

1 Introduction
The low-molecular weight carbohydrates are convenient substrates for the stereocontrolled synthesis of natural products, owing to the fact that they are inexpensive, enantiomerically pure, and available in many structural and configurational forms [1]. Since they bear many hydroxy groups of similar reactivity, possess too many stereogenic centers and do not have double bonds, they are usually transformed into simpler compounds where the above inconveniences are minimized while the merits of monosaccharides are fully retained. The α,β-unsaturated lactones 1-6, readily available from monosaccharides, represent particularly attractive compounds for organic synthesis owing to the presence of the electrophilic en-one fragment and only one or two stereogenic centers that are well defined (Fig. 1) [2,3].

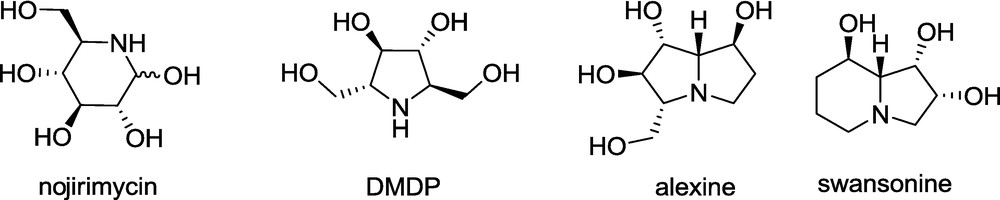
Many structurally diverse polyhydroxylated alkaloids have been isolated from natural sources such as plants or microorganisms. An apparent common structural feature of this group is a nitrogen-containing four- (azetidine), five- (pyrrolidine) or six-membered (piperidine) ring, or a related fused-ring system (pyrrolizidine, indolizidine, and quinolizidine) built up from the above monocycles, substituted with one or more hydroxyl groups (Fig. 2) [4–16]. The structural similarity of these compounds to monosaccharides, is reflected in the fact that they are also called iminosugars, and they have interesting biological activities, typically presented by the selective inhibition of glycosidases. Consequently, the iminosugars show antibacterial, antiviral, antitumor, or antidiabetic activities [4–15]. Therefore, the synthesis of iminosugars and their analogues has attracted the attention of many academic and pharmaceutical laboratories including our group [4–26].
For over 10 years our laboratory has been involved in the elaboration of the methodology of the iminosugar synthesis utilizing the unsaturated lactones 1-6 (Fig. 1) [17–26].
The present account focuses on the stereochemistry of the 1,3-dipolar cycloaddition of open-chain 7-11 [27], five-membered 10-14 [10,28a,29], and six-membered cyclic nitrones 15 [28b] and 16 [30] to both δ- (1-4) and γ-lactones (5, 6). Furthermore, we have researched the criteria for the rational selection of cycloaddition components in order to construct the desired structure and exercise full control over the configuration of the formed cycloadduct. Lastly, we wanted to provide a suitable demonstration of an application of this methodology in the synthesis of selected iminosugars. Special emphasis was placed on cycloadditions involving the five-membered ring nitrones 10-13.
Isoxazolidines, readily available from olefins and nitrones via the 1,3-dipolar cycloaddition, represent particularly attractive starting materials for the synthesis of a variety of iminosugars that exhibit biological activity [27,31,32]. The usefulness of the isoxazolidine approach to the iminosugars has been demonstrated by a large number of reported examples, since nitrones usually offer good regio- and stereoselectivity with the introduction of convenient functionalities [27,31,32]. The strategy usually involves hydrogenolysis of the N–O bond followed by the construction of the iminosugar skeleton via intramolecular alkylation or acylation of the nitrogen atom (Scheme 1) [33]. It should be noted, however, that the unsaturated chiral lactones are particularly attractive substrates in the synthesis of iminosugars since, in contrast to their acyclic congeners, they often provide a single adduct and they introduce stereogenic centers bearing the hydroxyl groups that the target molecule requires.
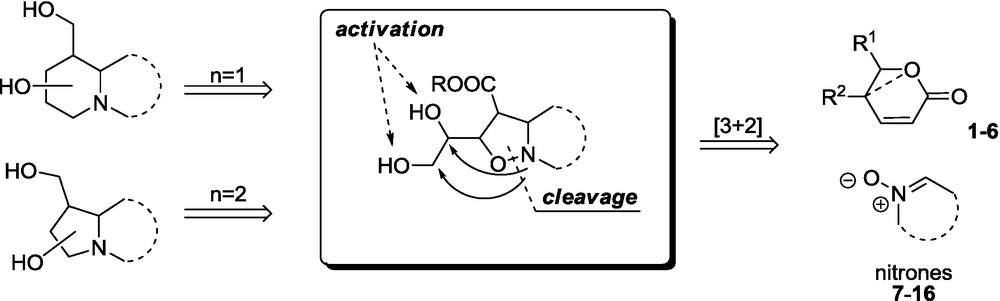
The present approach has an additional advantage, which makes it a powerful method for the iminosugars’ synthesis. As is demonstrated in Scheme 1, the intramolecular alkylation step can be easily controlled by a suitable activation of the available hydroxyl function, which eventually leads to the formation of either a pyrrolidine or piperidine ring. Depending on the type of nitrone, acyclic or cyclic, the strategy allows formation of monocyclic or bicyclic iminosugars.
2 Synthesis of enantiopure α,β-unsaturated lactones
An unsaturated δ-lactone fragment can be found in many natural products (Fig. 3), both simple such as parasorbic acid (17) [34] and osmunda lactone (18) [35], and even structurally more complex, such as anamarine (19) [36] and boronolide (20) [37]. Butenolides (unsaturated γ-lactones) [38] can also be found as fragments of many natural products (Fig. 3) such as isocladospolide (21) [39], corrosolin, (22) [40] and (−)-incrustoporin (23) [41].

In 1977, we proposed [2] a general, two-step methodology for the synthesis of δ-lactones 2-4. The method consists in the oxidation of the acetal center in 2,3-unsaturated glycosides 24 and 25 with hydrogen peroxide in the presence of molybdenum trioxide leading to the anomeric hydroperoxides 26 and 27. The subsequent elimination of water, using an acetic anhydride/pyridine mixture (Scheme 2) [2], leads to the final unsaturated lactone 2-4. Anomeric oxidation of the glycosides 24 and 25 and their other O-substituted derivatives can also be performed using hydrogen peroxide in dioxane in the presence of sulphuric acid [42,43].

The racemic compound rac-2 can be easily obtained from the Diels-Alder adduct of butyl glyoxalate and methoxy-butadiene by a reaction sequence which consists of the reduction of the ester group, protection of the hydroxyl group and anomeric oxidation as reported above [2,44].
The d-glycero-lactone 2 can be easily obtained from the d-erythro-lactone 3 by deoxygenation of the allylic position, followed by the migration of the double bond under the influence of an organic base [45]. It should be noted, as well, that the configuration of the C-5 carbon atom in lactones 3 and 4 can be reversed by a sequence of reactions which consist of opening of the lactone ring followed by the ring closure under Mitsunobu conditions [46].
In 1982, Lichtenthaler et al. [3] reported the direct oxidation of the glycols 28 with a 3-O-acyl substituent to the corresponding α,β-unsaturated δ-lactones 29 using anhydrous m-chloroperbenzoic acid in the presence of BF3/etherate. It has been shown that the oxidation proceeds via anomeric m-chloroperbenzoate esters. Lichtentaler's method [3] appears to have a general value and can be applied to the 2-substituted glycals as well. The one-step oxidation of glycols [3] seems to be superior to the stepwise transformation proposed by us [2]. In the case of lactones bearing different substituents at the O-4 and O-6 oxygen atoms, however, the hydrogen peroxide method, which uses inexpensive reagents, offers certain advantages over the commonly used Lichtentaler's [3] procedure. Numerous other methods of transformation of simple carbohydrates into the δ-enlactones [47], or formation of such lactones from a non-carbohydrate precursor are known [48].
The unsubstituted butenolide (5) and (S)-5-hydroxymethyl-(5H)-furan-2-one (6) and its O-substituted derivatives represent also valuable chiral starting materials for the synthesis of a variety of natural products [39,49]. Both compounds 5 and 6 are commercially available and can be easily obtained by several methods [50–52].
3 1,3-Dipolar cycloaddition of open-chain nitrones to δ and γ-lactones
Earlier we have shown [53] that the cycloaddition of nitrones derived from formaldehyde (7) or acetaldehyde (8) to the sugar enlactones 2 and 4 proceeded with a high regio- and stereoselectivity to yield exclusively anti, or endo-anti adducts, respectively (Scheme 3) [53]. Such high stereoselectivity is noteworthy, particularly since the less reactive nitrones with aromatic substituents when reacted with the same dipolarophiles gave mixtures of exo and endo adducts with significant preference for the exo one. The exo preference can be attributed to the interaction of the aromatic substituent of the nitrone with an electrophilic ester carbonyl group. The high preference of the anti addition to the terminal substituent of δ-lactone is a result of the axial approach phenomenon [54] of the nitrone oxygen atom to the conformationally stabilized dihydropyranone ring. This stereochemical pathway of cycloaddition is probably a consequence of the more advanced formation of the C–O bond of the cycloadduct than the C–C one [53]. In the case of γ-lactones, the anti preference can be explained by steric interaction of the terminal substituent of the lactone with the entering nucleophile, while the exo-/endo- selectivity is controlled by the same factors as for δ-lactones (Scheme 3) [55].

