

1 Introduction
The comparative explosion in the chemistry of dithiolate-bridged diiron carbonyls and their variously substituted analogues is, of course, driven by the hope that compounds related to the diiron subsite of the [FeFe] hydrogenase H-cluster, {2Fe}H, will provide the efficient catalysts needed for either production or consumption of dihydrogen. Well-defined electrocatalytic responses have been reported for many of these compounds [1] and an important goal of the chemical studies is the identification of materials that support rapid electrocatalysis at relatively modest overpotentials.
Rational catalyst design and optimisation is underpinned by an understanding of the reaction path. For the dithiolate-bridged diiron carbonyl compounds related to {2Fe}H electrocatalysis has been studied in most detail for Fe2(S(CH2)3S)(CO)6 (3S) [2,3]. Electrochemical and spectroelectrochemical (SEC) studies of electrocatalytic proton reduction of p-toluene sulfonic acid (HOTs) by 3S suggest a reaction path that involves a sequence of two one-electron, one-proton steps to give a two-electron, two-proton product that undergoes dihydrogen elimination. The relatively slow rate of dihydrogen elimination (ca. 5 s−1) is increased by further reduction [2,3]. Comparison between the reduction chemistry and electrocatalysis by 3S and phosphido-bridged analogues suggests that the reaction path for the two-electron, two-proton reaction involves hydrides that occupy sites opposite to the S (or P) bridging ligand [4]. The molecular details of the reaction have been explored by density functional theoretical (DFT) methods [5]. These studies support the proposed sequence of reduction and protonation reactions and the proposition that dihydrogen elimination from the two-electron, two-proton product of 3S is slow and rate limiting [2]. Importantly, when taken together with studies of the phosphido-bridged analogues [4,6], this work provided the first experimental evidence supporting the proposition that the reaction path involves protonation of the Fe–Fe bond. Whereas HOTs is a sufficiently strong acid to protonate 3S− (in CH3CN and THF), under the same conditions acetic acid (HOAc) is not a strong enough acid to protonate the one-electron reduced product and the reaction path for electrocatalysis by weak acids such as HOAc has been suggested to proceed through two one-electron reductions followed by two protonation steps [7]. Contrary to the general observation of one-electron reduction steps for bis(thiolate) and dithiolate-bridged diiron carbonyl compounds, the benzene-1,2-dithiolate analogue (BDT) undergoes a reversible two-electron reduction [8,9]. Very recent electrochemical and DFT studies of the electrocatalytic proton reduction reaction of HOAc with BDT have revealed that the reaction proceeds with significant structural reorganisation following two-electron reduction and it is at this level that the first protonation step occurs; further reduction and protonation lead to dihydrogen elimination [10].
Two issues related to electrocatalytic proton reduction have been raised by Evans and coworkers [11]. The first concerns the impact of direct proton reduction on the electrocatalytic response, a matter particularly important when a significant rate of reaction occurs with a high overpotential. Under these circumstances the choice of electrode material is of critical importance. For electrocatalytic proton reduction the overpotential presented by a mercury electrode would, in the absence of complicating chemistry, present the best choice. Owing to the high affinity of mercury for thiolates alternative working electrode materials need to be used for the study of H-cluster analogues and vitreous carbon presents a suitable alternative. The second issue concerns the impact of the acid strength (pKa) on the driving force for the reaction. Since the potential needed for an electrocatalytic reaction that follows an EC′ path (electrochemical then chemical steps preceding reaction to recover the starting complex) is largely determined by the redox potential of the catalyst, the overpotential will be lowest for electrocatalytic proton reduction by the weakest acid capable of supporting the reaction at a given rate.
The only substantive operational reason for using a relatively weak acid in a functional system is to avoid irreversible destruction of the catalytic material or, if supported, the synthetic or biological matrix in which it is contained. Probing mechanism and/or suppressing uncatalysed reaction pathways by using weak acids can be informative and many studies report experiments of this type that have been used to benchmark the activity of the newly reported compounds [7,12–18]. In this contribution we show that for a diverse range of thiolate-bridged diiron carbonyl compounds reduction gives stable products with a grossly different structure from that of the parent compound and that electrocatalysis of proton reduction with a weak acid source such as HOAc proceeds after further reduction of these structurally altered species. For this reason such experiments provide a poor and misleading criterion for relating the electrocatalytic activity to the structure and composition of the diiron parent compound and the relationship this may have to the diiron subsite of [FeFe] hydrogenase.
2 Results and discussion
When dealing with aqueous media the definition of “weak” and “strong” acids is well understood, however the application of these terms in the context of nonaqueous media is often ambiguous and requires qualification. In acetonitrile only acids such as triflic and perchloric acids are largely dissociated and p-toluene sulfonic acid (HOTs) with a pKa of ca. 8 is overwhelmingly in the undissociated form [19]. In the context of the electrocatalytic proton reduction reactions of bridged diiron(I) carbonyl compounds at the focus of this study “weak” and “strong” acids are operationally defined in terms of their ability to protonate the one-electron reduced form of the compound. In terms of this operational definition HOTs is an example of a strong acid and HOAc is a weak acid.
