

1 Introduction
The Michael reaction of indoles with α,β-unsaturated carbonyl compounds is one of the most important organic transformations and plays a key role in the total synthesis of complex natural products such as diolmycins [1] and hapalindole [2]. Many indole ligands with high-affinity for G-protein coupled receptors have been identified [3]. The hapalindole alkaloids are mostly obtained from the blue-green algae Hapalosiphon fontinalis [2] and exhibit potent antibacterial and antimycotic activity [4]. They are accessed by Friedel-Crafts Michael type addition of indole to electron-deficient olefins in the presence of either protic [5] or Lewis acids [6–8]. Existing methods of synthesis require metal catalysts and stoichiometric amounts of the reagents with cumbersome isolation procedures and longer reaction times. The extra expense and effort to process this inevitably generated toxic metal waste hinder them from being adopted for large scale synthesis. However, the acid catalyzed addition of indoles on olefin often requires careful control of the acidity to prevent side reactions such as dimerization, isomerization, and polymerization, etc. Furthermore, acidic conditions also prevent the use of acid-labile substrates. As a result, excess amounts of indoles or α,β-unsaturated carbonyl compounds are required to obtain high yields in many cases. In view of these drawbacks, the synthetic protocols utilizing new catalysts devoid of metals are becoming more important due to the growing concern for sustainable chemistry. The metal (ion)-free catalysis of organic reactions is a contemporary challenge that is just being taken up by chemists [9]. Recently, pentafluorophenylammonium triflate (C6F5N+H3.OTf̄; PFPAT) has received increasing attention as a water-tolerant Brønsted acid catalyst for organic synthesis demonstrating highly chemo-, regio- and stereoselective results [10]. Compared to conventional Lewis acids, it has advantages of water stability, recyclability, operational simplicity, strong tolerance to oxygen and nitrogen-containing substrates and functional groups, and it can often be used in catalytic amounts. In continuation of our investigations on the development of new synthetic methodologies [11], we herein report a new, convenient, mild and efficient procedure for Michael addition of indoles to α,β-unsaturated ketones in the presence of pentafluorophenylammonium triflate (PFPAT) as an effective and novel organocatalyst under mild reaction conditions (Scheme 1).

C3-Alkylation of indole with enone.
2 Results and discussion
First, we studied the reaction of 2-cyclohexen-1-one with indole (1:1 molar ratios) in order to optimize the reaction conditions with respect to temperature, time, and the molar ratio of PFPAT to the substrate. We found that 10 mol% of PFPAT was sufficient to obtain the corresponding 3-alkylated product in 95% yield within 5 h at room temperature in CH3CN using 2-cyclohexen-1-one (Table 1, entry1).
Pentafluorophenylammonium triflate catalyzed Michael addition of indoles to enones.
| Entry | Enone | Indole | Product | Yield %ref |
| 1 | 3a | 95[7d] | ||
| 2 | 3b | 90[8e] | ||
| 3 | 3c | 90[8e] | ||
| 4 | 3d | 92[7d] | ||
| 5 | 3e | 95[7d] | ||
| 6 | 3f | 90[8e] | ||
| 7 | 3g | 90[8e] | ||
| 8 | 3h | 92[8c] | ||
| 9 | 3i | 85[8c] | ||
| 10 | 3j | 92[8e] | ||
| 11 | 3k | 80[8e] | ||
| 12 | 3l | 85[8c] | ||
| 13 | 3k | 80[8e] | ||
| 14 | 3m | 90[8e] | ||
| 15 | 3n | 90[8c] |
With the reaction conditions being optimized, we extended the scope to a variety of different indoles and enones. Substituted indoles such as 5-methoxy, 5-bromo, and N-methyl derivatives participated well in this reaction (Table 1, entries 2, 6–7). In addition, various electron-deficient olefins such as benzylideneacetophenone, benzylideneacetone, methyl vinyl kethone, 1-cyclohex-2-enone and 1-cyclopent-2-enone were examined for this transformation. As shown in Table 1 (entries 1–15), all examples reacted smoothly at room temperature for 5 h, and the isolated yields were good in almost all cases (80–95%). The yields were not sensitive to the substrates employed. Nearly all reactions are similarly in isolated yields, and the reaction is clean with no formation of side products like dimers or trimers, which are normally observed by the influence of strong acids. We also examined the effect of solvents. While the best yield was obtained in acetonitrile, much lower yields were observed with other solvents (dichloromethane and toluene).
In addition, the PFPAT catalyst was easily removed from the reaction mixture after work-up, by washing with an aqueous NaOH solution to remove the CF3SO3H, followed by distillation under reduced pressure (C6F5NH2: bp 153 °C at 760 mmHg).
Mechanistically, the reaction proceeds via the activation of enone by PFPAT followed by indole addition on olefin. The resulting intermediate undergoes subsequent tautomerization to give the C3-alkylated product as shown in Scheme 2.
