1 Introduction
Over the past 20 years, much emphasis has been laid on the unique properties of polyoxoanionic ligands [1], and many catalysts resulting from the grafting of transition metal cations on polyoxometalates (POMs) have been synthesized [2]. In particular, because of the unique catalytic potential of ruthenium [3], its association with polyoxoanions has drawn considerable interest [4,5].
We, and others, have been particularly interested in the incorporation of ruthenium in a polyoxometalate by grafting of {Ru(η6-arene)}2+ or {Ru(DMSO)n}2+ (n = 1, 2, or 3) moieties on the polyoxoanionic framework [6–11]. A small number of these compounds have been tested in oxidation and racemisation catalysis [1e,12]. Unfortunately, only in one case has the effort been made to relate the catalytic activity to the intrinsic reactivity of the precatalyst [12c]. The result of the latter study was promising, thus showing that the investigation of the intrinsic reactivity of ruthenium derivatives of polyoxometalates is important in order to understand their catalytic activity. Moreover, a systematic investigation of the behaviour of these species could help finding the best experimental conditions which might lead to the formation of a catalytically active species.
Among the different compounds described, the [α-PW11O39{RuL3(H2O)}]5− compounds (L3 = η6-arene, (DMSO)3) have drawn the attention of our group (Fig. 1) [8a,b]. In these anions, the {RuL3}2+ fragment is linked to two non-equivalent oxygen atoms of the lacuna of [α-PW11O39]7−, and the ruthenium achieves an 18 electron configuration by coordination to an aqua ligand.
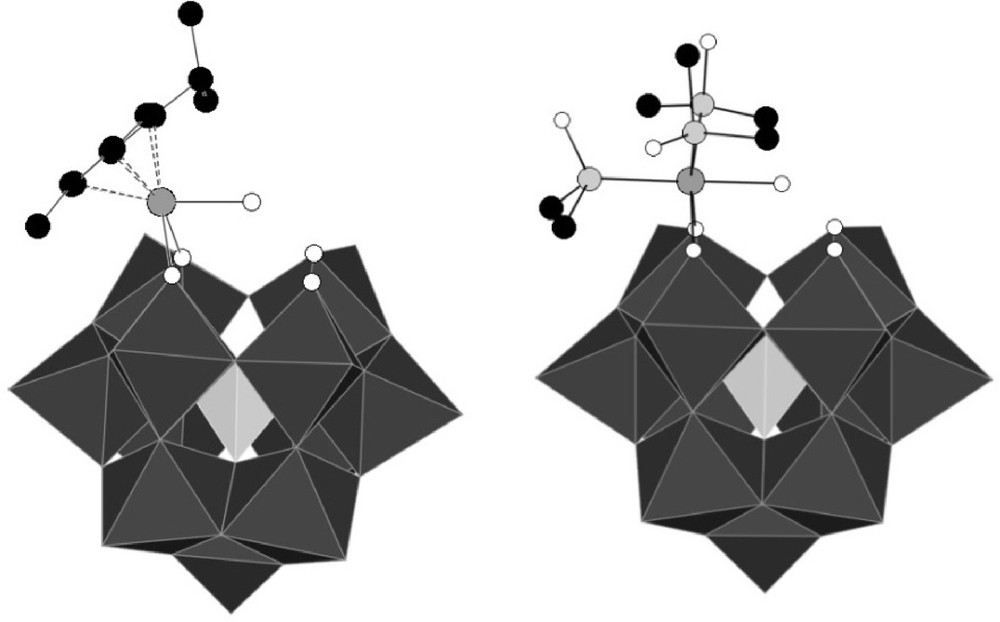
Polyhedral representation of the structures of [α-PW11O39{Ru(η6-cymene)(H2O)}]5− PW11Ru(pcym) (left) and [α-PW11O39{Ru(DMSO)3(H2O)}]5− PW11Ru(dmso)3 (right) (for reasons of clarity, hydrogen atoms are not depicted.).
On the one hand, we performed a computational study in order to account for the regiospecificity of the grafting of the ruthenium on the monolacunary Keggin anion [8b]. On the other hand, we started to investigate the intrinsic reactivity of these species, in order to see what options are available to release coordination sites around the ruthenium center. Indeed, in these compounds, the ruthenium is linked to a water molecule, which is generally considered as a labile ligand, and which could thus be substituted by a substrate molecule during a catalytic cycle. We have thus shown that under thermal activation, the [α-PW11O39{Ru(η6-arene)(H2O)}]5− anions transform into [{PW11O39Ru(η6-arene)}2{WO2}]8− complexes [8a], whereas the [α-PW11O39{Ru(DMSO)3(H2O)}]5− anion leads to the formation of [α-PW11O39{Ru(DMSO)}]5− (Fig. 2) [8b]. However, under conventional heating, both of these transformations are incomplete. Whereas alternative more efficient ways of synthesis of [α-PW11O39{Ru(DMSO)}]5− have been described [7,8a], the [{PW11O39Ru(η6-arene)}2{WO2}]8− anion remained a species difficult to synthesize. Moreover, evidence for structurally analogous [{PW11O39Ru(DMSO)}2{WO2}]8− has not been obtained.