The influence of weak and strong acids on the reduction chemistry of dithiolate-bridged diiron hexacarbonyl compounds is illustrated by the electrocatalytic and IR spectroelectrochemical (IR-SEC) properties of the ethylene (2S) and propylene (3S) linked dithiolates and the bis(thiolate) compound Fe2(μ-SEt)2(CO)6, SEt. The inclusion of SEt allows consideration of the case where rearrangement or partial dissociation of the dithiolate is more facile. The electrochemical properties of thiolate-bridged diiron carbonyl compounds have been examined by several workers and the cyclic voltammograms of the three compounds are shown in Fig. 1 [20,21]. The primary reduction wave (process I) is, at best, partly reversible for the dithiolates and is irreversible for the bis(thiolate) compounds. Process I is nearly reversible for CO-saturated solutions of 2S and 3S and this suggests a reaction path involving CO dissociation. A second reduction wave (process II) is most clearly defined for SEt; for 3S and 2S the peak current is less than half of that obtained for process I. In all three cases at least two different reductions can be discerned for processes II. The daughter products formed following reduction differ both in terms of their relative concentrations and re-oxidation potentials. It is noteworthy that there is a surprisingly large difference between the re-oxidation potentials for the daughter products of 2S and 3S.
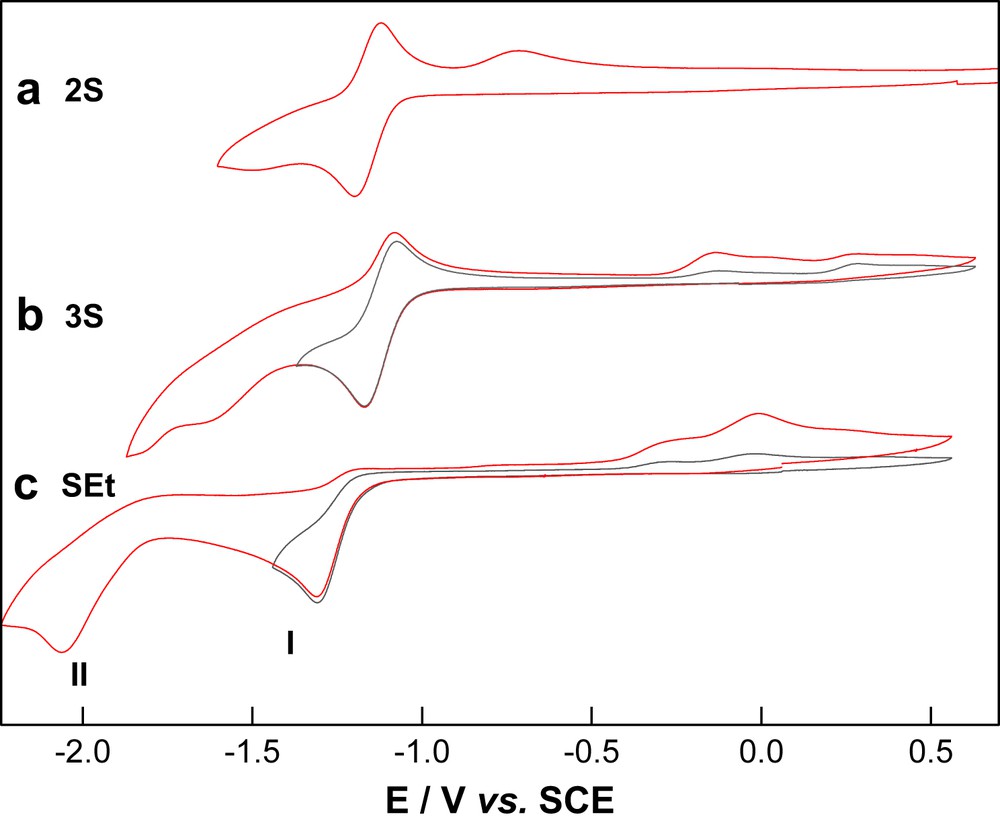
Cyclic voltammetry of 1 mM solutions of (a) 2S, (b) 3S and (c) SEt in THF (0.2 M TBA[PF6], vitreous carbon working electrode, scan rate 100 mV s−1).
