

1 Introduction
The availability of pure enantiomers is of primary importance in the pharmaceutical, agronomical, flavour and fragrance industries. The main advances in the development of enantiomerically pure compounds have been accomplished in the last years with the prosperity of preparative enantioselective separation methods [1] and enantioselective catalytic synthesis [2–4]. Among the various ways to produce enantiopure compounds, enantioselective homogeneous catalysis constitutes a very appealing strategy. The preparation of a good enantioselective catalyst implies the synthesis of an appropriate chiral ligand which is one of the most crucial steps, the structure of the ligand being often the result of the knowledge-based intuition or serendipity. The NCN pincer-ligand [2,6-(Me2NCH2)2–C6H3] and its metal complexes were first introduced by van Koten [5]. Since then, efforts have culminated in the construction of chiral C2-symmetrical pincer-ligands and complexes that offer promising utility in enantioselective catalytic synthesis [6]. Dependent on the choice of the metal and the donor atom, various reactions were catalyzed, such as aldol condensation [7], hydrogen transfer [8], allylic alkylations [9]. Although the concept of C2 symmetry has been very successful, there is no fundamental reason why C2-symmetric ligands should necessarily be superior to their non-symmetrical counterparts. For this purpose we deemed worthy to take up the preparation of an NCN chiral asymmetrical pincer-like ligand.
2 Results and discussion
The adopted strategy consisted in a three-step synthesis. As shown in Scheme 1, the preparation of the target ligand started with the methylation of the commercially available 3-aminoacetophenone by trimethylphosphate [10] (1, yield 35%).1 The 1-(3-(dimethylamino)phenyl)ethanone 2 is then enantioselectively reduced at 0 °C by the means of the chiral Ru-catalyst [11] (R,R)-TsDPEN-ruthenium p-cymene namely [((R,R)-(p-toluenesulfonyl)-1,2-diphenylethylene diamine) (η6-p-cymene)ruthenium] chloride to give the corresponding chiral alcohol in 99% enantiomeric excess (3, yield 98%).2 Next step consisted in the displacement of the hydroxyl group by nitrogen functionality.3 This reaction was achieved under Mitsunobu reaction conditions, hydrazoic acid being reacted with the chiral alcohol (4, yield 92%) [12] affording the azide compound.
Thereafter the resulting azide was hydrogenated into the respective amine in the presence of catalytic amounts of Pd/C in methanol (5, yield 98%).4 Pincer-ligand may be further methylated by formaldehyde, in the presence of sodium cyanoborohydride and zinc chloride5 (6, yield 95%) [13].
In general preparative methods for main-group organometallics are applicable to transition metals [14]. Metathesis (a reaction between an organolithium or magnesium reagent and a metal halide) and ortho-metallation have been widely used in our laboratory. Cycloruthenation of benzyl primary, secondary and tertiary amines [15] led to the preparation of enantioselective hydrogen transfer catalysts [16]. Ruthenation of our primary amine NCN ligand resulted in the formation of a diastereomeric mixture. For the major and minor isomers, cycloruthenation is indicated by the presence of a shifted doublet corresponding to the proton ortho to the Ru centre δ (ppm) = 7.65 (d, 1H, 9 Hz), 7.56 (d, 1H, 9 Hz). The characteristic signals of the arene ligand are found at 5.59 and 5.55 (s, 6H, C6H6). The metallated position was deduced from the ABC pattern of the spectra. These comprise a doublet with a great coupling constant δ (ppm) = 7.65 (d, 1H, 9 Hz), 7.56 (d, 1H, 9 Hz), a doublet of doublet with a great and a little coupling constants 6.63 (dd, 1H, 3JH–H = 9 Hz, 4JH–H = 3 Hz), 6.58 (dd, 1H, 3JH–H = 9 Hz, 4JH–H = 3 Hz), and a doublet with a little coupling constant 6.47 (d, 1H, 4JH–H = 3 Hz), 6.41 (d, 1H, 4JH–H = 3 Hz). As a conclusion Ru atom is found out of the pincer-ligand (7, Scheme 2). For that reason these complexes were not further characterized for the moment.6
The pincer-ligand was treated with butyl lithium to afford the lithiated ligand. To assess the lithiated position the organolithium compound was quenched with D2O. The pattern of the 1H NMR spectrum was analysed again. The spectrum displayed an ABC coupling scheme: a triplet at δ (ppm) = 7.26 (t, 1H, 3JH–H = 7.5 Hz), a doublet at 6.87 (d, 1H, 3JH–H = 7.5 Hz), and a second doublet at 6.81 (d, 1H, 3JH–H = 7.5 Hz). It was concluded that this time metallation occurred inside the pincer-ligand. The next step was a trans-auration which yielded the gold (I) aryl compound (9).7 It is to be noticed that such a complex opens the route to various transition-metal complexes [17].