4 1,3-Dipolar cycloaddition reactions of cyclic nitrones to unsaturated lactones
To expand our studies on 1,3-dipolar cycloadditions involving unsaturated lactones, we directed our attention towards the use of cyclic nitrones 10–16 in these reactions [56–62]. The attractiveness of cyclic dipoles as synthons in a target-oriented synthesis has been proven repeatedly [10,27,31,63]. We expected that the combination of both cyclic components should lead to a high stereoselectivity of the cycloaddition and, as depicted in Scheme 1, should also provide an attractive entry to the stereocontrolled synthesis of polyhydroxylated azabicycloalkanes with a potential bioactivity [17–26].
4.1 Cycloaddition involving δ-lactones
The cycloaddition reactions between chiral five-membered nitrones and γ- and δ-lactones provides an interesting example of single or double asymmetric induction, where the chirality elements of each reactant may influence stereoselectivity either in concert or in opposition. To reach the consistent picture of these reactions, we decided to include both the non-chiral nitrone 10, chiral 11-13, and racemic rac-14, as well as non-chiral lactones 1 and 5, racemic d,l-glycero rac-2, d-glycero 2, d-erythro 3, and d-threo 4 lactones [56–58].
Due to the significant steric hindrance of the 3-t-butoxy substituent, the lactone 1 reacts with nitrone 11 to give almost the pure adduct 35 in a high yield as a result of the exo approach of the dipole to the re-re side of the dipolarophile (Scheme 4) [57]. A similar high stereoselectivity was observed for the cycloaddition of lactone 1 and nitrone 12 to afford 37 (Scheme 5). In both cases, the lactone molecule approaches the nitrone anti to the 3-t-butoxy group [58].
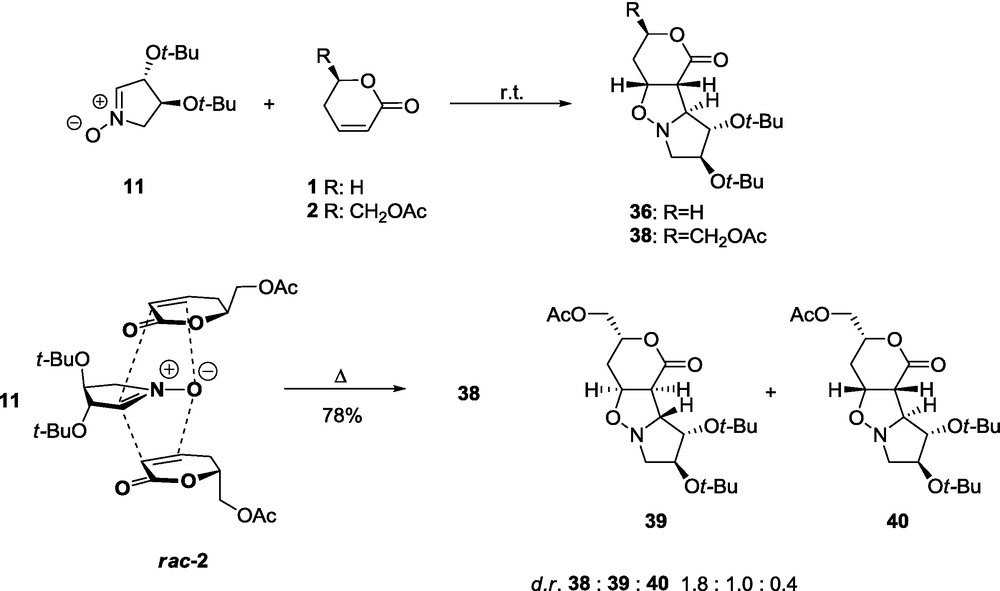
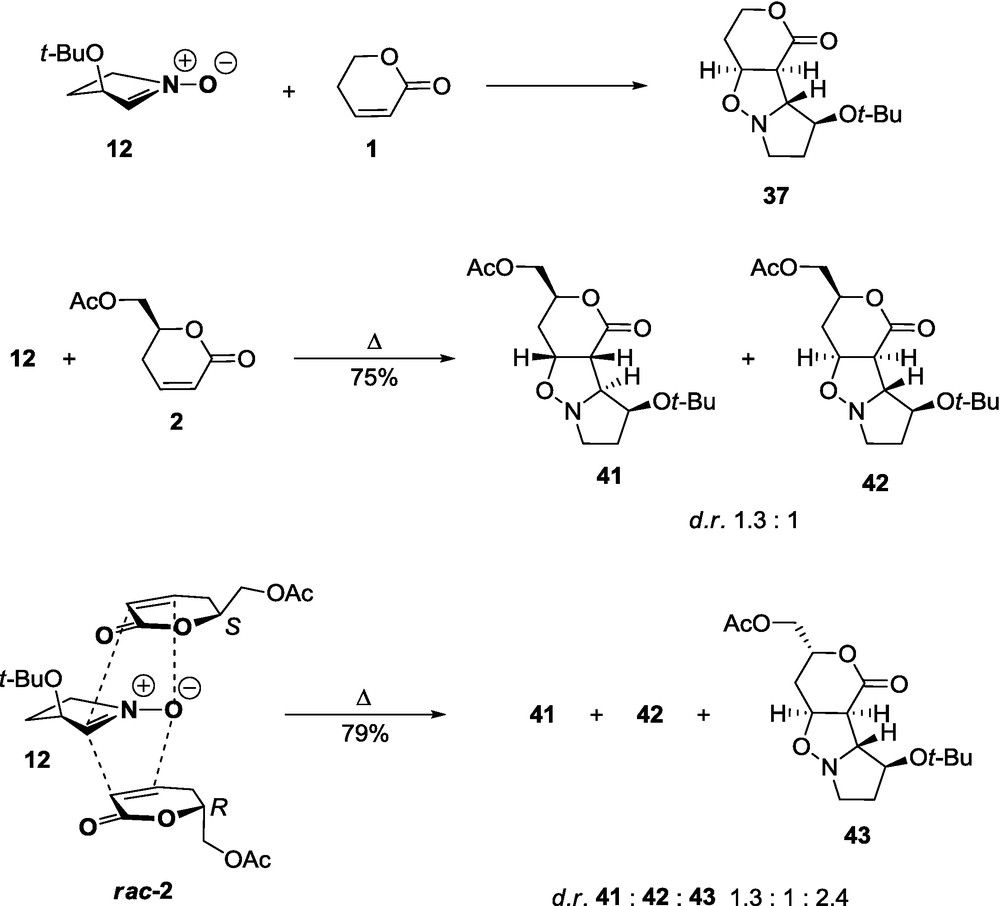
Introduction of chirality elements to both reactants influences the stereoselectivity, depending upon whether they constitute a matched or a mismatched pair. The nitrone 11 with d-glycero lactone 2, a matched pair, at room temperature produces a single product 38. On the other hand, the nitrone 11 with the racemate rac-2, under the reflux leads to a mixture of three adducts 38, 39, and 40. The last two of which are obtained from l-glycero lactone 2ent. The stereochemical relation of 11 and rac-2 is illustrated by a significant kinetic resolution of the racemate in the reaction of two molar equivalents of the lactone rac-2 with the nitrones 11 and 12 at room temperature. Since the l-glycero lactone 2ent (Scheme 4) is less reactive, it can be isolated with 77.4% e.e. (86%) [57]. An analogous situation is observed in the case of the reaction of 12 with d-glycero 2 and the D,l-glycero lactones rac-2. Reactants 12 and 2, the mismatched pair, under reflux afford two adducts 41 and 42 in a ratio of about 1.3:1. Reaction between 12 and the racemic lactone rac-2 under reflux gives three products 41, 42, and 43 in a ratio of about 1.3:1:2.4. At room temperature, a significant kinetic resolution is observed. The d-glycero lactone 2 (Scheme 5) is less reactive and could be isolated unreacted in a 93% yield (e.e. 81%) [58]. In the case of cycloaddition involving 11, the t-butoxy substituent at C-4 of the nitrone, which introduces a secondary effect in the matched pair 11 and 2, explains the lower kinetic resolution of the starting enantiomers rac-2. The same secondary effect of the 4-t-butoxy group in the nitrone is quite evident if one compares the proportion of diastereomers in the mismatched pairs 41:42 = 1:1.3 and 39:40 = 2.5:1, obtained in the case of the mismatched pairs 12:2 and 11:2ent [58]. The optically pure lactones 2 or 2ent can be obtained if the corresponding discriminating nitrones 11 and 12 are used in excess (at least 20%).
Due to the single asymmetric induction, the cycloaddition of the non-chiral nitrone 10 to d-glycero 2, d-erythro 3, and d-threo 4 lactones provides the basic information regarding the influence of lactone substituents on the direction of asymmetric induction. The cycloaddition of the nitrone 10 to lactones 2 and 4 yields, in each case, a single product, 44 or 45, respectively. The reaction of nitrone 10 with lactone 3 gives cycloadducts 46 and 47 in a ratio of 4.2:1, as a result of the exo approach of the dipole to both sides of the dipolarophile (Scheme 6) [56–58].

