], Y = PF6, SbF6), no over-oxidation products of the obtained diols were observed, fairly common side reaction under Upjohn dihydroxylation conditions in absence of IL. Moreover, the use of L47 instead of L46 (Fig. 5), allowed the reuse of catalyst (three runs without changing the catalyst behaviour), due to the in situ ligand dihydroxylation, giving L48. Sheldon group has later analysed the effect of ligand structure on recycling [101], showing the relationship between the ligand polarity and its capability to be immobilized in IL phase. Taking into account these results, Zhang and co-workers designed an ionic chiral ligand for Os-catalysed AD processes, L49 (Fig. 5), by mono-quaternisation of L47 [102]. In this case, up to five runs were possible without any loss of activity and enantioselectivity, under similar conditions used by Song [98].
4.3 C–C bond formation
4.3.1 Allylic alkylation
[C4C1im][PF6] was used as solvent for the enantioselective allylic substitution reaction of (E)-1,3-diphenyl-3-acetoxyprop-1-ene with dimethyl malonate (Scheme 34) catalysed by palladium species containing chiral ferrocenylphosphine ligands L16–L17 (Fig. 4) [103]. The products were obtained in good yields (up to 81%) and moderate ee (up to 74%). However, when L17 was used, the yields dropped after a first recycling run. Similar results were obtained using a biphasic [C4C1im][PF6]/toluene mixture and were attributed to ligand leaching from the IL during the extractions. This study was then extended to other chiral ligands (R)-L9, L15, L19, L20 (Fig. 4) [104]. In all cases, except for (R)-L9, recycling of the catalyst solution did not affect the enantioselectivities whereas productivities, as mentioned above, decreased with all the ligands used. This activity loss was very important in the case of (R)-L9 for which a decrease in enantioselectivity was also observed, thus strongly suggesting that this ligand is more easily extracted than the others. Optimization of the system containing ligand L19 showed that at 60 °C the product could be obtained with 98% of yield and 88% of ee.
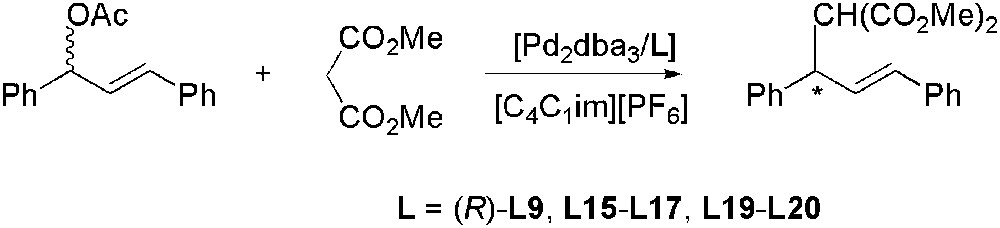
Pd-catalysed enantioselective allylic alkylation.
4.3.2 Allylation
The enantioselective In(III)-catalysed allylation of aldehydes by allyltributyltin has been studied in different ionic liquids. Benzaldehyde was chosen as substrate to test different chiral ligands in [C6C1im][Cl] (Scheme 35) [105]. The best results were obtained with L34 (88% yield, 48% ee) which was then chosen to test different ILs but none was as efficient as [C6C1im][Cl] in terms of enantioselectivities and yields. Changing to organic solvents (THF, dichloromethane) also led to low selectivities. Replacement of benzaldehyde by other substrates afforded, in [C6C1im][Cl], the corresponding expected allylation products with moderate ee for all the aromatic aldehydes whereas low values were observed for the aliphatic ones.
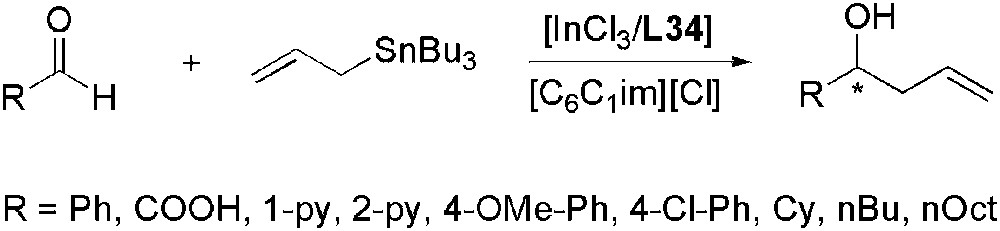
Enantioselective indium-catalysed allylation of aldehydes using L34 as chiral ligand.