The reduction chemistry of 3S has been shown to feature an unexpectedly rich chemistry that involves rearrangement of the CO and thiolato groups as well as unexpected dimeric (tetrairon) forms [2], some of which have been characterised by X-ray crystallography [22,23]. The reactivity and structure of the different species may be understood in terms of the strong tendency of the Fe centres to maintain a so-called “18 electron” configuration [24]. Unlike the analogous phosphido-bridged compounds, which undergo two-electron reduction reactions to yield electron precise electron configurations simply by the loss of the Fe–Fe bond [4], the thiolate-bridged compounds predominantly undergo reduction in one-electron steps. The chemical reversibility of process I is sensitive to the identity of the bridging ligand, generally being greater for dithiolates than for bis thiolates, and this is evident from the voltammetry shown in Fig. 1. Clearly a reaction path involving CO loss is implicated by these observations, however, the CO-depleted reduced species initially formed have not been identified. IR spectroelectrochemical (IR-SEC) studies of 3S suggest that reaction of 3S1− is fast relative to reduction at the electrode and the lifetime of the one-electron reduced product is inversely proportional to the concentration of 3S, suggesting a bimolecular reaction path [2]. Since CO dissociation from the one-electron reduced product would formally give an electron deficient species the involvement of a second such species would provide a path to a decarbonylated electron precise product. While the suggested structures of the singly decarbonylated tetrairon species shown in Scheme 1 are speculative it has recently been shown that one-electron reduction of the related compounds, Fe2(μ-pdt)(CO)5L, where L is a labile ligand, yields a tetrairon species (2SD) with two bridging CO ligands, which has been characterised by X-ray crystallography (Scheme 1) [23]. The studies of 2SD demonstrate that rearrangement between the diiron and tetrairon species may be fast on the electrochemical timescale. It is likely that the differing stabilities of the tetrairon species for ethanedithiolate and propanedithiolate compounds serves as an important factor in explaining the differing reaction pathways obtained during reduction of 2S and 3S.
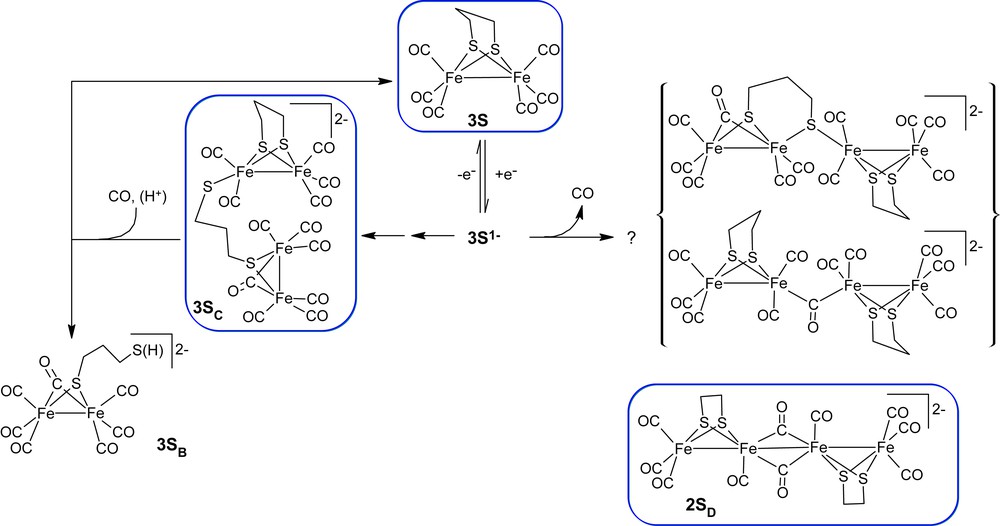
X-ray structures are available for 3S [25], 3SC [22] and 3SD [23].
The electrochemistry of 3S in the presence of HOTs has been reported and the reaction path considered in detail [2]. The electrocatalytic response of 2S with HOTs is given in Fig. 2a. Protonation of the one-electron product is reflected by a shift of process I to more positive potentials where the magnitude of the shift is related to the magnitude of the equilibrium and rate constants. Electrochemical simulation has shown that an electrocatalytic pathway involving two one-electron reduction/protonation sequences (ECEC′ reaction) satisfactorily accounts for the process occurring at more positive potentials [2] and a simulation based on the scheme given in Ref. [2] is shown in Fig. 2b. The rate limiting step, dihydrogen elimination from H23S, may be accelerated by further reduction and this accounts for a second process occurring at slightly more negative potentials at higher acid concentrations. DFT calculations suggest that such a reaction path is energetically feasible and also provide insights into the structures of the reduced species [5].