3 Conclusion
A chiral asymmetrical NCN pincer-like ligand has been synthesized in three steps. The chirality has been introduced at the level of the enantioselective reduction of 1-(3-(dimethylamino)phenyl)ethanone. Cycloruthenation by C–H activation led to a diastereomeric mixture of complexes where the ruthenium atom is found out of the pincer. trans-Auration via selective lithiation of the tertiary amine was achieved and opens the route to transition-metal complexes where the metal is found inside the pincer-ligand. Further work will explore the achievement of chiral receptors [18,19] and catalysts.
Acknowledgements
The Ministère de l'Education Nationale provided financial support for PhD fellowship to PSD.
1 1-(3-(Dimethylamino)phenyl)ethanone. A solution of 0.1 mol of 1-(3-aminophenyl)ethanone and 13.6 ml (0.113 mol) of trimethylphosphate was added to a 300 ml round-bottomed flask equipped with a side arm. The stirred reaction mixture was gradually heated to approximately 150 °C over 60 min. After 2 h at reflux, the reaction mixture was cooled to room temperature. A solution of 16.65 g of sodium hydroxide in 100 ml of water was added, and the mixture was stirred vigorously for 1.5 h to hydrolyze the phosphate ester. An additional 200 ml of water was added. The product was extracted with two volumes (150 ml) of ether. The combined ether extracts were dried over a mixture of anhydrous magnesium sulphate and sodium hydroxide pellets, filtered, and distilled through a Vigreux column. The residue was further distilled at reduced pressure. 1-(3-(Dimethylamino)phenyl)ethanone was collected as a yellow oil. Yield: 35%. 1H NMR (300 MHz, CDCl3) δ (ppm) = 7.32 (m, 3H, Ar), 6.96 (m, 1H, Ar), 3.00 (br s, 6H, NMe2), 2.59 (s, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ (ppm) = 26.8, 40.5, 111.2, 117.4, 117.6, 129.2, 137.9, 150.6, 198.9.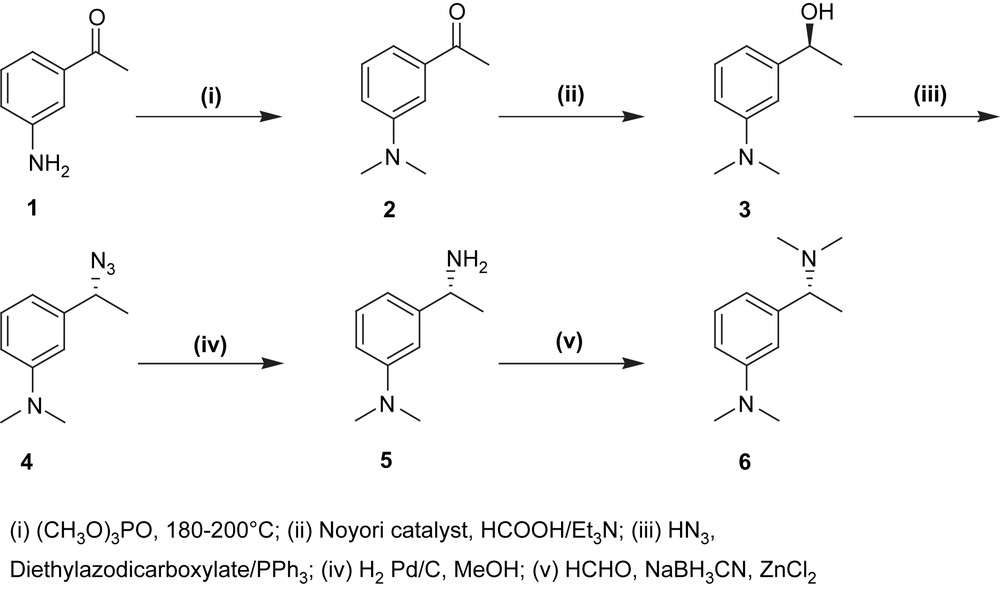
2 1-(3-(Dimethylamino)phenyl)ethanol. A mixture of 1-(3-(dimethylamino)phenyl)ethanone (14 mmol) and the Ru-catalyst (R,R)-TsDPEN-ruthenium p-cymene (46 mg, 1/200 eq.) in a 5:2 formic acid–triethylamine azeotropic mixture (7 ml) was stirred at 0 °C for 72 h. The mixture was diluted with water and then extracted with ethyl acetate. The organic solution was washed with aqueous NaHCO3 and brine. The organic layer was dried over magnesium sulphate, concentrated under reduced pressure to afford 2.56 g of violet oil. Yield: 98%. ee: 99%. 1H NMR (300 MHz, CDCl3) δ (ppm) = 7.23 (t, 1H, 3JH–H = 7.8 Hz, Ar), 6.79 (s, 1H, Ar), 6.73 (t, 1H, 3JH–H = 7.5 Hz, Ar), 6.68 (dd, 1H, 3JH–H = 8.4 Hz, 4JH–H = 2.4 Hz, Ar), 4.86 (q, 1H, 3JH–H = 6.4 Hz, CH), 2.96 (br s, 6H, NMe2), 1.87 (br s, 1H, OH), 1.50 (d, 3H, 3JH–H = 6.6 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ (ppm) = 25.1, 40.6, 70.5, 111.8, 113.9, 121.8, 129.1, 147.2, 150.6. ee has been measured by GC using a capillary column (Chiraldex β-PM, 50 m × 0.25 mm).