(S)-BINOL was also tested as ligand for this reaction in various ILs (Scheme 36) [106]. In the model reaction using benzaldehyde as starting material, the best results were obtained with [C6C1im][PF6] which afforded the homoallylic alcohol in moderate yield (62%) and relatively good enantioselectivity (70%). ILs containing a chloride as anion did not show any enantioselectivity. The reaction was then extended to other aldehydes. The best enantioselectivity was obtained with trans-4-phenyl-3-buten-2-one. Recycling tests showed that, even after long reaction time, no reaction occurred anymore. Analysis of the ether extracts showed the presence of BINOL. The inactivity of the recycled solution was thus attributed to catalyst hydrolysis leading to its deactivation.
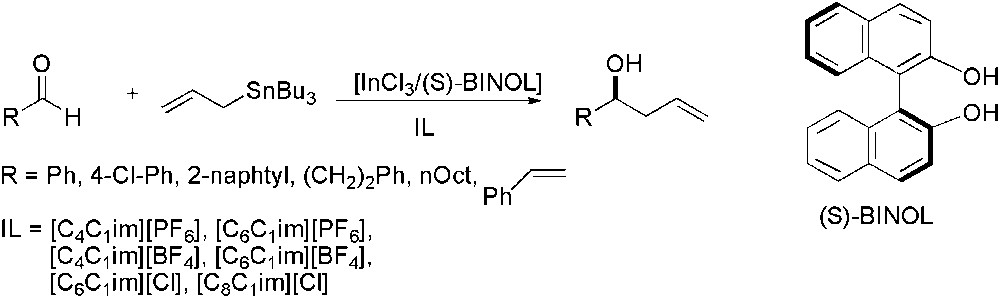
Enantioselective indium-catalysed allylation of aldehydes using (S)-BINOL as chiral source.
The same reaction was studied using In catalyst containing pybox derivatives L38–L43 as ligands (Scheme 37) [107]. Initial experiments carried out in neat [C6C1im][PF6] gave poor results. Addition of CH2Cl2 as co-solvent improved the performance of the catalytic system. With all the ligands tested, the reaction of benzaldehyde afforded the allylation product in good yields and with ee up to 88%. Different substrates were then screened, obtaining comparable results, even in the case of aliphatic aldehydes. The best enantioselectivity was observed for 4-Cl-benzaldehyde (94% ee). Recycling experiments evidenced that IL phase containing the chiral catalyst can be reused four times without any significant decrease in terms of yield and just a slight decrease of the ee values.
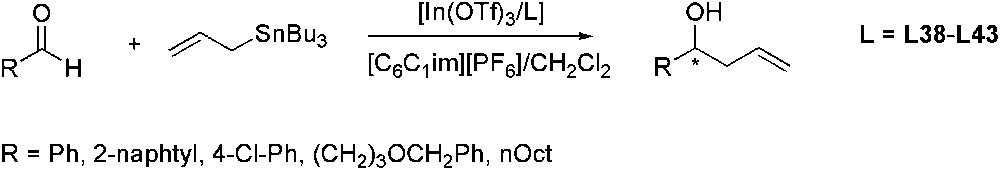
In-catalysed enantioselective allylation of aldehydes using L38–L43 as chiral ligands.
In/L40 catalytic system was also applied to the allylation of ketones in [C6C1im][PF6]/CH2Cl2 (Scheme 38) [108]. Reaction conditions were optimized using acetophenone as substrate and the allylation product was obtained in 82% yield with an enantiomeric excess of 65%. These values decrease only slightly after the fourth recycling run of the ionic liquid phase. The reaction was then extended to different ketones. In all cases, good yields were obtained and ee as high as 93% in the case of 1-indanone.