The high preference for the anti addition of the nitrone 10 to the terminal acetoxymethyl group in the lactones 2-4 is in accordance with our previous observations [56]. This preference can be explained assuming the axial approach of the nitrone oxygen atom and, as a consequence, the formation of the C–O bond prior to the formation of the C–C bond (Scheme 5). The terminal acetoxymethyl substituent serves as an anchor to stabilize the oH5 conformation [64,65]. The lower stereoselectivity, observed in the case of the addition of 10 to the d-erythro lactone 3, gives an indication of the importance of the steric requirements of the 4-O-acetyl substituent in the lactone (Scheme 6) [56].
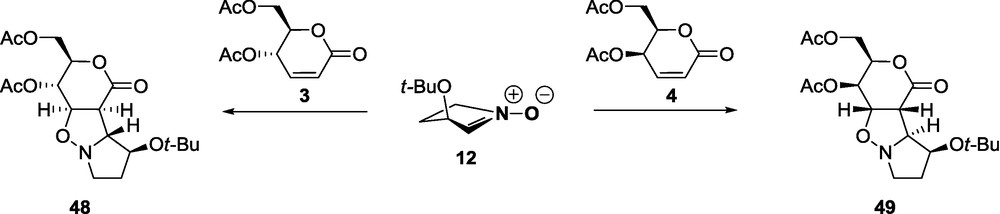
The significant steric hindrance of the 3-t-butoxy substituent in nitrones 11 and 12 and the terminal acetoxymethyl group in lactones 2 and 4 is demonstrated in the cases of a single asymmetric induction (1 with 11, or 12, and 10 with 2, or 4) and the cases of double asymmetric induction where the reactants form matched pairs (2 and 4 with 11, and 2ent with 12) as the exclusive formation of a single cycloadduct. Introduction of additional chirality elements to both reactants influences the stereoselectivity depending on whether the reactants are a matched or a mismatched pair. In the case of a matched pair, the influence of 4-t-butoxyl in the nitrone or 4-acetoxy in the lactone does not significantly change the stereoselectivity. In the case of a mismatched pair, however, the introduction of an additional acetoxy substituent at position 4 of the lactone shifts the direction of the cycloaddition depending on the configuration at C-4 of the lactone. Indeed, the addition of nitrone 12 to d-erythro lactone 3 proceeds exclusively syn to the acetoxymethyl group to form adduct 48, whereas the addition of nitrone 12 to d-threo lactone 4 proceeds exclusively anti to that group and affords 49 [56] as a sole product (Scheme 7). These results demonstrate that the direction of asymmetric induction and consequently the configuration of newly formed stereogenic centers, could be controlled by the proper selection of substrates [56–58].
It has been shown that the racemic lactone rac-2 can be separated into enantiomers using the nitrones 11 [56] and 12 [57]. This prompted us to investigate the opposite case when an enantiomerically pure lactone is applied to the resolution of a racemic nitrone. As a model, racemic nitrone 14, readily available from d-arabinose [6,10j], has been chosen [60]. Its enantiomerically pure form can also be obtained; however, its preparation is not trivial, since the synthesis requires an intramolecular alkylation of the corresponding oxime [10l–o,29].
As expected, the nitrone 14 forms a matched pair with lactone 4, whereas its enantiomer 14ent forms a mismatched pair. The cycloaddition of the lactone 4 to 2 equiv. of the racemate rac-14 at room temperature after 6 days results in an apparent kinetic resolution to provide exclusively the adduct 50 in a 82% yield and the unreacted nitrone 14ent (53% yield) with a 64% e.e. (Scheme 8). As shown before, the optically pure nitrone 14ent can be obtained if an excess of lactone 4 is used. The mismatched pair involving 14ent and 4 could react via both the exo and the endo modes. In boiling toluene the lactone 4 with 1 equiv. of enantiomerically pure nitrone 14ent showed the formation of two exo products, adducts 53 and 54, in the ratio of about 2:1, in a 74% yield. Similarly to the previously investigated reactions involving six-membered lactones, the endo product is not formed [60].

4.2 Cycloaddition reactions involving γ-lactones
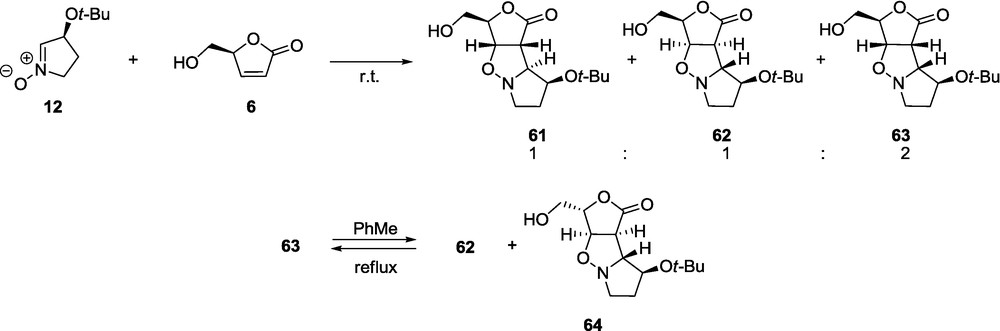
The 1,3-dipolar cycloaddition of nitrones 11-13 and their enantiomeric forms 11ent-13ent, as well as the racemic 14 to the α,β-unsaturated γ-lactones, such as non-chiral 5 and the d-glycero 6 proceeds more readily than reported for δ-lactones to provide kinetically controlled products [59,60]. Upon heating and with an extension of the reaction time, however, the reversibility of the cycloaddition can be observed and the presence of more thermodynamically stable products can be detected [60]. Moreover, in the case of lactone 6, a partial racemization may occur and, consequently, adducts derived from 6ent are formed. Contrary to the corresponding additions involving δ-lactones [56–58], where only the exo approach of reactants is observed, γ-lactones can add nitrone in both the exo and the endo modes. The high preference of anti addition to the terminal hydroxymethyl group in lactone 6 and to the 3-t-butoxy group of the nitrone is observed; the 4-t-butoxy substituent plays a secondary role. The endo addition of the reactants is energetically more demanding than the exo addition and occurs if none of the substituents in lactone or nitrone hinders such an approach. Due to the presence of complex steric interactions, a single product is formed in two cases only, 11/6 and 12ent/6 [59,60].
The single asymmetric induction in cycloaddition of the chiral nitrone 12 to the non-chiral lactone 5 has been reported by Brandi's group in a review article [66]. The reinvestigation of this reaction under our standard conditions in toluene, at room temperature showed the formation of two cycloadducts exo-anti 56 and endo-anti 57 in a ratio of about 3:1, respectively. The same reaction in boiling toluene revealed the presence of the exo-syn adduct 58 after 48 h. Further extension of the reaction time led to the reduction of the concentration of the endo compound 57. After 5 days the proportion of 56:57:58 was found to be 6:1:6 whereas after 7 days it was 9:1:14 (Scheme 9). At the same time, the yield of the reaction decreased to 65 and 56%, respectively. It shows that the adduct exo-syn 58, which was not observed under the kinetic control of the reaction, is thermodynamically the most stable one [59,60].

Based on common knowledge, the higher stability of 58 compared to 56 and 57 is unexpected since the formation of adducts syn to the t-BuO group should certainly be less favorable and indeed the calculated activation energy of the transition states supports this conclusion [61,62]. However, the detailed investigation based on NMR and X-ray analysis supported by computational methods revealed that the adduct 58 has an additional stabilizing effect which decreases its energy. The second order perturbation theory analysis of the Fock matrix in a NBO basis [62] shows the stabilizing effect between the lone pair of the oxygen atom of the 3-t-butoxy group and the relatively acidic proton H4a located in the α-position to the electron-withdrawing group (nO→σ*C−H, 0.6 kcal/mol, Fig. 4). Such an extra stabilization effect could account for the experimental observation. The interaction between H4a and the oxygen atom of the t-BuO group is also observed in the NMR spectra. The signal of the H4a proton is located downfield for 58 in comparison to its position for both 56 and 57 [62].

Extra stabilization interaction in the adduct 58: A) Calculated lowest energy conformation; B) Solid state conformation by X-ray.
Introduction of two anti located t-butoxy substituents to the nitrone molecule (11) eliminates the possibility of endo additions to 5 due to a steric interaction of one or the other t-butoxy substituent with the lactone ring. Consequently, two exo adducts 59 and 60 are obtained in a ratio of about 93:7, respectively (Scheme 10). The high preference for the anti addition of the lactone 5 to the 3-t-butoxy group of the nitrone 11 is similar to that observed for the addition to six-membered lactones [59,60].
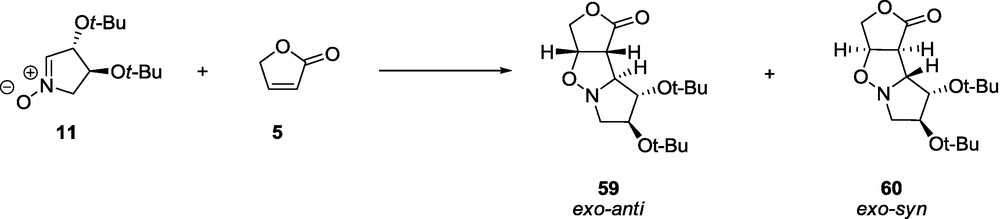
The mismatched pair, the nitrone 12 and the lactone 6 give three cycloadducts: exo-syn 61, exo-anti 62, and endo-anti 63 with respect to the 3-t-butoxy group of the nitrone in a ratio of about 1:1:2, respectively (Scheme 11). The main product of the cycloaddition between 12 and 6 (endo-adduct 63) when refluxed in a toluene solution for 24 h demonstrated the reversibility of the reaction (Scheme 11). The postreaction mixture shows the unreacted endo adduct 63 and two new products: an exo-anti adduct 62 and a cycloadduct 64 in a ratio of about 8:1:3, respectively. We found that further heating leads to a significant decomposition of all of the products. Addition of triethylamine accelerates the racemization of lactone 6 and leads (after 8 days of reflux) to the formation of a mixture of 64 (derived from the matched pair, 12 and 6ent) and 62 in a ratio of about 2.7:1, respectively, in a low yield [59,60].
Cycloadditions involving γ-lactones [59,60] proceed with a lower diastereoselectivity, as compared with the cycloaddition to the six-membered congeners [56–58]. A single adduct is formed only in the case of the matching pair when the endo approach of the reactants is hindered. Cycloadducts 65 and 66, readily available from the d-glycero lactone 6 and nitrones of l-(+)-tartaric acid 11 and d-(+)-malic acid 11ent respectively, are particularly attractive. The adduct 65, easily accessible from relatively inexpensive substrates, is a convenient starting material for the synthesis of polyhydroxy pyrrolizidines or indolizidines. The key stereogenic centers (bridgehead carbon atoms) are constructed during the cycloaddition step. The other stereogenic centers can be easily epimerized or removed (Fig. 5).