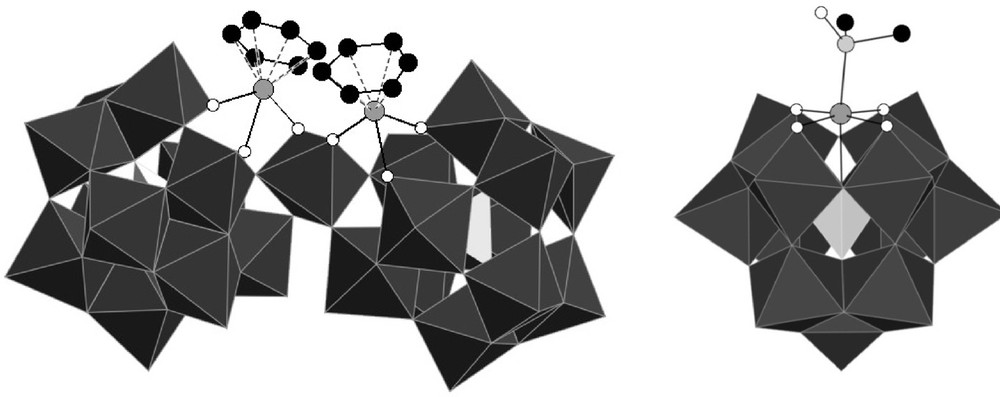
Polyhedral representation of the structures of [{PW11O39Ru(η6-C6H6)}2{WO2}]8− {PW11Ru(benz)}2(WO2) (left) and [α-PW11O39{Ru(DMSO)}]5− PW11Ru(dmso) (right) (for reasons of clarity, hydrogen atoms are not depicted.).
In order to better understand the reactivity of [α-PW11O39{RuL3(H2O)}]5− complexes, we performed a detailed investigation of the formation of [{PW11O39Ru(η6-arene)}2{WO2}]8−. In this article, we thus describe a high yield synthesis of [{PW11O39Ru(η6-p-cymene)}2{WO2}]8−, and also report on the formation under analogous synthetic conditions of a new {Ru(DMSO)3}2+ derivative of a polyoxotungstate, [{PW11O39Ru(DMSO)3}2{(WO2(H2O))2O}]8−, in which a {(H2O)O2WOWO2(H2O)}2+ bridge ensures the connection between two {PW11O39Ru(DMSO)3}5− subunits. Throughout the article, compounds [α-PW11O39{Ru(η6-arene)(H2O)}]5−, [{PW11O39Ru(η6-arene)}2{WO2}]8−, [α-PW11O39{Ru(DMSO)3(H2O)}]5−, [α-PW11O39{Ru(DMSO)}]5−, and [{PW11O39Ru(DMSO)3}2{(WO2(H2O))2O}]8− will be referred to as PW11Ru(arene), {PW11Ru(arene)}2(WO2), PW11Ru(dmso)3, PW11Ru(dmso), and {PW11Ru(dmso)3}2(W2O5), respectively.
2 Experimental
2.1 Reagents
K7[α-PW11O39]·14H2O [13], K10[P2W20O70(H2O)2]·22H2O [13], [Ru(p-cymene)Cl2]2 [14], fac-[RuCl2(DMSO)3(DMSO)] [15] complexes were prepared as described in the literature. All other reagents were obtained from commercial sources and used as received.
2.2 Syntheses
2.2.1 Synthesis of Cs8-xKx[{PW11O39Ru(p-cymene)}2{WO2}] (Cs8-xKx-{PW11Ru(arene)}2(WO2))
To a solution of K7[α-PW11O39]·14H2O (0.819 g, 0.26 mmol, 1 eq) in 9 mL of distilled water, were subsequently added 248 μL of a 1 mol L−1 solution of Na2WO4 (0.25 mmol, 1 eq), 0.078 g of [Ru(p-cymene)Cl2]2 (0.13 mmol, 0.5 eq), and 13 mL of distilled water. The pH of the mixture was adjusted to pH = 3.0, by adding a few drops of 37% HCl. The suspension was refluxed for 16 h, during which time the ruthenium precursor progressively dissolved. After having been left to cool at room temperature, CsCl (0.344 g, 2.04 mmol, 8 eq) was added to the solution, leading to the precipitation of an orange solid, which was separated by filtration (0.5 g), and identified as a fairly pure mixed potassium/cesium salt of [{PW11O39Ru(p-cymene)}}2{WO2}]8− based on comparison with the NMR and IR spectroscopic data previously reported for the potassium salt [8a].