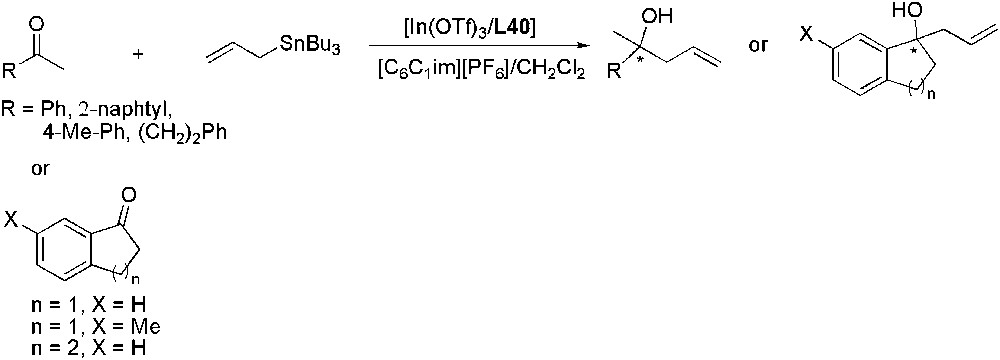
In-catalysed enantioselective allylation of ketones.
4.3.3 Cyclopropanation
Asymmetric Cu-catalysed cyclopropanation between alkenes and ethyl diazoacetate in ILs, has been mainly studied by Mayoral group (Scheme 39) [109]. They have demonstrated that ionic Cu/bis(oxazoline) catalysts (L35 and L36, Fig. 5) immobilized in ILs ([C3C1im][NTf2], [C3C1im][BF4], [(C8)3C1am][NTf2]) can be more robust than those related systems supported on anionic solids. This behaviour is due to the catalyst system's stability in IL medium, in contrast to ligand decoordination and consequently, enantioselectivity loss observed for immobilized catalysts onto solid supports [110]. However, they observed a reduction in enantioselectivity after each recovery cycle due to ligand leaching. In order to avoid it, and without addition of an extra amount of ligand, they have modified its structure, preserving the part responsible for the asymmetric induction unchangeable. Therefore, Cu/aza-bis(oxazoline) catalysts (L37, Fig. 5) could be recycled up to eight times without loss of activity or selectivity (enantiomeric excesses in the range 80–98% for both cis and trans isomers) in [C3C1im][OTf] [111]. In addition, Davies and co-workers have stated the effect of imidazolium IL's purity and the nature of IL anion ([C4C1im][Y], Y = OTf, PF6, NTf2, BF4, Cl, Br) in styrene cyclopropanation with ethyl diazoacetate using Cu/bis(oxazolines) systems, mainly when hydrophilic ILs were used [112].
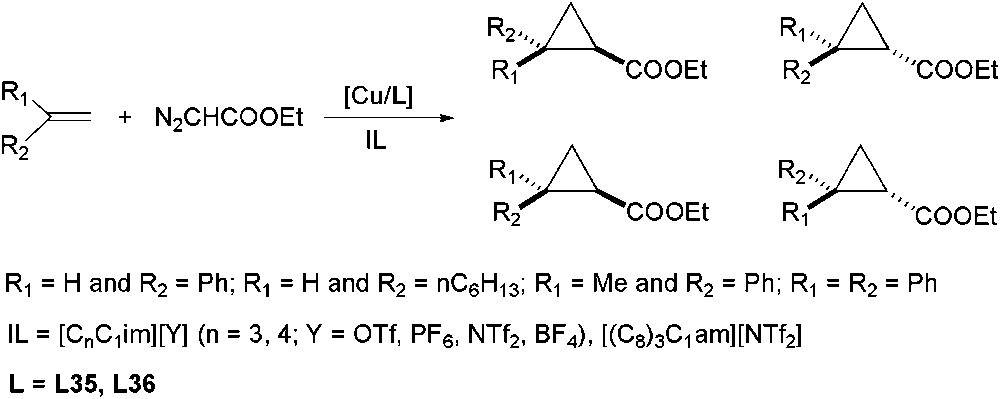
Cu-catalysed asymmetric cyclopropanation using chiral bis(oxazoline) ligands.