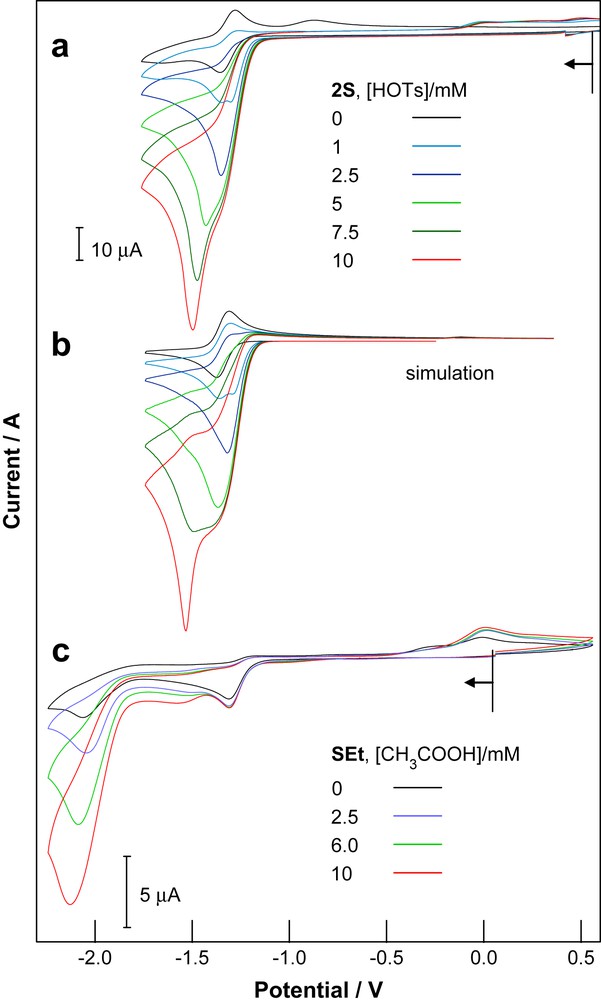
Electrocatalytic proton reduction by 2S and SEt. Cyclic voltammetry of (a) 2S (1 mM) and HOTs (0–10 mM); (b) digital simulation based on the electrocatalytic scheme described in Ref. [2]; (c) SEt (1 mM) and HOAc (0–10 mM). Experimental data were obtained from THF solutions with 0.2 M TBA[PF6] as supporting electrolyte and either a 1 mm (SEt) or 2 mm (2S) diameter vitreous carbon working electrode at a scan rate of 100 mV s−1.
The involvement of tetrairon species in the electrocatalytic reaction is reflected by the differing effects of CO on the reduction chemistry of 2S and 3S [2,26]. Whereas additional CO improves the reversibility of process I, by eliminating formation of decarbonylated products, in the presence of acid CO addition to the tetrairon species 3SC is sufficiently rapid to compete with electrocatalysis (Scheme 1). It is important to note that electrocatalytic proton reduction by 2S is not inhibited by CO and this has been explained in terms of the different tetrairon product formed following reduction [26].
A strong electrocatalytic response is obtained for solutions of HOAc with a wide range of hydrogenase model compounds, where the results obtained for SEt are given in Fig. 2c. Since SEt1− is not protonated by HOAc there is no shift of the potential of process I to more positive values. While the main electrocatalytic wave appears to be associated with process II additional waves develop at potentials intermediate between processes I and II. The background current due to direct reduction of HOAc at the carbon electrode makes a negligible contribution to the response at potentials more positive than −1.9 V and contributes less than 10% of the current at −2.2 V. As noted by Evans and coworkers the redox potential for the reduction of the respective acids depends on their pKa and for acetonitrile solutions the EHA0 value of HOTs is 0.71 V more positive than that of HOAc [11]. Consequently, the overpotential for reduction of the acid is lower for the reactions with HOAc.
A key question that has largely been avoided in previous studies of electrocatalytic proton reduction of weak acids by hydrogenase model componds is the form of the catalytic species responsible for the reaction. Superficially the assignment of the two reduction waves of SEt to sequential electron transfer reactions and thus formation of SEt2− leads to an electrocatalytic sequence in which dihydrogen elimination follows two protonation steps (EECC′ reaction). While studies of electrocatalytic proton reduction of HOAc by hydrogenase model compounds commonly assign EECC′ reaction paths, several aspects of the voltammetry sit uncomfortably with such an assignment. The most important of these is the attribution of process II to the Complex1−/Complex2− couple. In simple terms the irreversibility of the SEt/SEt1− couple would suggest that process II is due to chemically altered reduced species, moreover comparison between the electrochemistry of bis thiolate- and dithiolate-bridged compounds shows that the value of Ipc for process II is not related to the reversibility of process I. A recent detailed study by Capon et al. demonstrates clearly that while the primary reduction wave of thiolate-bridged diiron compounds related to 3S has many of the characteristics of a one-electron process, the second reduction is also involved [27]. In these cases the electrochemistry is complicated by competing chemical reactions and a slow rate of heterogeneous electron transfer. It was shown that observation of a distinct electrochemical response for the two redox waves requires the collection of voltammograms at scan speeds greater than 0.5 V s−1 and in these cases the second reduction occurs ca. 0.2 V more negative than the first [27].