2.2.2 Synthesis of {PW11Ru(dmso)3}2(W2O5)
2.2.2.1 From [α-PW11O39]7−
To a solution of K7[α-PW11O39]·14H2O (0.819 g, 0.26 mmol, 1 eq) in 9 mL of distilled water, were subsequently added 248 μL of a 1 mol.L−1 solution of Na2WO4 (0.25 mmol, 1 eq), 0.122 g of fac-[RuCl2(DMSO)3(DMSO)] (0.25 mmol, 1 eq), and 1.5 mL of distilled water. The pH of the mixture was adjusted to pH = 3.0, by adding a few drops of 37% HCl. The solution was refluxed for 16 h, during which time the colour of the medium switched from yellow to dark brown. After having been left to cool at room temperature, KCl (0.650 g, 8.72 mmol, 35 eq) was added to the solution, leading to the progressive formation of a brown solid. After 24 h of stirring at room temperature, the precipitate was separated by filtration, dried with Et2O, and identified as a fairly pure sample of K8[{PW11O39Ru(DMSO)3}2{(WO2(H2O))2O}]·12H2O (K8–{PW11Ru(dmso)3}2(W2O5)·12H2O) (0.289 g, η ≈ 32%). IR (KBr, cm−1): 3016 (w), 2924 (w), 1420 (w), 1320 (w), 1299 (w), 1125 (sh), 1098 (m), 1052 (m), 1040 (m), 1020 (w), 957 (s), 900 (m), 856 (sh), 834 (s), 809 (s), 775 (sh), 745 (sh), 706 (m), 620 (m), 516 (w), 429 (w), 365 (m), 263 (w). 1H NMR (D2O): δ = 3.25, 3.59, 3.60, 3.75, 3.81, 3.87 ppm (all singlets with equal intensities)1. 31P NMR (D2O): δ = −12.05 ppm. 183W NMR (H2O/D2O): δ = −231.5, −179.4, −170.2, −149.0, −134.3, −131.6, −131.1, −127.5, −109.5, −101.8, −98.8, −95.8 ppm (all singlets with equal intensities). Anal. Calcd for C12H64K8P2W24O103Ru2S6: C, 2.05; H, 0.92; S, 2.73; P, 0.88; Ru, 2.87; W, 62.69; K, 4.44. Found: C, 2.24; H, 1.25; S, 2.73; P, 0.86; Ru, 2.53; W, 65;77; K, 4.792.
2.2.2.2 From [P2W20O70(H2O)2]10−
To a solution of K10[P2W20O70(H2O)2]·22H2O (0.940 g, 0.165 mmol, 0.5 eq) in 10 mL of distilled water, were added 0.162 g of fac-[RuCl2(DMSO)3(DMSO)] (0.330 mmol, 1 eq), and 2 mL of distilled water. The pH of the mixture was adjusted to pH = 3.0, by adding a few drops of 37% HCl. The solution was refluxed for 20 h, during which time the colour of the medium switched from yellow to dark brown. After having been left to cool at room temperature, 0.492 g of KCl (6.60 mmol, 20 eq) were added (without precipitation) to the solution, which was then left to evaporate at 25 °C. After a few days, a small number crystals of K7H[{PW11O39{Ru(DMSO)3}}2{(WO2(H2O))2O}]·10H2O (K7H–{PW11Ru(dmso)3}2(W2O5)·10H2O). X-Ray-data: monoclinic, P 21/n, a = 23.345(2), b = 35.664(6), c = 25.817(3) Å, β = 93.113(9)°, V = 21463(5) Å3. The quality of these crystals was suitable for structure resolution.
Alternatively, when a greater quantity of KCl was added (0.738 g, 9.90 mmol, 30 eq) to the mixture of K10[P2W20O70(H2O)2]·22H2O and fac-[RuCl2(DMSO)3(DMSO)] after the 20 h reflux, a precipitate formed after several hours of stirring at room temperature. After filtration of the precipitate, which mainly contains the potassium salt of {PW11Ru(dmso)3}2(W2O5), small crystals formed in the filtrate upon slow evaporation at room temperature over a few days. X-Ray-data: monoclinic, C 2/c, a = 23.689(4), b = 35.384(7), c = 27.085(5) Å, α = γ = 90°, β = 90.756(8)°, V = 21463(5) Å3. A partial structure determination has confirmed the presence of {PW11Ru(dmso)3}2(W2O5), but a complete determination could not be achieved for these crystals due to the severe crystalline disorder.
2.3 Physical methods
2.3.1 Spectroscopic characterization
The 1H (300.13 MHz, TMS) and 31P (121.5 MHz, external 85% H3PO4) NMR spectra were obtained in solution on a Bruker Avance II 300 spectrometer equipped with a QNP probehead. The 183W NMR spectrum was recorded in a 10 mm OD tube on a Bruker DRX 500 apparatus, operating at 20.8 MHz. The concentrated solution required for 183W NMR was prepared by dissolving 800 mg of the potassium precipitate of {PW11Ru(dmso)3}2(W2O5) in a minimum volume of an aqueous saturated solution of LiClO4, filtering off the precipitate of KClO4, and adding 10% D2O for field frequency lock. Chemical shifts were measured with reference to an external solution of Na2WO4 in alkaline D2O.
2.3.2 X-ray crystallography
Crystal structure data for K7H-{PW11Ru(dmso)3}2(W2O5)·10H2O were collected, using a combination of φ and ω scans, on a Nonius Kappa-CCD diffractometer (Table 1) with graphite monochromated Mo-Kα (0.71073 Å) radiation. Measurements were performed at 250 K. Unit cell parameters determination, data collection strategy and integration were carried out with the Nonius Eval-14 suite of programs [16a]. The data were corrected for absorption by a multiscan method [16b]. The structure was solved by direct methods with SHELXS-86 [16c], refined by full least-squares on F2 and completed with SHELXL-97 [16d]. Graphics were carried out with DIAMOND [16e]. W, Ru and S atoms were refined with anisotropic displacement parameters. The H atoms identified on difference Fourier maps were included by using a riding model.