4.3.4 Others
The asymmetric Diels–Alder reaction of an oxazolidinone and cyclopentadiene catalysed by platinum complexes containing diphosphine ligands, was carried out in various ILs (Scheme 40) [113]. Reactions were faster and significantly higher enantioselectivities (up to 94%) were obtained in ILs than in dichloromethane. Several recycling runs of the ionic liquid phase were performed, showing that after the third run no significant change in ee values was found and only a small decrease in activity was observed.
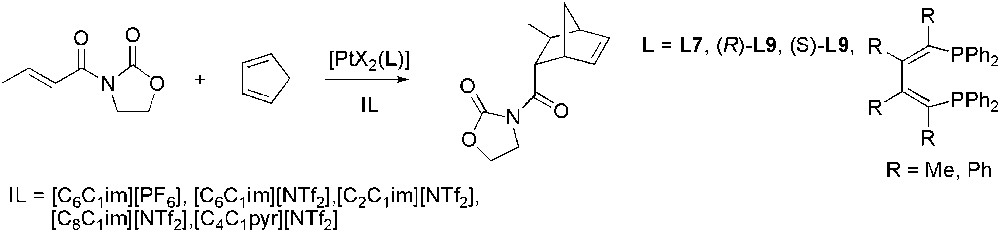
Pt-catalysed asymmetric Diels–Alder reaction.
Leitner's group has investigated the enantioselective co-dimerization of styrene with ethylene to give 3-phenyl-but-1-ene, using Wilke's catalyst under IL/scCO2 solvent conditions in a continuous-flow apparatus, by uninterrupted product extraction with compressed CO2 (Scheme 41) [114]. In particular, they used [C3C1im][Y] and [C4C1py][Y] (Y = NTf2, BF4, BARF, Al(OC(CF3)2Ph)4), obtaining high selectivities (ee up to 90%) for imidazolium cations with BARF as anion, due to its low nucleophilicity and consequently low coordination strength towards the metal centre.
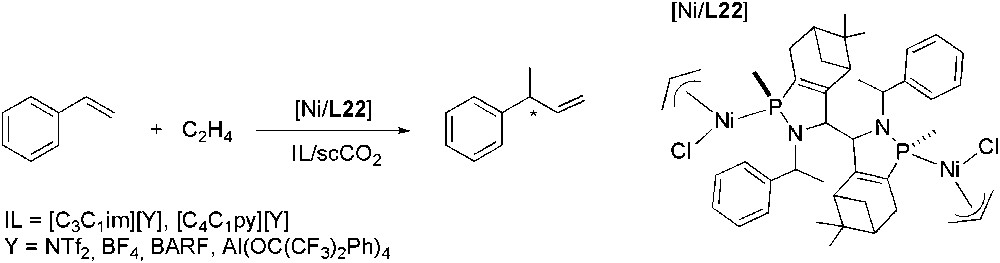
Ni-catalysed asymmetric hydrovinylation of styrene.
Imidazolium-based ILs were used as solvent for the copolymerization of CO and propene using palladium complexes containing various chiral diphosphines (Scheme 42) [115]. Addition of a small quantity of methanol was, however, necessary since in neat ionic liquid, no polymer formation was observed. If compared to the same reaction carried out in dichloromethane/methanol, the use of ILs resulted in higher productivities and formation of polyketones of higher molecular weight. An influence of the alkyl chain of the cation and the nature of IL on region-regularity, molecular weight, polydispersity, productivity and optical rotation of the copolymer was reported.