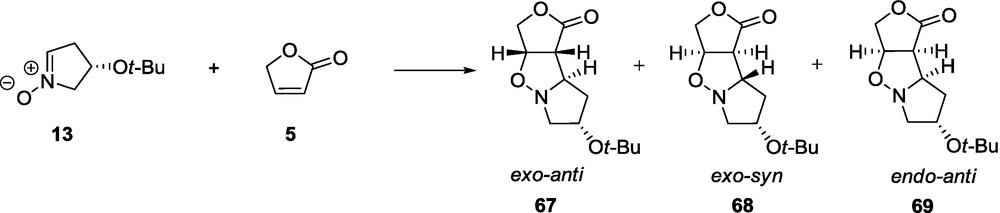
The contribution of the nitrone 4-t-butoxy substituent to the direction and magnitude of the asymmetric induction is demonstrated by the diastereoselectivity of the cycloaddition of the nitrone 13 to the lactones 5, and 6. The shift of the t-butoxy substituent in the nitrone from the position 3 to position 4 (compound 13) changes the preference of the reactants approach. In the reaction with 5, three products exo-anti 67, exo-syn 68, and endo-anti 69 are obtained in a ratio of 35:53:12, respectively (Scheme 12) [59].
The unexpected preference for the exo-syn adduct 68 could be explained, based on the assumption that the conformation of nitrone 13 in the transition state of cycloaddition resembles the ground-state conformation of 13 which should be described as an envelope 4E with the nitrone part located in the flat part of the ring. Such geometry is reflected in the 1H NMR spectrum of 13 (Scheme 13) and to a certain extent, in the crystalline state of 68 [59].
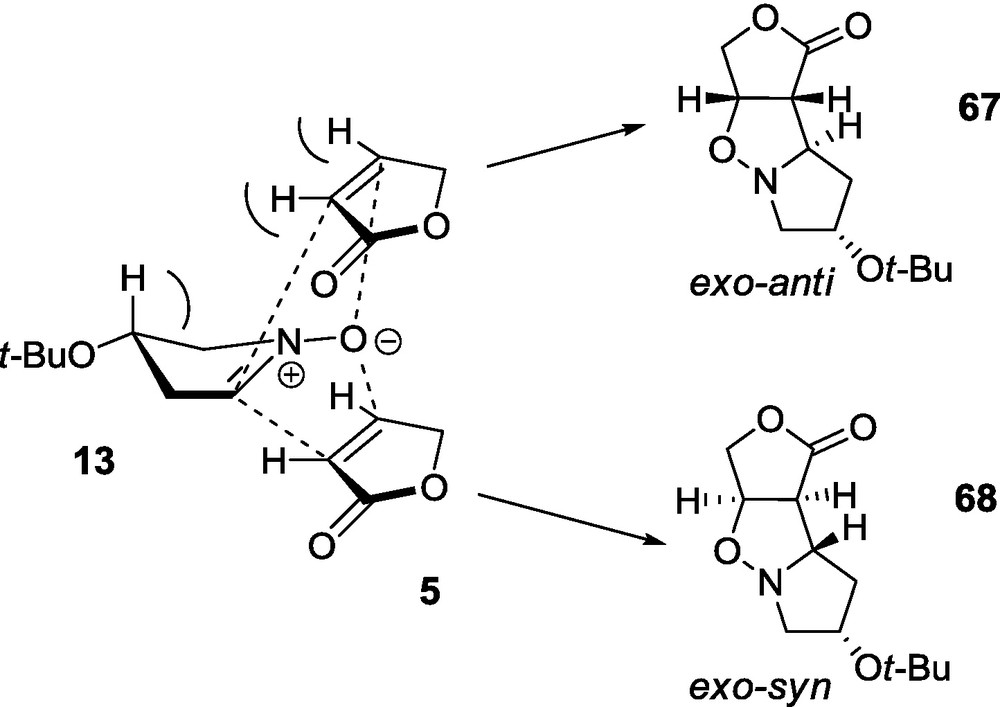
As expected, the cycloaddition of nitrone 13 to the lactone 6 affords two adducts, exo-syn 70 and exo-anti 71 in a ratio of about 21:79, respectively (Scheme 14). In comparison to the cycloaddition of 13 and 5, the presence of the additional hydroxymethyl group in the lactone molecule causes the disappearance of the endo adduct and the exo-anti adduct 71 becomes the main product. At the same time, the removal of the 3-t-butoxy substituent from the nitrone, in addition to the disappearance of the endo addition, causes a significant increase of the exo-anti addition (71) [59].
The cycloaddition of the nitrone 13ent to 6 leads to the formation of the exo-syn product 72 as the main component of the postreaction mixture (73%). Since the steric interactions that hinder the endo approach are removed, compound 72 is accompanied by the endo-anti adduct 73 (27%). The hydroxymethyl group in the lactone influences the direction of the asymmetric induction to a higher degree than the 4-t-butoxy substituent in the nitrone (Scheme 15) [59].

Bearing in mind the above results of the cycloaddition of nitrones 10-13 to the γ-lactones 5 and 6, one could expect that the cycloaddition of lactone 6 to the racemic nitrone rac-14 under kinetic control should lead to the interesting kinetic resolution providing two adducts: the exo adduct with 14 and the endo one with its enantiomeric form 14ent [60]. This reaction has been carried out using the TBDPS protected lactone 74 and rac-14 to provide at room temperature a mixture of products 75, 76, and 77 in a ratio of about 71:23:6, respectively, in 68% overall yield (Fig. 6). For the matched pair (74 and 14) only adduct 75 is formed. For the mismatched pair, 6 and 14ent, however, the endo adduct 76 is accompanied by the exo one 77 in a ratio of about 4:1, respectively. Under reflux for 7 days, the content of the endo adduct 76 raises significantly and only traces of compound 77 are detected. The yield of the reaction, however, drops to 49%. The extension of the reaction time leads to progressive diminishing of the yield while a concurrent racemization of γ-lactones additionally complicates the composition of the postreaction mixtures providing partially racemic products. For this reason, the γ-lactones are not effective for the enantiomer discrimination of racemic nitrones [60].

4.3 Cycloaddition reactions involving cyclic six-membered nitrones
Finally, to provide a better view of the stereochemistry of the cycloaddition of unsaturated lactones to cyclic nitrones, we extended our studies to investigate cases where the cyclic six-membered nitrones 15 and 16 were applied. The reaction of 15 with achiral lactone 1 has been conducted by Cid and de March [67] to show the exclusive formation of exo adduct 78 (Scheme 16).

In our studies, we subjected the nitrone 15 to the 1,3-dipolar cycloaddition reaction with lactones 2-4 [22]. The reaction between 15 and 2 provides a single adduct 79 as a result of the exo-anti approach of reactants [22]. The 1H NMR spectrum of 79 at room temperature displays lines characteristic of definite single nitrogen invertomers 79a and 79b in a ratio of about 2:1. In toluene-d8 at 100 °C, owing to the fast inversion process the 1H NMR spectrum of adduct 79 shows the presence of averaged signals only, but with substantial line broadening [22]. The same phenomena have been observed by Cid and de March for compound 78 [67].
A single product was also isolated from the reaction of nitrone 15 with the d-galacto lactone 4. As observed for the other adducts, compound 80, at ambient temperature, displays the presence of two invertomers 80a/80b in a ratio of about 1.25:1 [22].
As could be expected, a lower diastereoselectivity is observed for the reaction of nitrone 15 with the lactone 3 due to the reverse effect of both substituents in the dipolarophile on the asymmetric induction. In this case, two adducts are obtained (81 and 82) in the ratio of about 3:2; the exo-anti approach to the terminal acetoxymethyl group dominates. As in previous cases, both adducts exist as two nitrogen invertomers [22].
An additional substituent in the nitrone ring (compound 16) decreases the conformational lability of the six-membered ring and therefore all of the adducts do not show the presence of definite invertomers [68].
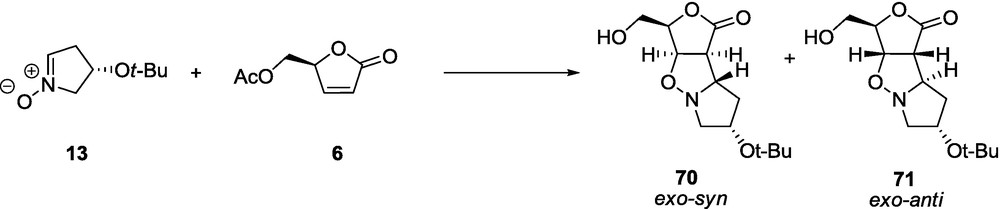
The reaction of nitrone 16 with butenolide 5 affords a mixture of two exo products 83 and 84 in a ratio 92:8 (Scheme 17); the anti-approach product, with respect to the BnO group, dominates [68].
As was already demonstrated [59,60], the cycloaddition reactions between γ-lactones and five-membered nitrones are reversible and their stereochemical outcome depends on the reaction conditions. Such behavior has never been observed for reactions involving δ-lactones regardless of the nitrones’ type [56–58]. A reversibility of the cycloaddition has been, however, observed for the 1,3-DC of γ-lactones (5,6) with the six-membered nitrone 16 [68]. For example, the exo-anti adduct 83 under reflux in toluene solution gives a mixture of both compounds 83 and 84 in a ratio 2:1 after 4 days. This phenomenon can be explained by a similar interaction that was found for the adduct 58 [68], albeit, for 84 an extra stabilization is weaker due to a larger conformational lability of the six-membered ring.
These [68] as well as previous studies [59–62], lend us to conclude that the reversibility of cycloaddition is determined by the ring size of the lactone component. It seems to be reasonable to conclude that this phenomenon is related to the reactivity of both types of lactones but it is the domain of the more reactive five-membered ones. Obviously, it is not out of the question that the cycloadditions involving δ-lactones are also reversible but an equilibrium state has not been experimentally recorded. Such a possibility, however, cannot be categorically excluded.
Replacement of the achiral lactone 5 by its chiral analogue 6 does not change significantly the outcome of the reaction and again two products 85 and 86 are obtained (Scheme 17) [68]. As in the case of 5 and 16, the major product is assigned to be an exo-anti adduct 85 and the minor one an exo-syn 86; formation of a endo adduct is not observed.
4.4 1,3-Dipolar cycloaddition of five-membered nitrones to δ- and γ-lactones: theoretical studies
As was highlighted in section 4.1, the reactions involving six-membered lactones proceed with a high diastereoselectivity which is manifested in the exclusive formation of exo-adducts [56–58]. In contrary, five-membered ones react with a lower diastereoselectivity affording a mixture of products; in this case we have observed the formation of endo adducts, as well [59,60]. Similar observations have been noticed by Cid et al. [67]. Moreover, it was found that the stereochemical pathway of cycloadditions involving γ-lactones is more complicated due to the reversibility of the reaction, which has never been observed for the δ-lactones.
Observed differences in reactivity and stereocontrol of both types of lactones encouraged us to perform a deeper analysis of the studied reactions from the theoretical point of view, applying computational methods.
As the simplest models, the reactions displayed in Scheme 18 were chosen [61,62,67]. Calculations carried out using DFT methods at the B3LYP/6-31 + G(d) level theory show that the overlap of the oxygen atom orbital of the nitrone with the C-β orbital of the lactone and the carbon atom orbital of the nitrone and the C-α of the lactone should be preferred for both five- and six-membered ring lactones [61]. These theoretical predictions are in agreement with the experimental observation; the formation of only one type of regioisomer is observed for the reactions shown in Scheme 18.