Crystal data and structure refinement parameters.
| K7H-{PW11Ru(dmso)3}2(W2O5)·10H2O | |
| Empirical Formula | C12K7O101P2Ru2S6W24 |
| Mr/g.mol−1 | 6902.48 |
| Color | Orange |
| Temperature/K | 250 |
| Crystal System | Monoclinic |
| Space Group | P 21/n |
| a/Å | 23.345(2) |
| b/Å | 35.664(6) |
| c/Å | 25.817(3) |
| α/° | 90 |
| β/° | 93.113(9) |
| γ/° | 90 |
| V/Å3 | 21463 |
| Z | 8 |
| ρcalcd/g.cm−3 | 4.272 |
| μ/mm−1 | 26.407 |
| θmin–θmax/° | 2.04–25.06 |
| h,k,l max | 27,42,30 |
| Reflcns collected | 189215 |
| Reflcns reported | 37659 |
| obsd reflcns (I > 2σ(I)) | 20845 |
| refined param | 1643 |
| R(int) | 0.1037 |
| R | 0.0823 |
| Rw (all data) | 0.1938 |
| goodness of fit S | 1.085 |
| Δρ(max/min)/e.Å−3 | −3.53/3.23 |
3 Results and discussion
3.1 Formation of [{PW11O39Ru(η6-arene)}2{WO2}]8− ({PW11Ru(arene)}2(WO2))
In a previous study [8a], we had shown that by reacting [Ru(η6-arene)Cl2]2 (arene = benzene, toluene, p-cymene) and [α-PW11O39]7− in water at 100 °C, [α-PW11O39{Ru(η6-arene)(H2O)}]5− (PW11Ru(arene)) first formed, and then evolved partially into [{PW11O39Ru(η6-arene)}2{WO2}]8− ({PW11Ru(arene)}2(WO2)). The formation of {PW11Ru(arene)}2(WO2), which consists of two {PW11O39Ru(η6-arene)}5− subunits connected by a {WO2}2+ bridge, had not been anticipated. It can be formally expressed by Eq. (1), where the additional tungstate ions possibly result from the decomposition of some [α-PW11O39]7− anions in situ3:
| (1) |
More recently, an alternative way of preparing these {PW11Ru(arene)}2(WO2) compounds with a higher yield was reported by Nomiya et al. [10c]. In this case, [PW11O39]7− was first formed in situ by degradation of [PW12O40]3−, before adding [Ru(benzene)Cl2]2, lowering the pH, and precipitating the resulting product. By this method, the tungstate fragments released in the first step are still available for the last step.
Considering Eq. (1), we looked at the possibility of improving the formation of {PW11Ru(arene)}2(WO2) by deliberately adding tungstate to the reaction mixture. A mixture of [α-PW11O39]7−, [Ru(η6-p-cymene)Cl2]2, and [WO4]2− in a 2:1:2 ratio was refluxed at pH = 3.0; after 16 h, {PW11Ru(pcym)}2(WO2) is the only species detected in the mother liquor according to 31P NMR. It is noteworthy that a slight excess of [WO4]2− is necessary for the quantitative formation of {PW11Ru(pcym)}2(WO2) in solution. This compound was then isolated by precipitation, by adding CsCl.
All in all, these different studies demonstrate that {PW11Ru(arene)}2(WO2)-type structures can be formed from PW11Ru(arene), by working under acidic conditions and in the presence of tungstate ions, in agreement with Eq. (1).
3.2 Formation of [{PW11O39Ru(DMSO)3}2{(WO2(H2O))2O}]8− ({PW11Ru(dmso)3}2(W2O5))
Previously, the progressive formation of [α-PW11O39{Ru(DMSO)}]5− (PW11Ru(dmso)) was observed upon refluxing an aqueous solution of [α-PW11O39{Ru(DMSO)3(H2O)}]5− (PW11Ru(dmso)3) [8b]: the water molecule and two DMSO ligands are substituted by oxygen atoms of the lacuna. Given the investigation performed on the synthesis of {PW11Ru(arene)}2(WO2), we tried to see whether isostructural {PW11Ru(dmso)3}2(WO2) derivatives could be obtained. These efforts led to the characterization of a new species, {PW11Ru(dmso)3}2(W2O5), which has been obtained by two alternative methods.
On the one hand {PW11Ru(dmso)3}2(W2O5) was obtained from a mixture of [α-PW11O39]7−, fac-[RuCl2(DMSO)3(DMSO)], and [WO4]2−. Initially, a 2:2:1 mixture was refluxed at pH = 3.0 for 24 hours. At this stage, according to the 31P NMR spectrum of the mother liquor, several compounds were present in solution: two major species (δ(P) = −12.05 and −12.25 ppm), PW11Ru(dmso)3 (δ(P) = −11.03 ppm), and two minor species (δ(P) = 0.54 ppm – phosphate ions – and δ(P) = −6.69 ppm)4. The chemical shifts of the major species are shifted towards lower frequency with respect to PW11Ru(dmso)3. Since the chemical shifts of {PW11Ru(arene)}2(WO2) anions are also shifted towards lower frequencies in comparison with those of PW11Ru(arene) [8a] (at variance with that of PW11Ru(dmso) found at −10.89 ppm), we supposed that the signals at −12.05 and −12.25 ppm might correspond to species in which {PW11O39{Ru(DMSO)3}5− subunits are associated through oxotungstic bridges.