The details of the chemical species formed during the course of the redox reaction are most clearly delineated by recourse to spectroscopy. Currently IR-SEC studies are available for the reduction of 2S and 3S [2,18]. The extension of these studies to SEt is important given the relative prominence of process II. The initial phase (4 s) of reduction of SEt at a potential encompassing the first reduction wave (−1.4 V) leads to the depletion of the starting material (negative bands at 2073, 2036 and 1994 cm−1) and its conversion into new species with ν(CO) bands at 2018, 2014, 1974, 1952, 1941, 1925, 1888, and 1728 cm−1 (Fig. 3a). At longer times an additional low intensity ν(CO) band grows into the spectrum near 1870 cm−1. When the potential is then set to less negative values (−1 V) there is partial recovery of SEt (10–15%) and this is associated with the depletion of a subset of the growth bands (Fig. 3b). The remaining set of growth bands has a profile that closely resembles the spectrum of a previously characterised product with bridging thiolate and CO groups, 3SB (Scheme 1). On this basis a product with a similar structure, SEtB, is formed following reduction of SEt. This assignment is confirmed by observation of the EPR spectrum of the oxidised form of SEtB [28]. Application of potentials more positive than +0.2 V results in oxidation of SEtB with formation of SEt (Fig. 3c). The recovery of SEt (ca. 75%) following reduction–oxidation cycle is poorer than that for the analogous dithiolate-bridged compounds 2S and 3S [2,18], and the final spectra (Fig. 3c) include broad bands near 2080, 2105 and 1980 cm−1 due to unidentified reaction products. It is important to note that at no stage through the reductions are bands attributable to the one-electron reduced product of SEt evident in the spectra. This observation is consistent with the irreversible character of process I for SEt.
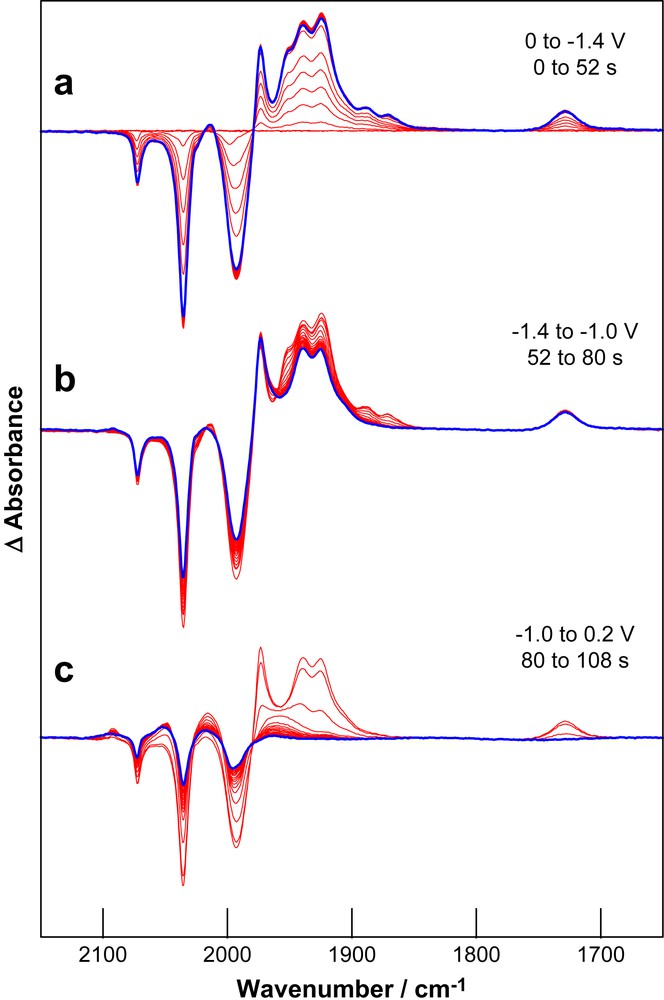
Differential absorption IR-SEC spectra of SEt (1 mM, 0.1 M TBA[ClO4], CH3CN, Ar) during (a) reduction through process I, (b) re-oxidation at mild potentials and (c) re-oxidation at +0.2 V. The last spectrum of each group is highlighted.
Application of a reducing potential to a solution of SEt sufficient to access process II results in a more complicated set of time dependent spectral changes (Fig. 4a). Coincident with the depletion of SEt there is an immediate growth of a set of bands attributable to SEtB, however, this species is unstable at the applied potential and the intensity of the growth bands decays to give a final spectrum with weak bands near 1900 and 1820 cm−1 and a strong band at 1745 cm−1. Re-oxidation at mild potentials results in depletion of the band at 1745 cm−1 with minimal recovery of SEt and a set of broad bands centred on 1900 cm−1 (Fig. 4b,c). Re-oxidation at more positive potentials (+0.4 V) results in recovery of about 30% of the starting material leaving weak residual growth bands between 2050 and 2000 cm−1 (Fig. 4d). Clearly the reduction of SEtB is associated with process II where the irreversibility of the spectral changes is consistent with the irreversible electrochemical behaviour. The main reduction product of the reaction is tetracarbonyl ferrate, [Fe(CO)4]2−, a common product formed following reductive degradation of iron carbonyl complexes [29,30]. The assignment of the band at 1745 cm−1 to this species is confirmed in experiments conducted using CO saturated solutions. In this case there is a simpler distribution of products and the re-oxidation process proceeds with the observation of bands at 1913, 1887 and 1866 cm−1 which are consistent with the formation of [Fe2(CO)8]2− [28].