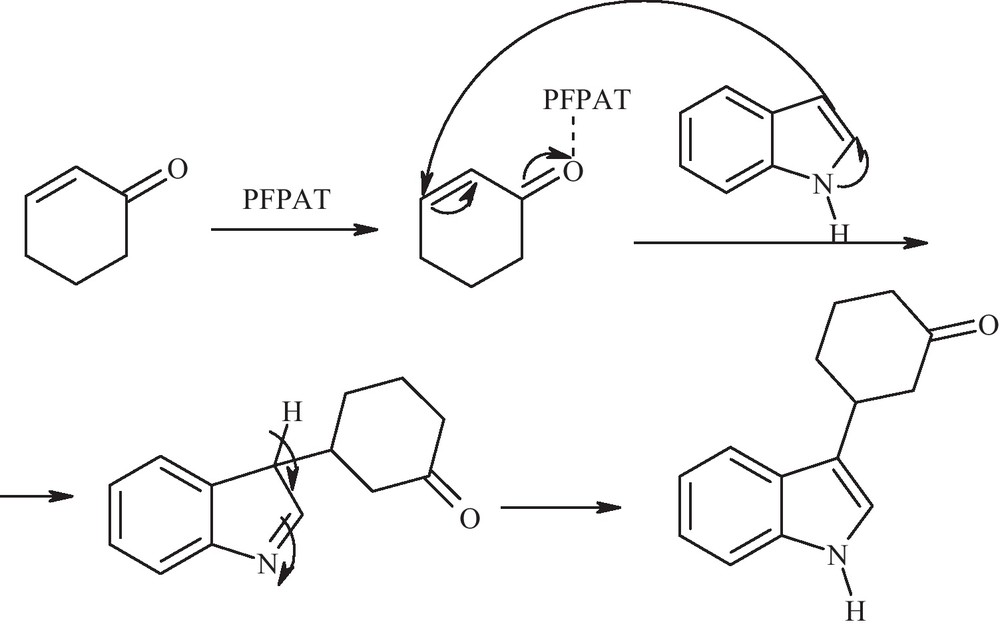
A plausible reaction mechanism.
In summary, we describe a mild, clean and efficient protocol for the conjugate addition of indoles to α,β-unsaturated ketones using pentafluorophenylammonium triflate (PFPAT) as a new organocatalyst. In contrast to the existing methods using potentially hazardous catalysts/additives, the present method offers the following competitive advantages:
- (i) PFPAT is easy to prepare from commercially available pentafluoroaniline and triflic acid;
- (ii) short reaction time;
- (iii) ease of product isolation/purification by non-aqueous work-up;
- (iv) no side reaction;
- (v) low costs and simplicity in process and handling and;
- (vi) the corresponding 3-alkylated indoles are produced by an environmentally benign process.
3 Experimental
3.1 Typical experimental procedure
A mixture of indole (1 mmol), α,β-unsaturated ketone (1 mmol), and PFPAT (0.04 g), was stirred at r.t. in a vial. After completion of the reaction as indicated by TLC, the organic phase was washed with 1 M NaOH aqueous solution (1 ml). The separated organic phase was evaporated under reduced pressure to give a crude residue, which was purified by recrystallization (solid product) or by column chromatography (hexane–EtOAc). Products were characterized by comparison of their physical and spectral data with those of authentic samples. Spectroscopic data for selected examples follow:
- • 3a: 1H NMR (400 MHz,CDCl3): δ= 1.75–2.04 (m, 3H), 2.20 (m, 1H), 2.35–2.55 (m, 2H), 2.61 (m, 1H), 2.77 (m, 1H), 3.42 (m, 1H), 6.92 (d, 1H, J = 2.0 Hz), 7.08–7.61 (m, 4H), 8.17 (br s, 1H, NH);13C NMR (100 MHz, CDCl3): δ= 24.9, 31.8, 36.0, 41.6, 48.1, 111.4, 119.1, 119.4, 119.6, 120.5, 122.2, 126.2, 136.5, 212.2; IR (KBr): 3337, 3179, 2904, 2853, 1706, 1638, 1443 cm−1; MS (EI): m/z = 213;
- • 3d: 1H NMR (400 MHz, CDCl3): δ = 3.72 (dd, J = 7.6, 7.6 Hz, 1 H); 3.81 (dd, J = 6.8, 6.8 Hz, 1 H), 5.07 (t, J = 7.2 Hz, 1 H), 6.96 (s, 1 H), 7.01 (t, J = 7.4 Hz, 1 H), 7.14 (q, J = 7.2, 7.2 Hz, 2 H), 7.23–7.31 (m, 3 H), 7.35 (d, J = 7.2 Hz, 2 H), 7.40–7.44 (m, 3 H), 7.53 (t, J = 7.4 Hz, 1 H), 7.93 (d, J = 7.2 Hz, 2 H), 7.97 (bs, 1 H); 13C NMR (100 MHz, CDCl3): δ = 198.6, 144.2, 137.0, 136.6, 133.0, 128.6, 128.4, 128.1, 127.8, 126.6, 126.3, 122.1, 121.4, 119.5, 119.4, 119.2, 111.1, 45.2, 38.1; IR (KBr): 3462, 3078, 3056, 3024, 1669, 1597, 1580, 1490, 758, 746, 703,692 cm−1; MS (EI): m/z = 325.