As shown in Fig. 7, the low energy difference of ∼0.4 kcal/mol between the exo and endo adducts for δ-lactones might suggest that under thermodynamic control the formation of an equimolar mixture of both adducts should be taken into consideration. This is, however, not the case since the significant energy difference between the transition states (endo versus exo 3.0 kcal/mol) causes the reaction to be kinetically controlled providing exo adducts only. The reversibility of the reaction has never been observed experimentally. In the case of γ-lactones the situation is the opposite, the relatively small energy difference between the transition states (endo versus exo 0.7 kcal/mol) compared to a 2.0 kcal/mol energy difference between the endo and exo adducts causes the reaction to be under thermodynamic control and an asymmetric transformation might occur and a more stable adduct should be formed [61]. This effect can be experimentally observed since upon extension of the reaction time under reflux, the reversibility of the cycloaddition is observed [60]. The low stability of the nitrone causes, however, a dramatic decrease of the reaction yield, practically excluding such an asymmetric transformation. Hence, the progress of the reaction is usually stopped after the disappearance of substrates leading to the mixture of adducts.
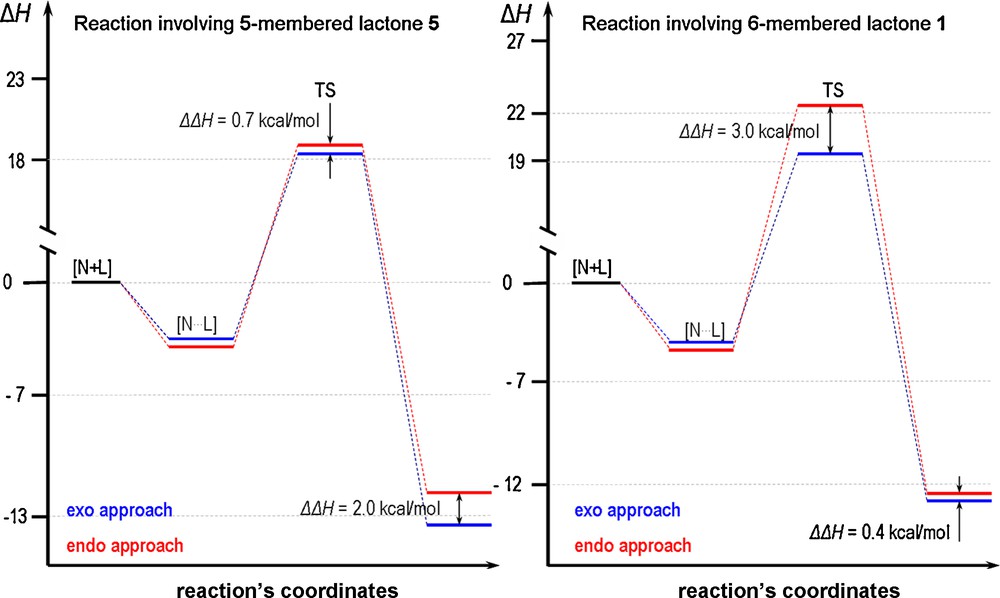
Energy diagrams of the cycloaddition reactions involving five-membered nitrones and δ- and γ-lactones.
To explain the experimentally observed different reactivity of γ- and δ-lactones, especially the ability to form endo-adducts by the former and lack of that ability exhibited by the latter, we have analyzed more carefully the geometries of both of the endo transition states [61]. We have found unfavorable interactions between the hydrogen atoms of the nitrone and the δ-lactone in the transition state (Fig. 8). The distance between the indicated hydrogen atoms is 1.96 Å, and this value is close to that at which a bond is formed. A similar interaction can also be observed for a transition state involving the γ-lactone. However, in this case, such an interaction is less strong since the corresponding distance amounts to 2.60 Å (Fig. 8) [61]. The unfavorable interaction in the endo approach is larger for the transition state involving a six-membered lactone which causes an increase of the endo transition state energy and thus, consequently, increases the diastereoselectivity of the reaction (strong preference for the formation of the exo product). However, during the reaction involving a five-membered lactone the steric interaction in the endo approach is smaller and thus the exo/endo diastereoselectivity is lower.
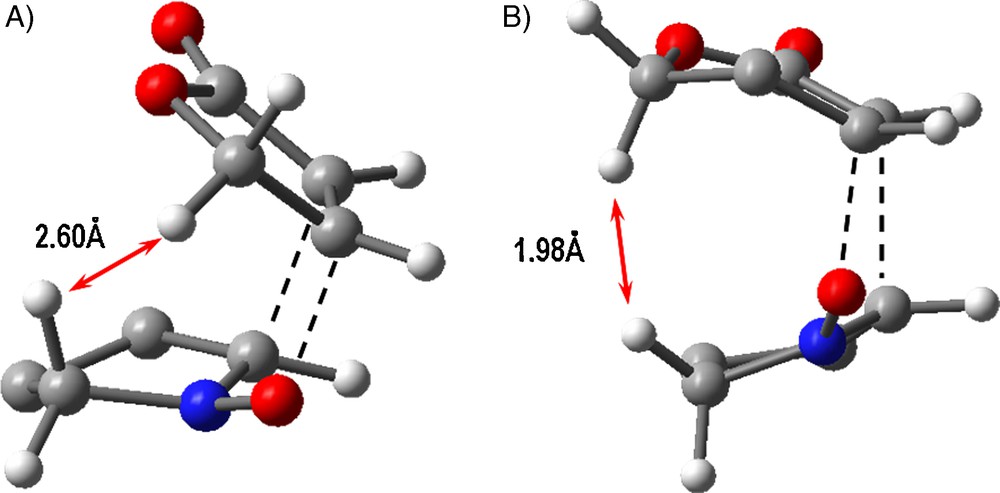
Unfavorable interaction in the endo transition states of cycloaddition involving five-membered nitrones and: A) γ-lactone; B) δ-lactone.
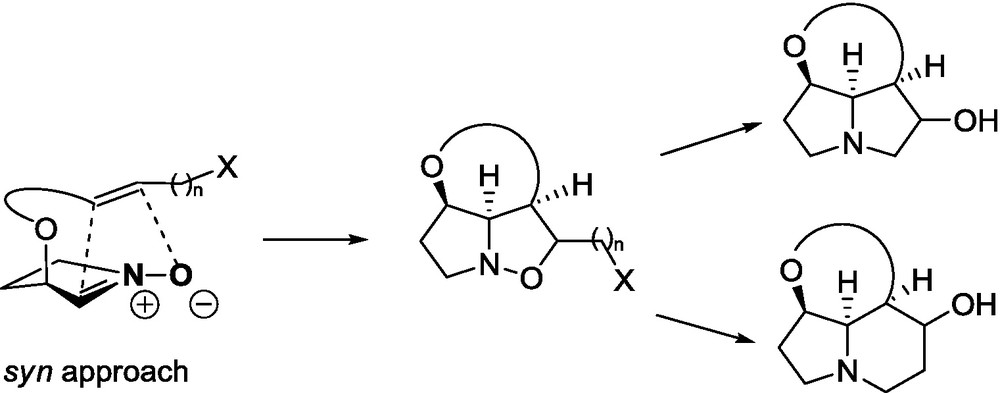
The explanation of the endo/exo selectivity in 1,3-dipolar cycloadditions, based on experimental observations, has been provided by Garcia Ruano et al. [69]. It was attributed to the favorable coulombic interactions between the nitrone, positively charged and the lactone, negatively charged, at the corresponding endo transition state. This interaction has a lower energy for the folded and larger six-membered ring lactones than for the flat γ-ones. Therefore, in the case of the addition of cyclic five-membered nitrones to δ-lactones the endo addition has not been observed. This explanation corresponds well with our calculations carried out using DFT methods. Recently reported by Langlois’ group [70], the endo approach of oxazoline N-oxide 87 to unsaturated δ-lactone 88 and to other α,β-unsaturated esters in the mismatched pair is explained by the interaction between the positively charged iminoether fragment in the nitrone and the negatively charged lactone carbonyl group (Fig. 9).