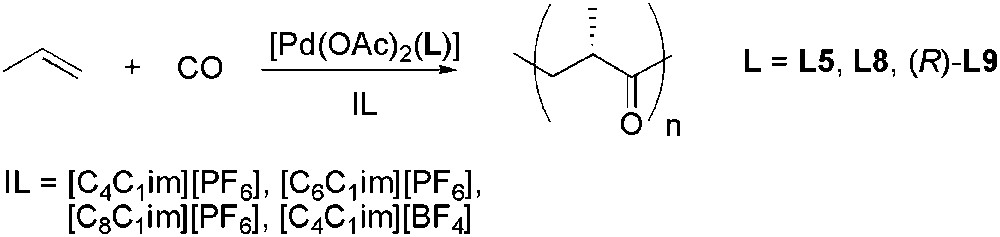
Stereoselective palladium-catalysed CO/propene copolymerization.
The catalytic enantioselective Michael reaction was described using palladium complexes with BINAP derivative ligands ((R)-L9, (R)-L11, (R)-L12) [116]. The reaction of a β-ketoester with methyl vinyl ketone in [C4C1im][OTf] gave the Michael adduct with new carbon–carbon bond with 82% yield and 84% ee after 18 h at 0 °C (Scheme 43). The second reaction cycle gave the same results but the third showed a considerable decrease in the reaction rate. However, in the presence of acidic benzenesulfonimide the enone was activated, causing acceleration of the reaction and the reuse of the catalyst was then possible at least five times.
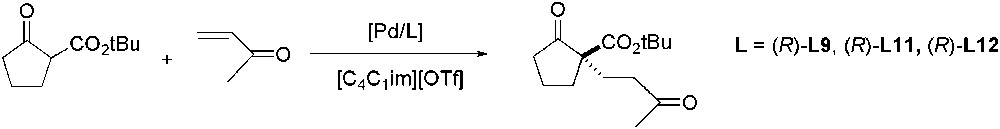
Pd-catalysed asymmetric Michael reaction.
Chiral platinum complexes catalysed the asymmetric reaction between various alkenes and ethyl- or phenylglyoxal in [C2C1im][NTf2] (Scheme 44) [117]. For each catalyst, the ee obtained in IL was higher (up to 95%) or at least, comparable to those obtained in dichloromethane. The variation in ee with time was studied in both IL and dichloromethane. It was evidenced that in the case of the organic solvent, there was a gradual decrease in ee as the reaction proceeds whereas it remained constant in [C2C1im][NTf2]. A possible origin for this observation could be the faster racemisation of the catalyst in dichloromethane. Recycling of the ionic liquid phase afforded after three runs the same enantioselectivity but yields decreased. Since no traces of the catalyst were detected in the ether extract, this was attributed to catalyst deactivation rather than to its leaching.
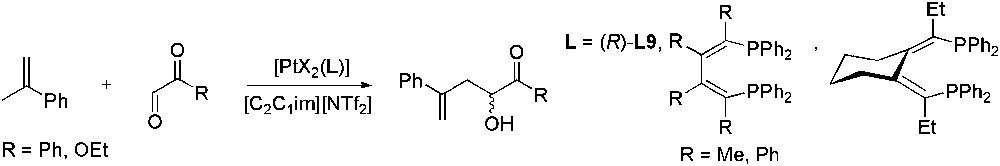
Pt-catalysed enantioselective carbonyl-ene reaction.
The CIL [(2-Me)C4C1im][PF6] was also used as reaction medium for metal-catalysed reactions. In 2003, the Heck oxyarylation reaction was studied using chiral phosphine ligands, L3, L4, (R)-L9 and L24 (Scheme 45) [118]. However, very poor enantioselectivity was observed (up to 10%), probably due to the formation of an achiral intermediate.
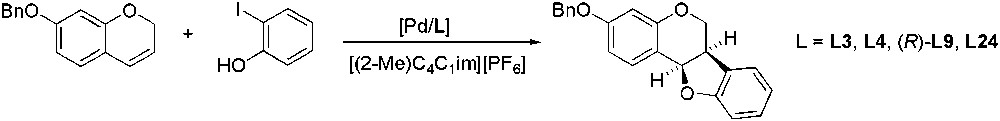
Pd-catalysed enantioselective Heck oxyarylation.