3 3-(1-Azidoethyl)-N,N-dimethylbenzene amine. To a stirred solution of 1-(3-(dimethylamino)phenyl)ethanol (2.56 g, 15.5 mmol), triphenylphosphine (4.87 g, 18.6 mmol, 1.2 eq.), and hydrazoic acid (11.8 ml of 1.69 M, 1.3 eq.) at 0 °C, diethyl azodicarboxylate (8.5 ml, 18.6 mmol, 1.2 eq.) was added drop wise. The mixture was stirred for 2 h at ambient temperature, diluted with water and extracted with ethyl acetate. The organic extracts were dried, evaporated to afford 2.6 g of a yellow residue. Yield: 92%. 1H NMR (300 MHz, CDCl3) δ (ppm) = 7.26 (m, 1H, Ar), 6.75 (m, 3H, Ar), 4.57 (q, 1H, 3JH–H = 6.9 Hz, CH), 2.98 (br s, 6H, NMe2), 1.53 (d, 3H, 3JH–H = 6.9 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ (ppm) = 21.4, 40.8, 61.6, 110.1, 111.1, 112.8, 129.4, 141.9, 150.2.
4 3-(1-Aminoethyl)-N, N-dimethylbenzene amine. To a solution of 3-(1-azidoethyl)-N,N-dimethylbenzene amine (2 g, 10.5 mmol) in methanol (75 ml) was added Pd/C (10%, 30 mg), and hydrogen was allowed to bubble through the solution for 24 h. The solution was filtered through Celite, concentrated under vacuum to afford yellow oil 1.74 g. Yield: 98%. Free base: 1H NMR (300 MHz, CDCl3) δ (ppm) = 7.21 (t, 1H, 3JH–H = 7.8 Hz, Ar), 6.71 (m, 2H, Ar), 6.63 (dd, 1H, 3JH–H = 8.4 Hz, 4JH–H = 2.4 Hz, Ar), 4.06 (q, 1H, 3JH–H = 6.6 Hz, CH), 2.94 (br s, 6H, NMe2), 1.61 (br s, 2H, NH2), 1.39 (d, 3H, 3JH–H = 6.6 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ (ppm) = 25.5, 40.4, 51.7, 109.9, 110.9, 113.8, 128.8, 148.9, 150.9; bis chlorhydrate: 1H NMR (300 MHz, D2O) δ (ppm) = 7.63–6.56 (m, 4H), 4.57 (q, 1H, 3JH–H = 6.8 Hz, CH), 3.24 (br s, 6H, NMe2), 1.60 (d, 3H, 3JH–H = 6.8 Hz, CH3); Anal. Calcd for C10H16N2·1.7 HCl: C, 53.09; H, 7.89; N, 12.38. Found: C, 53.05; H, 7.73; N, 12.78.
5 3-(1-(Dimethylamino)ethyl)-N,N-dimethylbenzene amine. To a stirred solution of 3-(1-aminoethyl)-N,N-dimethylbenzene amine (1.26 g, 7.68 mmol) in methanol (12 ml) containing 37% aqueous formaldehyde (2 ml, 24 mmol) at room temperature was added a solution of sodium cyanoborohydride (483 mg, 7.68 mmol) and zinc chloride (1.25 g, 9.2 mmol) in methanol (12 ml). After the reaction mixture was stirred at room temperature for 24 h, the solution was taken up in a 0.1 N NaOH and most of methanol was evaporated under reduced pressure. After the aqueous solution was extracted with dichloromethane, the combined extracts were washed with water and brine, dried over anhydrous magnesium sulphate, thereafter evaporated to dryness and filtered over alumina to afford yellow oil 1.4 g. Yield: 95%. Free base: 1H NMR (300 MHz, CDCl3) δ (ppm) = 7.18 (t, 1H, 3JH–H = 7.3 Hz, Ar), 6.62–6.69 (m, 4H, Ar), 3.16 (q, 1H, 3JH–H = 6.6 Hz, CH), 2.95 (br s, 6H, NMe2), 2.22 (br s, 6H, NMe2), 1.38 (d, 3H, 3JH–H = 6.6 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ (ppm) = 20.0, 29.7, 40.7, 42.8, 66.9, 111.8, 111.9, 116.1, 129.1, 150.7; bis chlorhydrate: 1H NMR (300 MHz, D2O) δ (ppm) = 7.65–7.58 (m, 4H, Ar), 4.50 (q, 1H, 3JH–H = 6.6 Hz, CH), 3.21 (br s, 6H, NMe2), 2.77 (br s, 3H, NMe), 2.64 (br s, 3H, NMe), 1.65 (d, 3H, 3JH–H = 6.6 Hz, CH3); 13C NMR (75 MHz, D2O) δ (ppm) = 15.1, 40.2, 40.3, 46.1, 65.3, 120.6, 121.9, 130.1, 131.6, 136.5, 143.1.