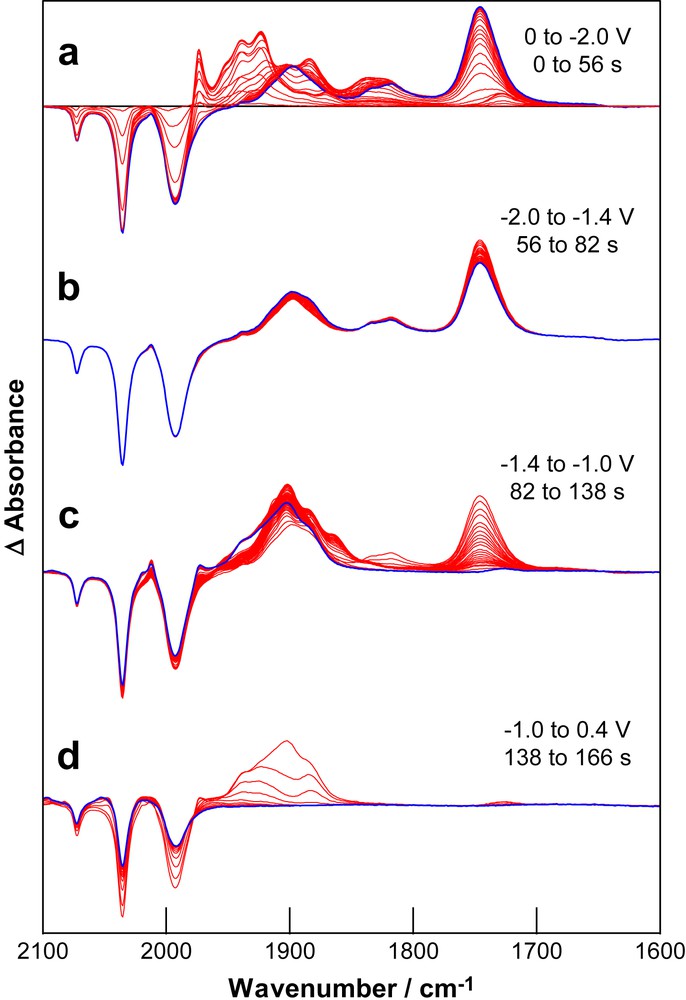
Differential absorption IR-SEC spectra of SEt (same solution as for Fig. 3) during (a) reduction through process II and re-oxidation at (b) −1.4 V, (c) −1.0 V and (d) +0.4 V. The last spectrum of each group is highlighted.
The spectral changes obtained following reduction of 3S in the presence of HOAc at potentials sufficient to access process I are shown in Fig. 5 and at potentials sufficient to access process II in Fig. 6. Under mild conditions the reaction proceeds to give similar products in the presence and absence of HOAc and the primary reduction product is 3SB. Despite the greater relative stability of 3S1− over SEt1− there is no sign of formation of 3S1− during the early stage of the reaction (Fig. 5a). In the absence of additional CO other reduction products are generated in low concentration and this is reflected by the features apparent in the spectra at 1900 and 1955 cm−1. It is interesting to note that during the formation of 3SB there is depletion of bands due to HOAc at 1730 and 1755 cm−1. Once the electrosynthesis in the thin layer of solution between the working electrode and the IR-transmitting window is complete there is no further depletion of HOAc (Fig. 5b). Application of a re-oxidising potential (+0.5 V) results in substantial (>90%) recovery of 3S together with near-quantitative recovery of HOAc (Fig. 5c). The reversible interconversion of HOAc and OAc1− is due to an acid-base reaction with the dissociated thiolate formed during generation of 3SB (Scheme 1).