Endo transition state of cycloaddition involving six-membered lactone 88 and oxazoline N-oxide 87.
In the transition state of cycloaddition reactions to both the γ- and the δ-lactone, the C–O distance is shorter than the C–C distance. Such a situation is in agreement with the previously studied 1,3-dipolar cycloadditions to open-chain nitrones [71]. If we take into consideration that C–O bonds are more advanced than C–C bonds, it means that the first step can be envisioned to some extent as a Michael addition (oxygen of nitrone acts as a nucleophile). In one of our previous papers [56], we have postulated more advanced C–O bond formation, bearing in mind a high preference of the anti approach of a nitrone to the terminal substituent of the δ-lactone. Such a stereochemical preference has been noticed as a general trend for all conjugated additions to δ-lactones [54].
Our theoretical studies on 1,3-dipolar cycloadditions of nitrones and unsaturated lactones [61,62] reproduce very well the experimental observations such as the regioselectivity, the endo/exo selectivity and the reversibility of reactions involving γ-lactones. This allows us to explain not only the observed phenomena, but also to plan and predict the result of a more complicated reaction involving a single or double asymmetric induction, where the chirality elements of each reactant may influence stereoselectivity either in concert or in opposition, as in matched and mismatched pairs. Consequently, this permits us to plan the target-oriented synthesis of iminosugars via a proper selection of cycloaddition components, lactone and nitrone.
5 Synthesis of selected iminosugars
Brandi's 1,3-dipolar cycloaddition methodology [10] has been expanded by us to pyrrolidine and piperidine related iminosugars. For a detailed study leading to piperidine and pyrrolidine related iminosugars the lactones 74, 89, 90 and the nitrone 7 have been selected. The cycloaddition reactions using lactones 74 and 89, performed under standard procedures, yield only the single cycloadducts d-ribo 91 and d-gulo 92, respectively. In the case of lactone 90 two products are formed, l-allo 93 and l-manno 94 in a ratio of about 2.5:1 (Fig. 10) [18].
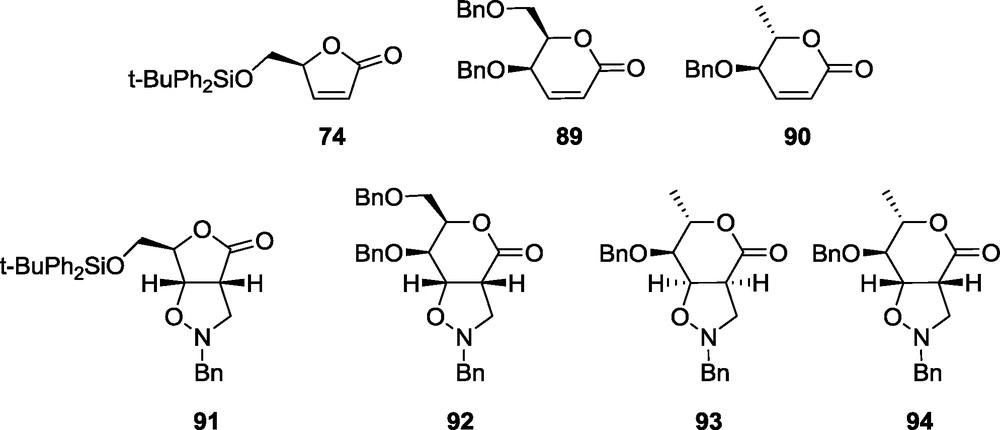
Cycloadducts 91-94 were subjected to a sequence of reactions consisting of the reduction of the lactone fragment to the diol, suitable protection (double acetylation or isopropylidene formation), hydrogenolysis of the N–O bond, desilylation, mesylation and intramolecular nucleophilic substitution to provide, depending on the site of mesylation, pyrrolidine 95 or piperidine 96 closely related to isofagomine 97 (Scheme 19) [72]. A similar transformation sequence applied to the adduct 92 affords the iminosugar having the l-gluco configuration 97 (Scheme 19) [18].
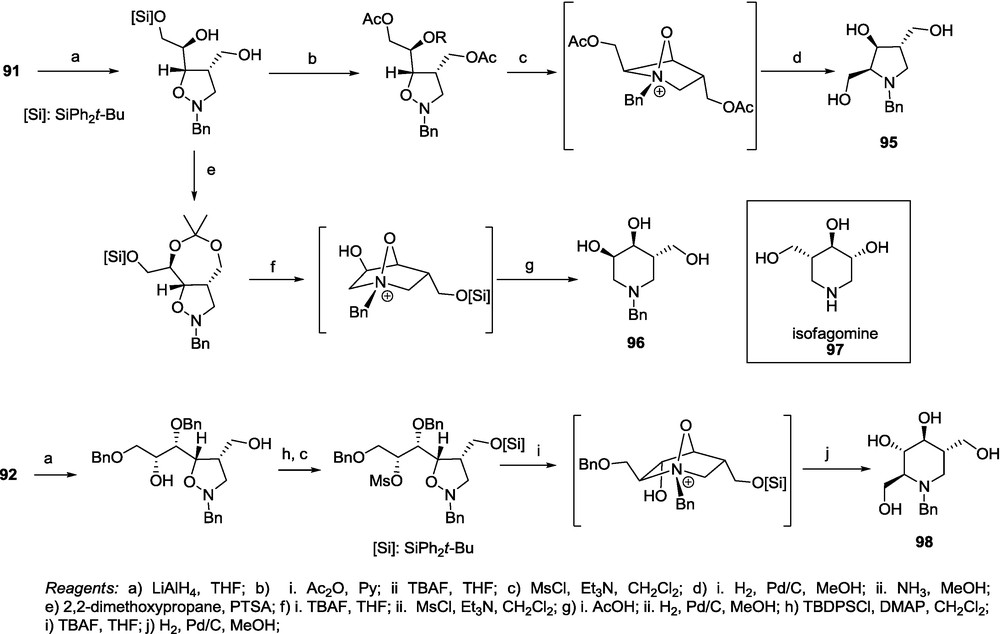
The same transformation of adducts 93 and 94 gives the corresponding d-galacto 99 and d-ido 100 iminosugars (Scheme 20) [18].

With the exception of transformations leading to 96, the reaction sequence involves the inversion of the configuration at the carbon atom that is assigned the d- or the l-conformation series of the starting sugar lactone. In all cases, exchange of substituents at the C-2 carbon atom occurs, thus from adduct d-ribo 91–l-xylo 95, or d-arabino 96, from d-gulo 92 - l-gluco 98 (Scheme 19), from l-allo 93 - d-galacto 99 and from l-manno 94 - d-ido compound 100 (Scheme 20) are obtained (Scheme 21) [18].
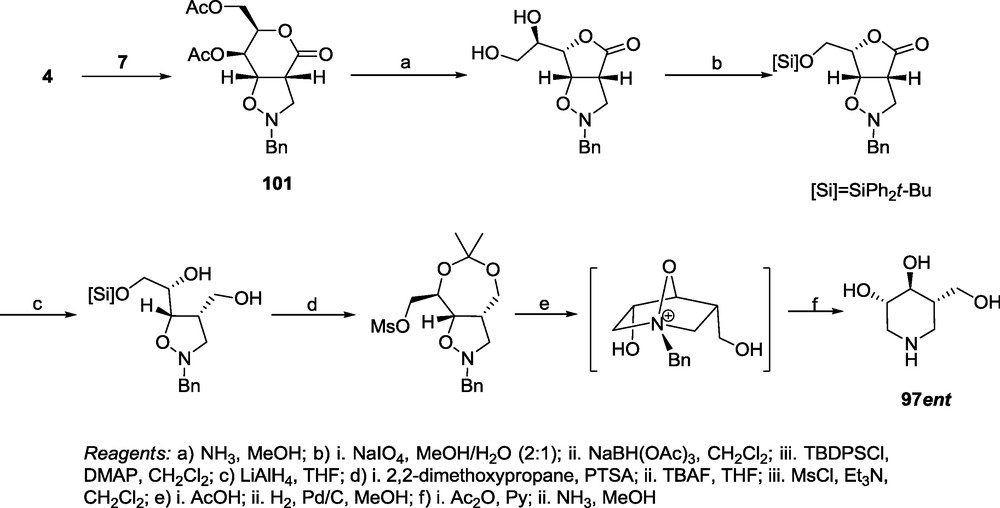
Cycloadduct 101 obtained by the 1,3-dipolar cycloaddition of N-benzyl nitrone 7 to the d-threo lactone 4 was subjected to a rearrangement to the γ-lactone and then to a sequence of reactions displayed in Scheme 21, to provide the (–)-isofagomine (97ent) [21], the enantioner of (+)-isofagomine (97) which has been found to be one of the most potent β-glucosidase inhibitors synthesized by a Danish group [72].
Application of a 1,3-dipolar cycloaddition of Brandi's nitrones 11 and 12 [10a,73] and the lactones 1-6 demonstrates the usefulness of the above methodology for the preparation of polyhydroxylated pyrrolizidines and indolizidines. The adduct 36, available preparatively with an excellent stereoselectivity, was transformed into lentiginosine 102 and 7-hydroxylentiginosine 103 (Scheme 22) [17].
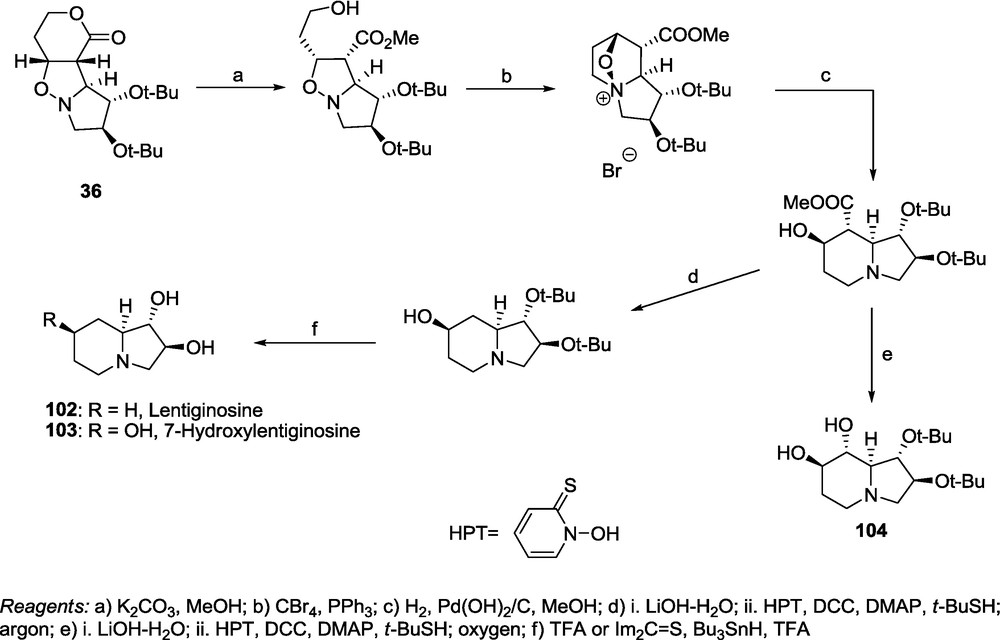
Barton's decarboxylation procedure [74] applied in the presence of oxygen provided an oxidative decarboxylation leading to the regioisomer of castanospermine 104 with the retention of configuration (Scheme 22) [17].
The same reaction scheme applied to adduct 49 opens an easy access route to analogs of castanospermine 105 and australine 106 (Scheme 23) [19,20]. The configuration of the stereogenic centers at C-1, C-6, C-7, C-8 and C-8a of castanospermine 105 is the same as the configuration at C-6, C-2, C-1a, C-5a and C-5b of the adduct 49. Therefore, the removal of the terminal CH2OAc, followed by the hydrogenolysis of the N-O bond and intramolecular alkylation of the nitrogen atom should provide the 8-homoindolizidine 107 [20]. However, the configuration of the stereogenic centers at C-1, C-5, C-6, C-7 and C-7a of australine 106 is different from the configuration at C-6, C-2, C-1a, C-5a and C-5b of the adduct 49 only at the stereogenic center of the C-2 carbon atom. However, bearing in mind that alkylation of the nitrogen atom, in order to form the pyrrolizidine skeleton, should proceed with an inversion of configuration, the adduct 49 represents an excellent substrate for the synthesis of 7-homoaustraline 108 [19]. Both syntheses were completed to provide the expected indolizidine 107 and pyrrolizidine 108 [19,20].