4.4 Miscellaneous
4.4.1 Asymmetric catalytic allylic amination
The first asymmetric catalytic allylic amination in an ionic liquid has been recently described by Lyubimov and co-workers using chiral P-donor ligands L26–L29 (Scheme 46). Cationic palladium complexes containing these ligands were tested as catalysts for asymmetric amination of racemic (E)-1,3-diphenyl-3-acetoxyprop-1-ene with bis(n-propyl)amine in [C4C1C1im][BF4] [119]. The highest enantioselectivity was achieved with Pd/L28 system. In the first catalytic cycle, 84% ee and quantitative conversion were obtained, however, the second cycle suffered from a considerable drop in both activity and selectivity. Catalyst involving BINOL-based phosphoramidite L29, showed comparable asymmetric induction in IL to that observed in THF or CH2Cl2.
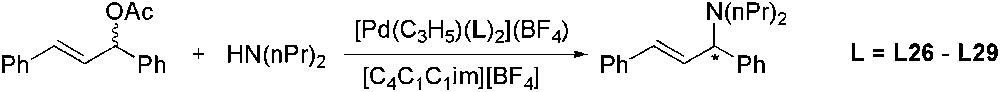
Pd-catalysed asymmetric amination of (E)-1,3-diphenyl-3-acetoxyprop-1-ene.
4.4.2 Asymmetric aminohalogenation
Because haloamines are important building blocks in organic and medicinal chemistry, the aminohalogenation reaction has become an active topic. The substitution of classical solvents by an IL and most particularly by [C4C1im][BF4] allowed the sterospecific aminohalogenation of cinnamic esters with good yields (76–81%) [120]. Later, the same team described the asymmetric aminohalogenation of α,β-unsaturated N-acyl 4-alkyloxazolidinones, which did not proceed in organic solvents but did in IL (Scheme 47) [121]. The success of this reaction has been largely attributed to the high polarity of ILs because during the reaction an ionic intermediate is plausibly formed and IL could accelerate this formation. Acceptable chemical yields (60–72%) and ee (up to 75%) were obtained with a good scope of substrates.
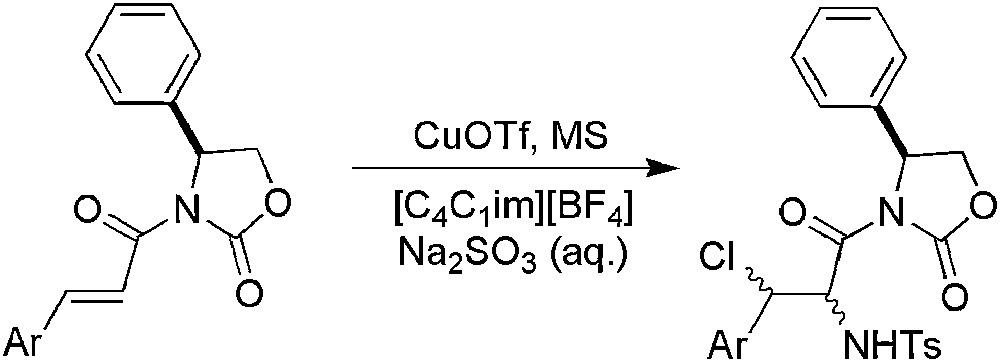
Cu-catalysed aminohalogenation of α,β-unsaturated N-acyl 4-alkyloxazolidinones.
4.4.3 Asymmetric addition of alkynes to imines
The enantioselective preparation of propargylamines by addition of alkynes to imines with the tridentate chiral ligand bis(oxazolinyl)pyridine L39 and Cu(I) complex has been recently described [122]. Higher yields were achieved at 40 °C than at room temperature in [C4C1im][NTf2] (88% versus 74% at rt), but some erosion on the enantioselectivity occurred (88% versus 94% at rt), as also observed in water or toluene [123]. Moreover, Cu(OTf) gave considerably higher enantioselectivity than CuBr. In addition, the IL hydrophobicity and the substituent on 2 position of imidazolium cation (H or alkyl group) played an important role on the ee (27% with [C8C1im][PF6] versus 76% with [C4C1C1im][PF6]) (Scheme 48). Regarding the anion structure, no appreciable change on the enantioselectivity was observed for NTf2, BF4 or PF6.