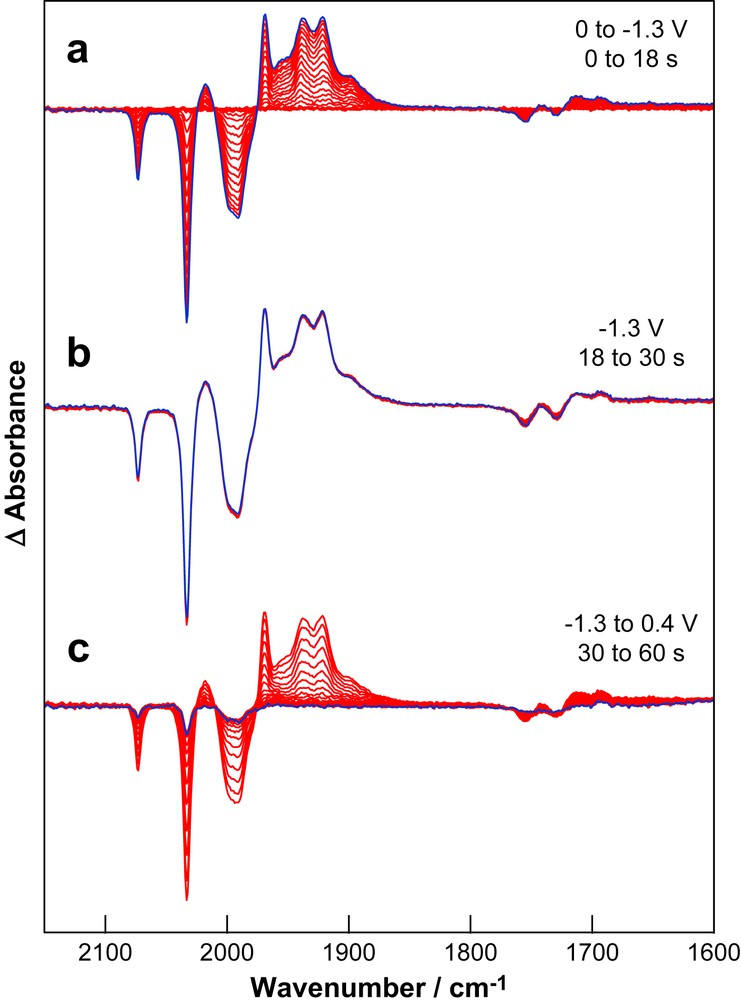
Differential absorption IR-SEC spectra of a solution of 3S (1 mM) and HOAc (20 mM) in THF (0.2 M TBA[PF6]) during (a) reduction (process I), (b) time dependent changes following reduction and (c) re-oxidation at 0.4 V. The last spectrum of each group is highlighted.
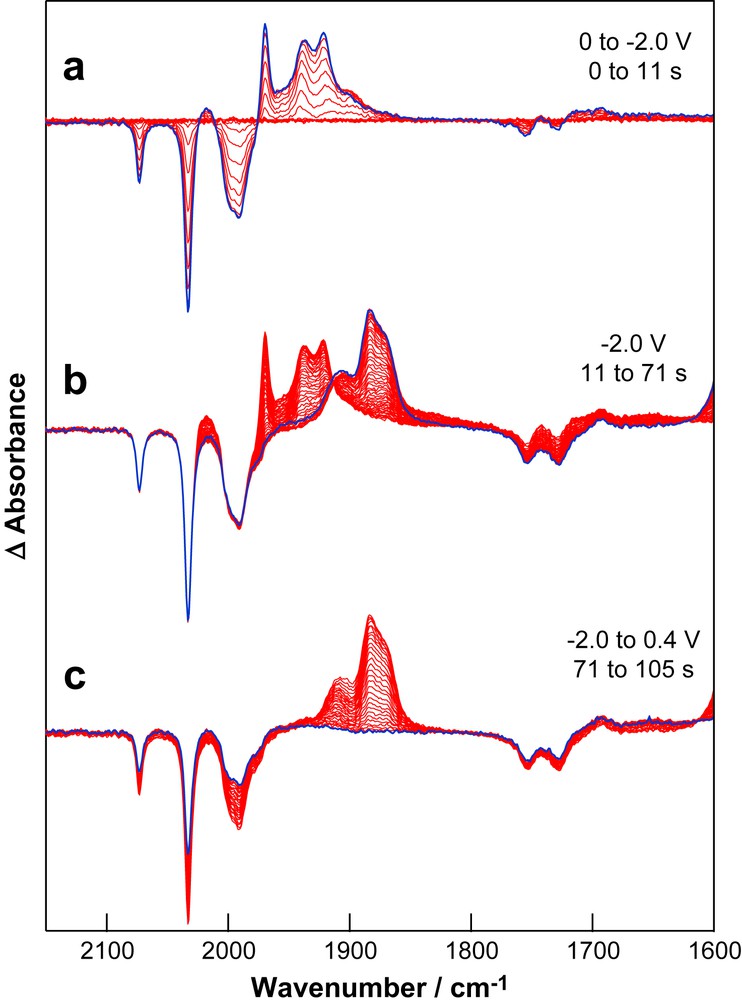
Differential absorption IR-SEC spectra of the solution of 3S used for Fig. 5 during (a) reduction (process II), (b) time dependent changes following reduction and (c) re-oxidation at 0.4 V. The last spectrum of each group is highlighted.
Experiments conducted with an applied potential sufficient to access process II give spectral changes corresponding to the reduction of 3S to give 3SB as the initial product (Fig. 6a). Following exhaustive depletion of the neutral compound, 3SB is replaced by more highly reduced products with bands at 1907, 1883, 1871 cm−1 in the ν(CO) region together with bands at 1590 and 1373 cm−1 (Fig. 6b). Through this period there is continued depletion of HOAc. Application of a potential of +0.5 V is accompanied by the loss of the growth features together with recovery of ca. 40% of the neutral compound and partial recovery of HOAc (Fig. 6c). The generation of dihydrogen during the reduction process is implied by the relative magnitudes of the depletion bands due to 3S and HOAc, together with the diminished extent of recovery of HOAc during re-oxidation. While it is not possible at this stage to comment on the form of the highly reduced species the low wavenumber features are consistent with the protonation of highly reduced metal carbonyl species to give formyl adducts. While these experiments do not allow identification of the catalytic species, it is clear, however, that reduction beyond 3SB is required in order to achieve proton reduction.