The high stereoselectivity of formation of the cycloadduct 49, which creates a proper substrate for the synthesis of castanospermine 105 and australine 106, is worthy of note since the 1,3-dipolar cycloaddition of an olefin syn to the protected oxygen atom at C-3 of the five-membered ring nitrone can be under taken generally as an intramolecular process (Scheme 24) [75]. The competition between both substituents in the lactone 4 and 3-tert-butoxyl in the nitrone 12 in 1,3-dipolar cycloaddition creates a unique situation which results in the formation of a sole product 49 as a result of the syn approach to the C-3 substituent in the nitrone.
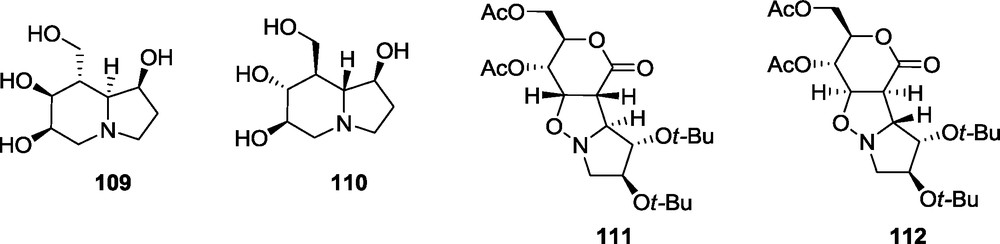
Synthesis of a potential mannosidase inhibitor 109 which can be indolizidine with a d-manno configuration around the piperidine ring, is not straightforward, according to a previous report [25]. The opposite configuration at C-4 of the d-erythro lactone 3, causes the preferential addition anti to the t-butoxy group of the nitrone to give, in the case of 3 and 12, the adduct 48. Therefore, the configuration of all stereogenic centers of the piperidine fragment of the indolizidine 110 would be reversed to that of 49 providing the l-gluco configuration [58]. The desired d-manno configuration of that fragment may be produced only if the d-erythro lactone 3 and nitrone 11 are used. However, the adduct 111, derived from the lactone 3 and nitrone 11 is accompanied by 10% of the alternative exo isomer 112 (Fig. 11) [58].
Although analogous cycloadditions involving γ-lactones proceed with a lower diastereoselectivity, compared to that of δ-lactones, due to the possible formation of endo adducts, in the case of 6 and 11 a single product 65 is formed [59]. Compound 65, subjected to the same transformations as applied for the synthesis of 8-homocastanospermine 107 and 7-homoaustraline 108, gives the indolizidine 113 [25] with a d-manno configuration around the piperidine ring, or the pyrrolizidine 114 [24] related to casuarine (115) [76] and its 3-epimer (116) (Scheme 25).

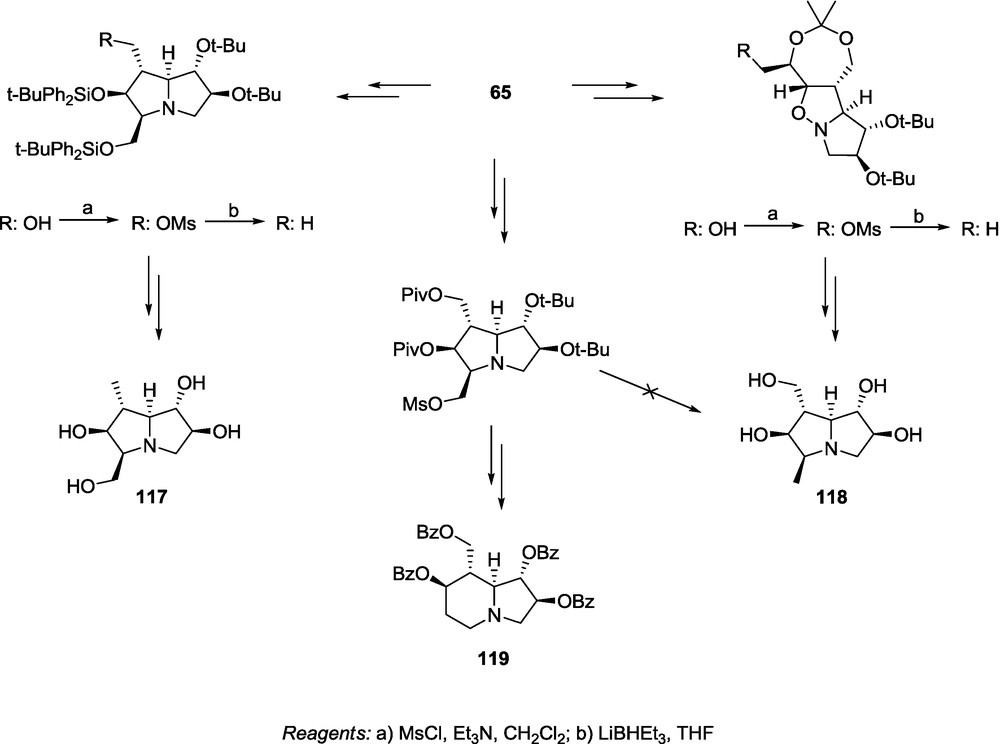
There are numerous polyhydroxylated alkaloids containing methyl group attached to the iminosugar skeleton [4,7,12–15,77]. Interesting biological properties of methyl-iminosugars encouraged us to propose a synthetic route to the methyl substituted pyrrolidines related to casuarine [24]. For this purpose, we reported a synthetic route to transform adduct 65 into the 1-methyl- or 3-methyl analogues of 3-epi-casuarine (117 and 118) (Scheme 26). The key step of that strategy was deoxygenation of the corresponding hydroxymethyl groups. It was performed by treatment of a proper mesylate with LiBHEt3. Depending on the position of the methyl group, the deoxygenation step can be done either at an earlier, or at a later stage of our standard transformation sequence [24]. In the case of 117, the deoxygenation was performed at the late synthesis step after formation of the pyrrolizidine skeleton, whereas for 118, we were forced to perform the reduction at an earlier step (isoxazolidine step) otherwise numerous side reactions, including ring rearrangement leading to 119, occurred (Scheme 26) [24].
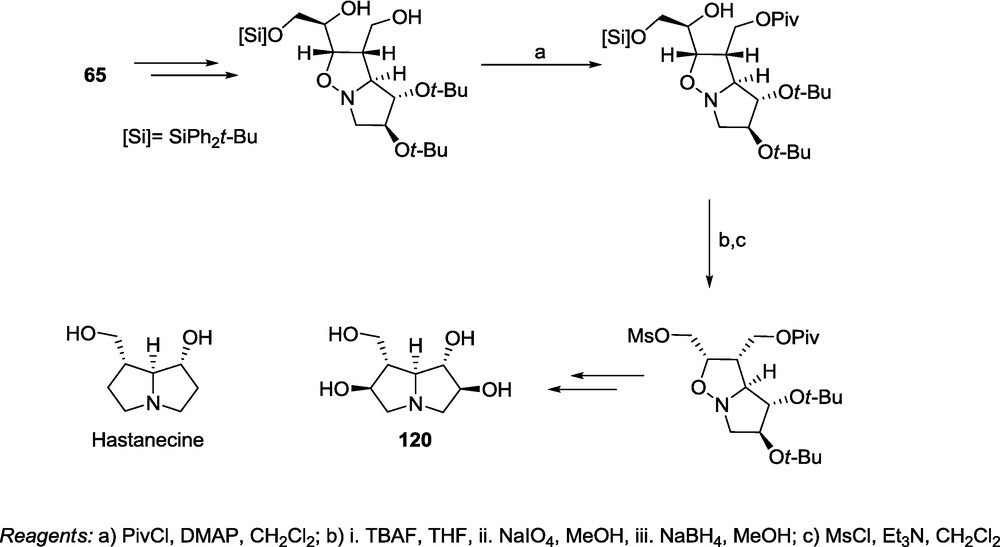
The same adduct 65 can be used for the synthesis of 2,6-dihydroxy-hastanecine 120 [23]. Transformations leading to the necine 120 are the same as used for the synthesis of pyrrolizidine 114 except for the removal of a terminal atom of the side chain diol (Scheme 27). In this case, cycloaddition provides two crucial stereogenic centers at the bridgehead carbon atoms C-7 and C-7a (Scheme 27).
Alkaloids containing the amino-pyrrolizidine and the amino-indolizidine motifs are widespread in nature (Fig. 12) [5,8,12–15,78]. Their potential bioactivity as antidepressant, analgesic, antiviral, antibacterial, or antitumor agents should make them particularly attractive synthetic targets for academic and industrial research laboratories.