Then, in order to achieve higher conversion of PW11Ru(dmso)3, the reaction was repeated in the presence of an extra equivalent of tungstate. After 15 minutes of reflux, four major species were present in the mother liquor: PW11Ru(dmso)3, and three other complexes (δ(P) = −11.20, −11.90 and −12.05 ppm). After 16 hours of reflux, three major species were present (δ(P) = −12.05, −12.20 and −12.25 ppm), while PW11Ru(dmso)3 was no longer detected5. These observations tend to show that after 16 hours, the major signals correspond to species resulting from the reaction of PW11Ru(dmso)3 with tungstate anions. At this stage, addition of KCl (25 eq) led to the progressive formation of a brown solid, which was separated by filtration and characterized by solution NMR: the 31P NMR spectrum shows only one major signal at −12.05 ppm6, while the corresponding 1H NMR spectrum displays six signals of equal intensity in the region which corresponds to DMSO ligands. All attempts to get single crystals suitable for X-ray crystallography from this precipitate were unfortunately unsuccessful.
On the other hand, {PW11Ru(dmso)3}2(W2O5) was obtained by reacting [P2W20O70(H2O)2]10− with fac-[RuCl2(DMSO)3(DMSO)]. After 20 hours of reflux of an aqueous solution containing a 1:2 mixture of K10[P2W20O70] and fac-[RuCl2(DMSO)3(DMSO)] at pH = 3.0, the 31P NMR spectrum of the mother liquor indicates the presence of three major species (δ(P) = 0.50, −12.05 and −12.25 ppm) and of one minor species (δ(P) = −10.05 ppm). The signal located at 0.50 ppm can be assigned to phosphate anions, which attests to the decomposition of some [P2W20O70(H2O)2]10− anions with release of oxotungstate fragments. The two other major signals display the same chemical shifts as those observed in the course of the synthesis from [PW11O39]7− (vide supra), thus suggesting that by working at the same pH and with similar n(Ru)/n(P) and fairly similar n(Ru)/n(W) ratios (Ru/W = 2/20 vs 2/24), the same species tend to form in the mother liquor.
By adding a small quantity of KCl to this solution, a few red crystals appeared after several weeks of slow evaporation at room temperature, whose IR spectrum was identical to that of the compound obtained from [PW11O39]7−, and which allowed a single crystal X-ray analysis. Crystals obtained upon addition of a larger quantity of KCl proved unsuitable for complete X-ray structure analysis (Section 2.2.2.2).
3.3 X-ray crystal structure analysis of K7H[{PW11O39Ru(DMSO)3}}2{(WO2(H2O))2O}]·10H2O
The asymmetric unit contains two crystallographically independent [{PW11O39Ru(DMSO)3}2{(WO2(H2O))2O}]8− anions, which only slightly differ in their interatomic distances and bond angles (Table 2). A polyhedral representation of one of these is depicted on Fig. 3, while ball and stick representations of both anions showing the full atom labelling scheme can be found in the supplementary material.
Interatomic distances and bond angles around the ruthenium and the ditungstate bridges in the two independent anions in K7H-{PW11Ru(dmso)3}2(W2O5)·10H2O. The labeling of the atoms corresponds to the one given in Fig. 4.
| Anion 1 | Anion 2 | |||
| Distance (Å) | ||||
| WO | 1.70 (4) | 1.72 (3) | 1.70 (4) | 1.73 (4) |
| WOH2 | 2.23 (4) | 2.19 (3) | 2.28 (4) | 2.28 (4) |
| WOL1 | 1.86 (4) | 1.85 (4) | 1.87 (4) | 1.87 (3) |
| WOL2 | 2.28 (4) | 2.05 (4) | 2.16 (5) | 2.16 (4) |
| WORu | 1.81 (3) | 1.75 (4) | 1.77 (3) | 1.67 (3) |
| WOB | 1.88 (4) | 1.94 (4) | 1.89 (4) | 1.90 (4) |
| RuOL3 | 2.18 (4) | 2.01 (4) | 2.07 (4) | 2.06 (4) |
| RuOL4 | 2.43 (4) | 2.17 (3) | 2.09 (3) | 2.10 (3) |
| RuORu | 1.95 (4) | 2.07 (4) | 2.02 (3) | 2.13 (3) |
| RuS | 2.224 (17) | 2.224 (17) | 2.24 (2) | 2.206 (17) |
| 2.274 (15) | 2.262 (17) | 2.25 (2) | 2.237 (18) | |
| 2.251 (16) | 2.287 (17) | 2.27 (2) | 2.245 (17) | |
| Ru···Ru | 8.84 | 8.65 | ||
| Angles (°) | ||||
| WOBW | 145.1 (19) | 146 (2) | ||
| WOBRu | 163 (2) | 161.1 (19) | ||
| 164 (2) | 165.3 (18) |
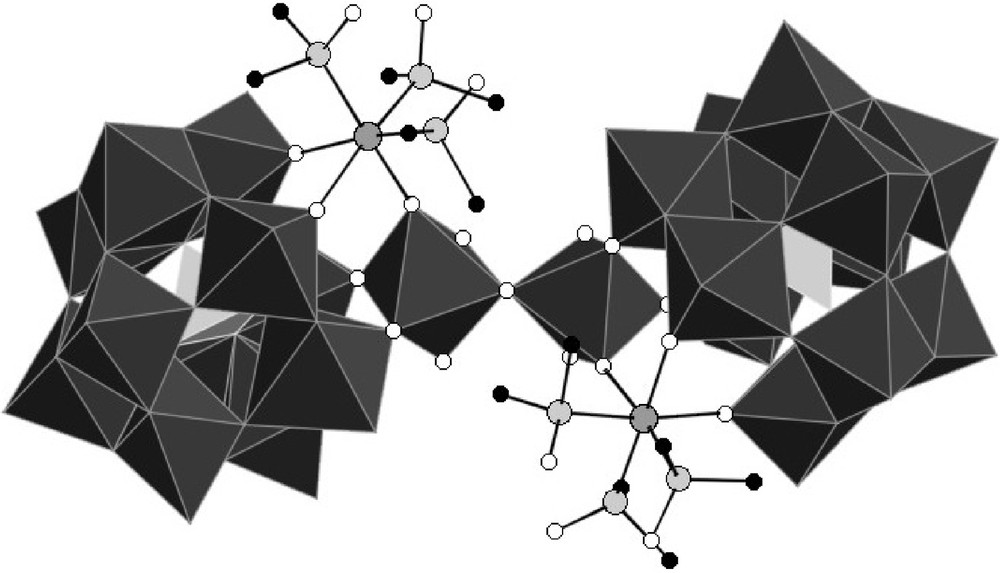
Polyhedral representation of [{PW11O39Ru(DMSO)3}2{(WO2(H2O))2O}]8−.