3 Conclusion
Consideration of the electrochemical response of bis(thiolate) and dithiolate-bridged diiron carbonyl compounds casts grave doubt on the simple EECC′ electrocatalytic path most commonly quoted for reactions where weak acids are involved. The rapid scan voltammetry reported by Capon et al. [27] would suggest that had such a scheme applied then the purportedly active two-electron reduced species would be formed at potentials more positive than process II. This conclusion is further supported by analysis of the electrocatalytic reduction of HOAc by BDT [10], a complex exhibiting a reversible two-electron couple [8,9]. The IR-SEC results reported herein confirm that reduction of the neutral bis thiolate (SEt) and dithiolate (2S and 3S) bridged diiron(I) carbonyl compounds at mild potentials (process I) generates species analogous to 3SB (Scheme 1) and the course of this reaction is not altered by the presence of HOAc. Process II does not correspond to the Complex1−/Complex2− couple but is associated with reduction of species analogous to 3SB. In the absence of additional CO the stoichiometry of the reaction demands formation of additional iron-based products and these also may contribute to the electrocatalytic response. It is important to note that while the catalysts are interesting in their own right, the structure of the electrocatalytic species is more likely to be related to 3SB than 3S and it is therefore highly unlikely that this chemistry is related to that of the H-cluster. If the purpose of the study of the electrocatalytic reactions of H-cluster model compounds is to cast light on the reaction chemistry of the biological system then reactions involving weak acids are at best of dubious value since there is no basis for assignment of the structure of the electrocatalytic species. Paradoxically, the reaction is less complicated when strong acids are used since in this case protonation of the one-electron reduced product engages a reaction path which, according to DFT calculations [5], leads to dihydrogen elimination from dithiolate (or bis thiolate) bridged diiron compounds.
4 Experimental
Unless otherwise stated chemicals (Aldrich) and high-purity gases (BOC) were obtained from commercial sources and used without further purification. The diiron hexacarbonyl compounds Fe2(μ-S(CH2)2S)(CO)6 2S, Fe2(μ-S(CH2)3S)(CO)6 3S and Fe2(μ-SEt)2(CO)6 SEt, were prepared by literature methods [31], and confirmed to be pure by spectroscopic and electrochemical analyses. All manipulations of solutions used in SEC measurements were carried out under a dinitrogen or argon atmosphere using standard Schlenk techniques or through the use of a VAC glovebox. Tetrahydrofuran (HPLC grade) was filtered through activated neutral aluminium oxide (Brockmann I, 150 mesh, Aldrich) to remove peroxides and pre-dry, then distilled from sodium benzophenone ketyl [32]. Acetonitrile was distilled over calcium hydride. Tetra-n-butylammonium perchlorate (TBA[ClO4]) and tetra-n-butylammonium hexafluorophosphate (TBA[PF6]), used as supporting electrolytes, were prepared and purified using standard procedures [33]. Caution! Perchlorate salts are potentially explosive. Solutions containing TBA[ClO4] as a supporting electrolyte should not be allowed to evaporate to dryness.
Cyclic voltammetry experiments were controlled using an Autolab PGSTAT30 potentiostat with GPES software and were carried out in a one-compartment glass cell using a 1 mm diameter vitreous carbon working, platinum counter, and jacketed silver wire reference electrodes. Spectroelectrochemical (SEC) experiments were conducted using a purpose built cell previously described [34], and employed a 3 mm diameter vitreous carbon working electrode, silver pseudoreference electrode, and platinum foil counter electrode. The potential of the reference electrode was estimated from the voltammetry of the solution under investigation and, by comparison with experiments conducted in the presence of ferrocene (Fc), all potentials are quoted relative to the Ag/AgCl reference electrode. Against this reference the Fc+/Fc couple occurs at +0.51 V in CH2Cl2 [35,36]. The applied potential was controlled using a PAR model 362 potentiostat. A Powerlab 4/20 interface (ADInstruments) using EChem V1.5.2 or Chart V4.12 provided a means of setting the applied potential and monitoring the potential and current responses during SEC experiments. Digital simulation of the voltammetry was conducted using the program DigiElch (version 4, DigiSoft) [37–39].
IR spectra were obtained using a Bio-Rad FT175C FTIR equipped with a Ge/KBr beamsplitter and narrow band MCT detector. Spectral subtraction and curve fitting were performed using Grams/32 AI software (Galactic).
Acknowledgment
SPB thanks the University of Melbourne and the University of East Anglia for the award and hosting of study leave during which the manuscript was prepared. The ARC (SPB), the BBSRC and EPSRC/Supergen V (CJP) are thanked for research funding and for the award of a postgraduate studentship to SJB.