However, there are only a few reports about the synthesis and biological activity of their polyhydroxylated analogues–amino-iminosugars [4,79]. This prompted us to check whether our strategy can be also applied to the synthesis of molecules containing the amino function attached to either a polyhydroxylated pyrrolizidine or indolizidine skeleton (Scheme 28).

The ammonolysis of a lactone moiety in the tricyclic cycloadducts 59 and 65 (derived from non-racemic five-membered cyclic nitrone 11 and unsaturated lactones 5 and 6) furnishes an amido function, which after a subsequent Hoffmann rearrangement, leads to a protected amino group directly attached to the bicyclic isoxazolidine skeleton [26]. The subsequent standard transformation as shown above, involving the cleavage of the N-O bond followed by the intramolecular N-alkylation, provides an access to the polyhydroxylated 7-aminopyrrolizidines 121, 122 and 8-aminoindolizidine 123, which are potential glycosidase inhibitors (Scheme 28) [26].
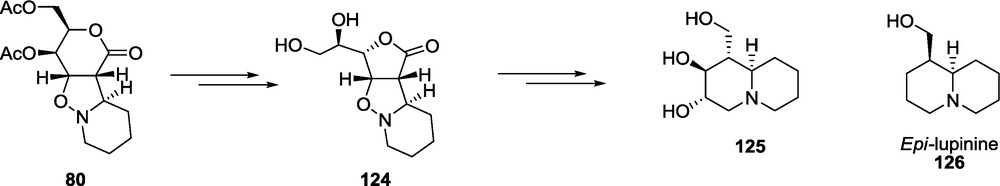
As stated previously, the 1,3-dipolar cycloaddition of the unsaturated d-threo-hexaldonolactone 3 and a six-membered cyclic nitrone 15 leads to a single adduct 80, which after rearrangement of the six-membered lactone ring to the five-membered lactone 124, can be transformed into the 1-hydroxymethyl-2,3-dihydroxyquinolizidine 125 related to lupinine 126 via a standard reaction sequence (Scheme 29). It should be pointed out, however, that the conformational lability of a nitrogen atom bearing an oxygen makes it difficult to follow the configuration of the adduct by NMR spectra [22] due to the broadening of the spectral lines.
6 Biological activity of synthesized compounds
Studies on the structure-bioactivity relationship of synthesized compounds become an integral part of our ongoing research program leading to iminosugars obtained via transformation of adducts of 1,3-dipolar cycloaddition reactions. The glycosidases’ inhibition tests were conducted against six commercially available enzymes: α-d-glucosidase from rice; β-d-glucosidase from almonds, α-d-mannosidase from jeak bean, α-l-fucosidase from bovine kidney, β-d-galactosidase and β-d-glucuronidase both from bovine liver. Some selected results are summarized in Table 1.
Selected results of bioactivity tests of selected iminosugars.
| Compound | IC50 values [mM]a | ||||||
| EN1 | EN2 | EN3 | EN4 | EN5 | EN6 | References | |
| 97ent | n.a. | 9.0 | n.a. | 8.3 | 15.0 | 12.0 | [21] |
| N-Ac-97ent | n.a. | 8.5 | 21.5 | 6.6 | 9.1 | 3.5 | [21] |
| N-Bn-97ent | n.a. | n.a. | 5.1 | n.a. | n.a. | n.a. | [21] |
| 107 | n.a. | n.a. | 26.8 | 1.6 | n.a. | n.a. | [20] |
| 108 | 20.0 | n.a. | n.a. | n.a. | n.a. | 24.0 | [19] |
| 113 | n.a. | n.a. | n.a. | 1.0 | 0.9 | 0.6 | [25] |
| 114 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | [24] |
| 117 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | [24] |
| 118 | n.a. | 13.0 | n.a. | n.a. | n.a. | n.a. | [24] |
| 120 | 9.9 | n.a. | n.a. | n.a. | n.a. | n.a. | [23] |
| 121 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | [26] |
| 122 | n.a. | n.a. | n.a. | 0.9 | n.a. | n.a. | [26] |
| 123 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | [26] |
| 126 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | [22] |
| 127 | n.a. | n.a. | n.a. | n.a. | n.a. | 8.0 | [80] |
| 128 | n.a. | n.a. | n.a. | n.a. | n.a. | 7.0 | [80] |
a The half maximal inhibitor concentration in mM.
(-)-Isofagomine 97ent and its N-substituted derivatives show a weak activity against tested enzymes. Amongst them, the N-benzylated derivative of 97ent is the most selective and interacts only with α-d-mannosidase [21]. In comparison, the natural (+)-isofagomine 97 is a potent inhibitor of β-d-glucosidase [4].
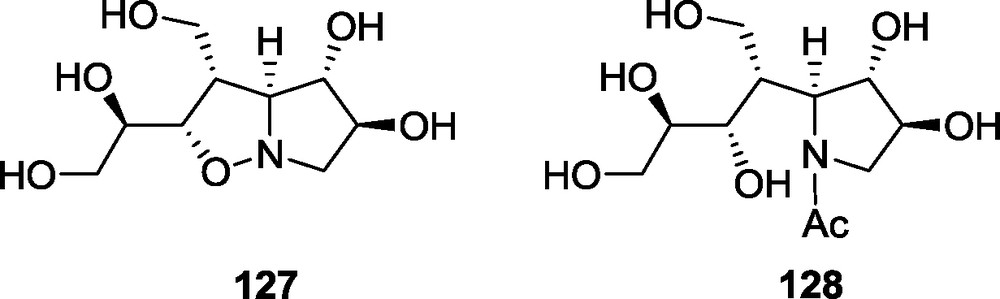
A weak inhibition was also noticed for bicyclic iminosugars. Both 8-homocastanospermine 107 [20] and the 7-homolog of australine (108) [19] have lower activity in comparison to the native molecules. Although compound 113 has a manno-configuration on the piperidine ring, it does not exhibit any significant activity against α-d-mannosidase, but is weakly active against other enzymes [25]. Interesting results were obtained for dihydroxy-hastanecine 120 [23] and its 3-methyl derivative 118 [24]. The former one showed activity against α-d-glucosidase, whereas the addition of a methyl group at position C-3 changed the selectivity and 118 became active against β-d-glucosidase. Some weak activity was also found for several intermediate compounds used in the synthesis of the iminosugars 107 and 108. The isoxazolidine 127, as well as the N-acetylated pyrrolidine 128, both derived from adduct 65, possess activity against α-l-fucosidase [80]. These compounds are intermediates in the synthesis of the iminosugars 113 and 114 (Fig. 13).
A lack of activity was observed for 3-epi-1-homocasuarine 114 [24], its 1-methyl derivative 117 [24], quinolizidine 126 [22] and the also amino-iminosugars 121 and 123 [26]. However, the amino-pyrrolizidine 122 displays a weak inhibition of β-d-galactosidase [26]. It is worthwhile to point out that there is not much known about the bioactivity of this relatively new subclass of iminosugars (amino-iminosugars). According to some biological data provided in the literature for monocyclic amino-iminosugars [4], one could expect that these compounds might possess glycoaminidases’ inhibition activity.
Although the tested compounds display only a weak inhibition of the tested glycosidases, the results obtained might provide an interesting basis for further structure-activity relationship studies.
7 Conclusions
In this account, we present the characteristic features of 1,3-dipolar cycloaddition of nitrones to readily available γ- and δ-lactones such as stereoselectivity, kinetic and thermodynamic control of reactions. The influence of structure and configuration of both components on the stereoselectivity of the cycloaddition was investigated and discussed. A special emphasis was placed on the cycloadditions involving five-membered ring nitrones since owing to their rigid conformation, the interpretation of experimental facts is straightforward and the obtained adducts offer a broad application for the synthesis of iminosugars having pyrrolizidine or indolizidine structures. For these reactions, the results of calculations carried out using DFT methods at the B3LYP/6-31 + G(d) level showed a very good agreement with the experimental facts.
Broad investigations show the influence of the substituents in the lactone and nitrone on the stereoselectivity of cycloaddition. It has been shown that the 3-t-butoxyl in the nitrone and the terminal oxymethyl group in the lactone play a decisive role in stereo selection. Other substituents may influence the stereoselectivity only in the mismatched pair situation of the primary groups. We have shown that cycloadditions involving δ-lactones are not reversible and proceed in the exo mode exclusively. In many cases, a single product is formed, therefore δ-lactones are particularly attractive in the target-oriented synthesis. The corresponding transformations involving γ-lactones are more complicated. As we have shown, the cycloadditions to γ-lactones proceed with a lower diastereoselectivity, as compared to the cycloaddition to six-membered ones because of the possible formation of endo adducts. Moreover, the reversibility of the cycloaddition, as well as racemization of the 5-substituted 2-(5H)-furanones, additionally complicates the composition of the postreaction mixtures and may lead to the formation of partially racemic products. Because of this, γ-lactones are less attractive in synthetic applications.
A proper selection of the readily available components of the 1,3-dipolar cycloaddition controls the absolute stereochemistry of the newly created stereogenic centers of the adduct. It allows us to design the desired configuration of the adduct and the synthesis of the core part of the target molecule. It is demonstrated that a standard reaction sequence involving hydrogenolysis of the N–O bond followed by alkylation of the nitrogen atom works well in many configurational arrangements.
Acknowledgements
This publication was financed by the European Union within the European Regional Development Fund, Project POIG.01.01.02.-14-102/09.