{PW11Ru(dmso)3}2(W2O5) can be described as the assembly of two {α-PW11O39Ru(DMSO)3}5− subunits through a {(WO2(H2O))2O}2+ bridge. The metal-oxygen distances within the two subunits are similar, and the anion has an overall pseudo-C2 symmetry, with the binary axis bissecting the {(WO2(H2O))2O}2+ group. In each {α-PW11O39{Ru(DMSO)3}5− entity, the ruthenium is linked to two non equivalent oxygen atoms of the lacuna of [α-PW11O39]7−,7 three DMSO ligands, and one oxo ligand of the {(WO2(H2O))2O}2+ bridge.
As depicted on Fig. 4, the environment around the tungsten atoms of the bridge, which will be referred to as Wp, is strongly distorted. Each tungsten is linked to the two remaining oxygen atoms of the lacuna of [α-PW11O39]7−, two other bridging oxygen atoms (ORu and OB), one terminal oxygen atom, and one aqua ligand. The latter is unambiguously identified by the corresponding WO bond length (Table 2), by comparison with other WVIOH2 distances reported in the literature [17].
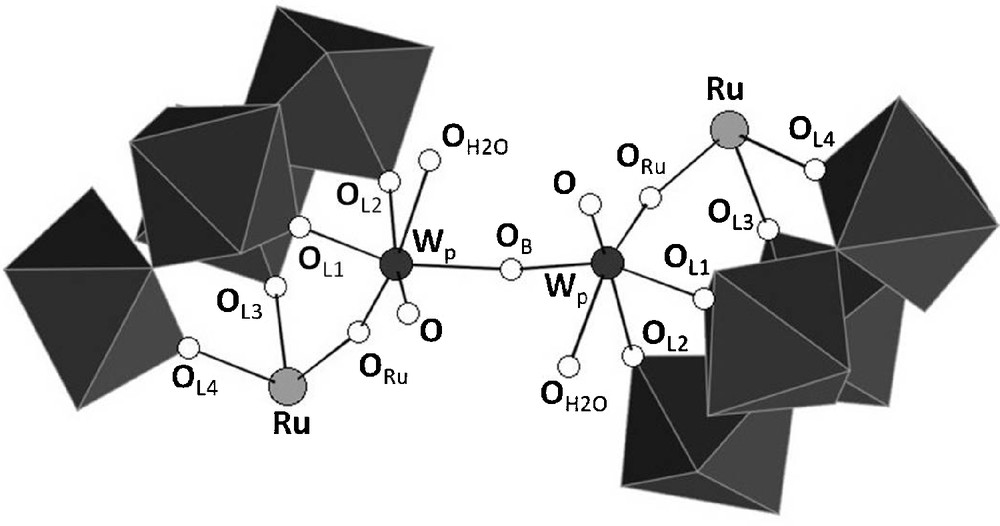
Representation of the {(WO2(H2O))2O}2+ bridge in {PW11Ru(dmso)3}2(W2O5) showing the labelling scheme used in Table 2.
The oxometallic frameworks of {PW11Ru(dmso)3}2(W2O5) and {PW11Ru(arene)}2(WO2) are clearly different, as shown in Fig. 5. Indeed, although they are both based on the association of two {α-PW11O39(RuL3)}5− subunits (L3 = η6-arene, (DMSO)3; Fig. 5 left), they contain different types of oxotungstic bridges: a small {WO2}2+ bridge is present in {PW11Ru(arene)}2(WO2), whereas a bigger {(WO2(H2O))2O}2+ bridge is found in {PW11Ru(dmso)3}2(W2O5). This leads to Ru···Ru distances between the two connected {α-PW11O39(RuL3)}5− fragments of ∼6.2 Å in {PW11Ru(benz)}2(WO2) [8a], and ∼8.7 Å in {PW11Ru(dmso)3}2(W2O5) (Table 2). The more sterically demanding {Ru(DMSO)3}2+ fragment probably does not allow the formation of [{α-PW11O39Ru(DMSO)3}2{WO2}]8−.