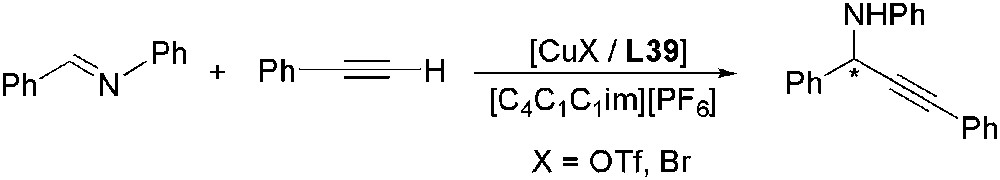
Cu-catalysed enantioselective addition of alkynes to imines.
4.4.4 Asymmetric hydroamination of activated olefins
Catalytic addition of morpholine to methacrylonitrile in different ILs was catalysed by [Ni(L21)(THF)](ClO4)2 (Scheme 49) [124]. In [C4C1im][BF4], enantioselectivities comparable to those obtained in THF were reached, but the activities were higher. Moreover, catalyst was less sensitive to air and moisture in IL.
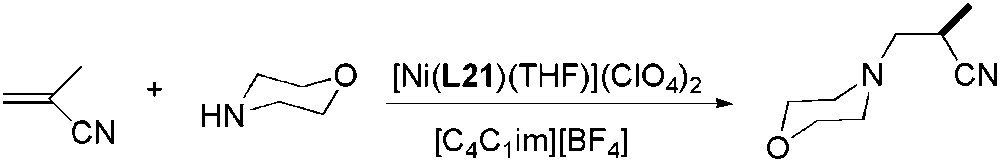
Ni-catalysed asymmetric hydroamination of methacrylonitrile with morpholine.
4.4.5 Asymmetric fluorination
The first organocatalysed enantioselective fluorination in ILs was reported by Cahard and co-workers, using a chiral quinine-based [N–F]+ reagent and silyl enol ethers as substrates [125]. The use of a chiral palladium complex with (R)-L12 ligand also gave very good ee (up to 94%) (Scheme 50) [116]. The reaction rate of the asymmetric fluorination of β-ketoesters was found to be dependent on the length of the alkyl substituent of the cation and on the anion used. The best results were reached using [C6C1im][BF4] as solvent (93% yield and 92% ee) and the catalyst could be reused 10 times without any loss of enantioselectivity.
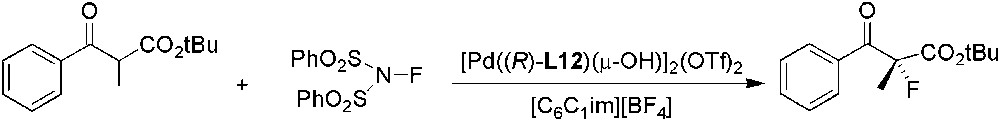
Pd-catalysed asymmetric fluorination of β-ketoesters.
Very recently, Kim et al. reported the catalytic enantioselective electrophilic fluorination of β-keto phosphonates using chiral palladium complex containing (R)-L9 ligand in IL (Scheme 51) [126]. [C4C1im][BF4] and [C4C1im][OTf] gave the best yields (95%) and enantioselectivities (93%) for β-keto phosphonate with R = Et and n = 1. In this case, catalyst recycling was also tested in [C4C1im][BF4], without changes in yield and selectivity after seven runs.