6 (RSRu) [(η6-C6H6)Ru(1,3-C6H3–NMe2–CHMeNH2)(NCCH3)] (PF6). To a suspension of [Ru(η6-C6H6)Cl2]2 (300 g, 0.6 mmol), NaOH (48 mg, 1.2 mmol) and KPF6 (442 mg, 2.4 mmol) in CH3CN (15 ml) was added the amine 5 (100 mg, 0.6 mmol) and the mixture was stirred at room temperature for 72 h. The resulting dark yellow suspension was filtered over Celite, concentrated in vacuo, and filtered over standardized Al2O3 using CH3CN as eluent. A yellow fraction was collected and concentrated in vacuo (305 mg, yield: 96%). 1H NMR (300 MHz, CD3CN) de = 7%, major δ (ppm) = 7.65 (d, 1H, 9 Hz, Ar), 6.63 (dd, 1H, 3JH–H = 9 Hz, 4JH–H = 3 Hz, Ar), 6.47 (d, 1H, 4JH–H = 3 Hz), 5.59 (s, 6H, C6H6), 5.30 (br s, 2H, NH2), 4.10 (q, 1H, 3JH–H = 6 Hz, CH), 2.86 (s, 6H, NMe2), 1.47 (d, 3H, 3JH–H = 6 Hz, CH3), minor δ (ppm) = 7.56 (d, 1H, 9 Hz, Ar), 6.58 (dd, 1H, 3JH–H = 9 Hz, 4JH–H = 3 Hz, Ar), 6.41 (d, 1H, 4JH–H = 3 Hz, Ar), 5.55 (s, 6H, C6H6), 4.45 (br s, 2H, NH2), 3.73 (q, 1H, 3JH–H = 6 Hz, CH), 2.84 (s, 6H, NMe2), 1.21 (d, 3H, 3JH–H = 6 Hz, CH3).
7 [Au 1,3-(C6H3–NMe2–CHMeNMe2(PPh3)]. To a solution of 6 (100 mg, 0.52 mmol) in 5 ml of pentane was added by syringe a 1.33 M solution of n-BuLi in hexane (0.39 ml, 0.52 mmol). After addition, stirring was continued for 16 h and completed lithiation was checked by deuteration of an aliquot of the reaction mixture. The reaction mixture was added, at −30 °C, to a suspension of [AuCl(PPh3)] (256 mg, 0.52 mmol) in 4 ml of diethyl ether and was allowed to warm to room temperature, and stirred for 6 h. The reaction mixture was quenched with water, filtered over Celite and extracted with diethyl ether. The extracts were dried over magnesium sulphate and evaporated to dryness. The residual cream-colored solid was washed with pentane affording 200 mg. Yield: 60%. Quenched organolithium compound: 1H NMR (300 MHz, D2O) δ (ppm) = 7.26 (t, 1H, 3JH–H = 7.5 Hz, Ar), 6.87 (d, 1H, 3JH–H = 7.5 Hz, Ar), 6.81 (d, 1H, 3JH–H = 7.5 Hz, Ar), 3.29 (q, 1H, 3JH–H = 6.5 Hz, CH), 2.77 (br s, 6H, NMe2), 2.05 (br s, 6H, NMe2), 1.29 (d, 3H, 3JH–H = 6.5 Hz, CH3); organogold complex: 1H NMR (300 MHz, CDCl3) δ (ppm) = 7.68–7.43 (m, 15H, PPh3), 7.08 (d, 2H, 3JH–H = 6 Hz, Ar), 6.97–6.90 (m, 1H, Ar), 4.01 (q, 1H, 3JH–H = 6 Hz, CH), 2.99 (s, 6H, NMe2), 2.23 (s, 6H, NMe2), 1.43 (d, 3H, 3JH–H = 6 Hz, CH3), 13C NMR (75 MHz, CDCl3) δ (ppm) = 22.4, 44.6, 46.7, 73.0, 128.8, 129.0, 129.1, 129.3, 130.8, 131.9, 134.0, 134.3, 134.5; 31P NMR (121 MHz, CDCl3) δ (ppm) = 45.5.