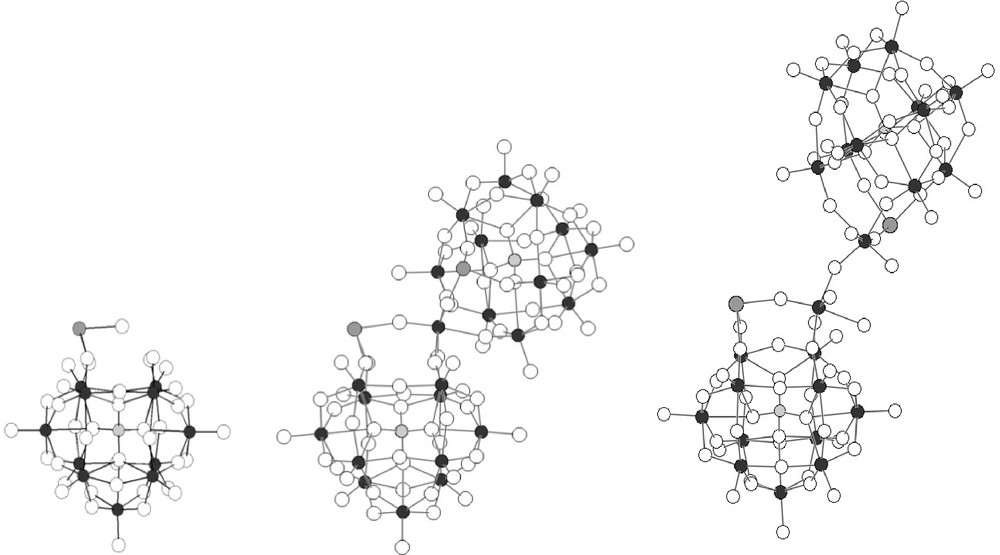
Oxometallic frameworks of PW11Ru(arene) and PW11Ru(dmso)3 (left), {PW11Ru(arene)}2(WO2) (middle) and {PW11Ru(dmso)3}2(W2O5) (right).
3.4 Solution NMR characterization of {PW11Ru(dmso)3}2(W2O5)
In solution, the 31P NMR spectrum of {PW11Ru(dmso)3}2(W2O5) only displays one signal (δ = −12.05 ppm), suggesting that the whole anion has C2 symmetry, with the C2 axis passing through the central oxygen atom of the {(WO2(H2O))2O}2+ bridge, in agreement with the solid state structure. On the 1H NMR spectrum, the observation of six singlets of equal intensity shows that the methyl groups are inequivalent, which means that there is no rotation around the RuS bond, as in the case of PW11Ru(dmso)3 [8b].
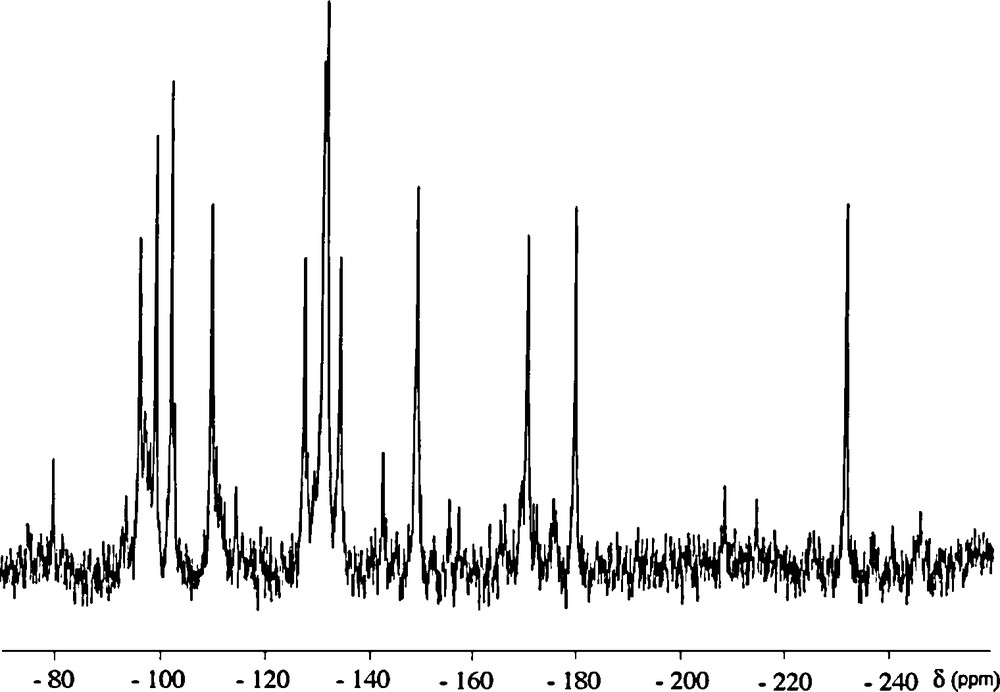
The 183W NMR spectrum of {PW11Ru(dmso)3}2(W2O5) was recorded on a solution prepared by addition of the potassium salt to a saturated aqueous solution of LiClO4, followed by filtration of precipitated KClO4. As depicted on Fig. 6, the spectrum mainly displays a twelve-line pattern, in agreement with the C2 symmetry of the whole anion8.