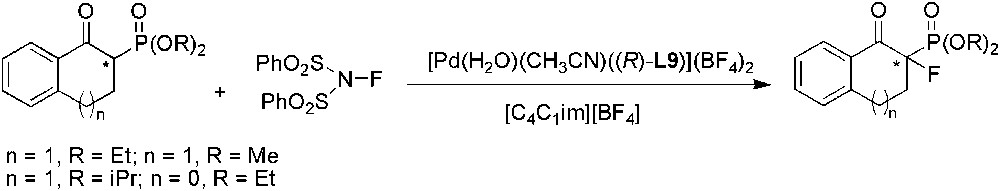
Pd-catalysed enantioselective fluorination of β-keto phosphonates.
4.4.6 Cyanosilylation of aldehydes
One of the highest-production volume compounds in asymmetric catalysis is mandelonitrile which is produced in multi-hundred ton scale. Garcia and co-workers have been particularly interested in recycling the chiral catalysts employed in cyanhydrine synthesis, vanadium–salen complexes. One of the strategies followed by these authors has been the use of N,N′-dialkylimidazolium ILs instead of dichloromethane in the cyanosilylation of aldehydes using trimethylsilyl cyanide (TMSCN) as nucleophile (Scheme 52) [127]. In this initial work they have obtained activities and selectivities comparable to those achieved in dichloromethane, but now the catalyst could be easily reused: for benzaldehyde, up to five times without any loss of activity and selectivity in [C3C1im][PF6]. Structural adaptations of vanadium complexes to make them more similar to ILs and less soluble in organic solvents, failed [128]. Actually, [VO(salen)@IL] complex modified by appending an imidazolium unit to make the system ‘ionophilic’, led to moderate selectivities (ee up to 57%), in contrast to the high enantiomeric excess achieved using the non-functionalized [VO(salen)] complex. This fact could be attributed to the interaction of the vanadyl group with the chloride anion present in the structure of [VO(salen)@IL] [129].
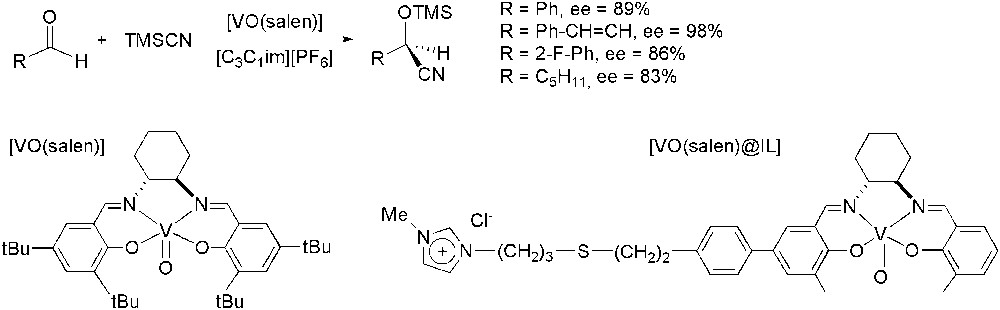
V-catalysed asymmetric cyanosilylation.
5 Final remarks
The chirality source for the enantioselective organic processes in IL medium comes from the solvent and/or the catalyst. Concerning the solvent, chiral ionic liquids lead to high enantioselectivities if coordinating groups are present in their structures [59,100] or if the solvent acts also as catalyst [61]. With regard to the catalytic species, most of them are those used in organic solvent, but catalyst leaching is sometimes detected during the work-up. To immobilize the catalyst in the IL phase, structural modifications can be done to decrease its solubility in organic solvents, usually by introduction of an ionic group [50,73,92,102,129], but in some cases a selectivity decrease is produced. Further work in this research axe remains to be done in order to improve the catalyst recycling by ligand modification and also the selectivity in new ionic liquids.
Acknowledgments
We thank CNRS and Université Paul Sabatier for financial support.
1 Enantiomeric ratio E is defined as E = ln[1 − c(1 + ee(product))]/ln[1 − c(1 − ee(product))], where c = conversion of racemic substrate and ee(product) = the enantiomeric excess of the product.
2 This methodology has also been employed by Itoh and co-workers using [C4C1im][(OsO2O-(CH2CH2O)10-C16H33)] and [C4C1C1im][(OsO2O-(CH2CH2O)10-C16H33)] and PCL.