183W NMR chemical shifts of diamagnetic Keggin oxotungstate anions and of their lacunary derivatives are generally higher than −200 ppm [18]. This is also the case for PW11Ru(arene) and {PW11Ru(arene)}2(WO2) [8a]. Thus, in the case of {PW11Ru(dmso)3}2(W2O5), the 11 peaks with chemical shifts between −90 and −180 ppm are assigned to the tungsten atoms of the {α-PW11O39Ru(DMSO)3}5− subunits, whereas the peak at −231.5 ppm can be assigned to the tungsten atoms of the {(WO2(OH2))2O}2+ bridge. Such a shielding can be related to the significantly different local geometry of bridging Wp [19]. It is noteworthy that, in polyoxotungstates, six-coordinated tungsten centres bearing an aqua ligand generally give rise to the lowest-frequency resonances [17].
4 Conclusion
In this article, the synthesis of polyoxoanions made of two {α-PW11O39(RuL3)}5− subunits (L3 = η6-arene, (DMSO)3) connected via oxotungstic bridges has been described. This study completes our investigation of the reactivity of [α-PW11O39{RuL3(H2O)}]5− [8a,b,e], as summarized in Scheme 1.
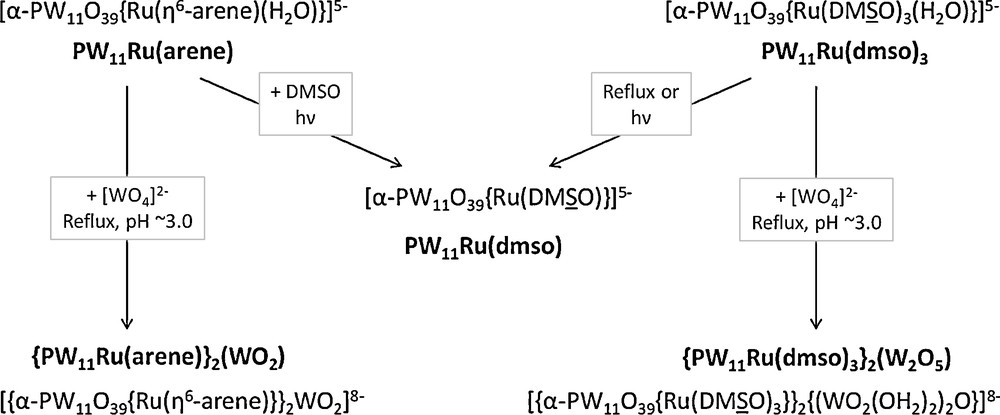
Summary of the reactivity of [α-PW11O39{RuL3(H2O)}]5− species (L3 = η6-arene, (DMSO)3).
On the one hand, an improved synthesis of [{α-PW11O39Ru(η6-arene)}2{WO2}]8− anions is proposed for arene = p-cymene. On the other hand, a new ruthenium-substituted polyoxotungstate, [{α-PW11O39Ru(DMSO)3}2{(WO2(H2O))2O}]8−, has been prepared by two alternative routes. To the best of our knowledge, it is the first polyoxotungstate that contains a bridging {W2O5}2+ unit. The difference between the two structural types is ascribed to the greater bulkiness of the {Ru(DMSO)3}2+ fragment. It should be remembered that through a computational study of the relative stabilities of the monolacunary Keggin anions and of their ruthenium-grafted derivatives, we had already underscored the influence of the steric bulk of the {RuL3(H2O)}2+ fragment [8e]. In that context, we wish to emphasize that Marie-Madeleine Rohmer was, together with Marc Bénard, the first to study the isomerism of organometallic derivatives of POMs and related compounds by computational methods [8f,g].
Acknowledgements
The authors are thankful to the CNRS and the University Pierre et Marie Curie for supporting this work.
1 In some cases, the signals located at δ(H) = 3.59 and 3.60 ppm overlap, to give only one signal of double intensity at 3.59 ppm.
2 The EDXS analysis of the precipitate leads to a n(Ru)/n(W) ratio of 12.5.
3 The minority signals observed on the 31P NMR spectrum of the mother liquor during the transformation of PW11Ru(arene) into {PW11Ru(arene)}2(WO2) could result from the degradation of part of the [PW11O39]7- anions, which leads to the release of oxotungstic fragments.
4 The corresponding 1H NMR spectrum is particularly complicated in the region in which the signals of the DMSO ligands appear.
5 At this stage, two very minor species are also detected (δ(P) = 2.27 and −13.05 ppm).
6 Signals of very weak intensity are also detected at δ(P) = 2.27, −11.21 and −12.95 ppm.
7 It is noteworthy that so far, no crystal structure of PW11Ru(dmso)3 has been obtained. For this anion, the regiospecific grafting of the ruthenium onto the lacuna has been deduced from the comparison of its IR spectrum with that of PW11Ru(pcym), and from a complete computational study of the coordination of {RuL3(H2O)}2+ (L3 = (DMSO)3, η6-arene) fragments on [PW11O39]7- [8b]. The crystal structure of {PW11Ru(dmso)3}2(W2O5) completely confirms these results. Indeed, given that {PW11Ru(dmso)3}2(W2O5) can form by reaction between two PW11Ru(dmso)3 entities, and considering the fact that the mode of grafting of the ruthenium is similar for PW11Ru(arene) and {PW11Ru(arene)}2(WO2), it appears that in PW11Ru(dmso)3, the metal is also linked to two non equivalent oxygen atoms of the lacuna.
8 A few signals of weak intensity, due to unidentified impurities, also appear on the spectrum.