

{kind=link}
1 Introduction
Electrochromism is the ability of a material to show reversible changes of optical properties, such as color and transparency, as a result of an electrochemical oxidation or reduction process [1,2]. Electrochromism was discovered by studying molybdenum trioxide [3,4] but there are some other inorganic and organic compounds that exhibit electrochromic properties. Most inorganic electrochromic materials are based on transition metal oxides of W [5,6], Ir [7], V [8], Ti [9–11] and Ni [12–18]. Organic electrochromic active materials are in most cases functionalized polymers [19–23] or smaller molecules supported on a functionalized transparent substrate [24–28] such as nanostructured titania [29]. Electrochromic organic and inorganic nanocomposites are also known [30]. In spite of recent progress, the choice of materials remains relatively limited by production costs. It is challenging to find materials that can change color by an electrochemical process, which can be then externally controlled, and show good reversibility for many coloration-bleaching cycles.
Nevertheless, the continuous increase of energy prices and the on-going economic shift towards renewable energy sources and technologies has led to a renewed interest for advanced and reliable electrochromic materials. Electrochromic windows that can switch between a transparent and colored mode represent an enabling technology for advanced “intelligent building” designs having high energy and environmental impact [6,31]. Using electrochromic windows and advanced insulation materials it is possible to continuously adjust, control and optimize the thermal balance of a modern building thus significantly reducing air conditioning and heating costs.
Besides the relatively limited choice of electrochromic materials, a key issue resides in the inability of most electrochromic materials to switch between a transmissive (bleached) state and light absorbing (light dimming) state without altering the spectrum of the transmitted light. For instance, tungsten trioxide switches from transparent to deep blue [5], while nickel oxide switches from transparent to brown. This color filtering may be disturbing for some users. Devices that can reversibly dim the transmitted light throughout the entire visible spectrum without any selectivity of the transmitted wavelengths are preferred.
One proven solution is the use of several electrochromic layers of materials so that the spectrum of the transmitted light is corrected and proportional dimming can be achieved at all wavelengths. In theory, using a principle analogous to subtractive color photography, an electrochromic device consisting of three thin films that can be colored red, green and blue can achieve this task [25,32–35]. When color intensities are balanced properly, such a device can switch between a bleached state and a totally absorbing (black) state. Even more, using three independent layers it should be possible to choose any color wanted in the colored state by simply adjusting the proportions between the three colors. However, in spite of the demonstrated principle of color subtraction such devices have not been commercialized yet, mainly due to the manufacturing difficulties and price of thin-film nanostructured electrodes.
Although the electrochromic properties of titania have been known for a while [9,36], the electrochromic properties of self-organized titania nanotubes fabricated by anodization have only been relatively recently demonstrated with respect to H+ and Li+ insertion reactions [37,38]. Self-organized titania nanotubes constitute a versatile, low cost functional material whose dimensions may be tuned in accordance to the electrochemical synthesis parameters. For instance it is possible to grow continuous layers of highly parallel titania nanotubes having diameters between 40–120 nm and lengths between 200 nm and 1000 μm. These nanotubes and some of their composites show good reversibility with respect to Li+ insertion [39–44] as well as relatively fast electrochemical H+ insertion [45]. When in their pristine form and for tube lengths below 20 μm, the nanotube layers are fully transparent in the visible spectrum with little light interference effects due to their dimensions that are far from the optical wavelengths. Anodic titania nanotubes have already been used in conjunction with other electrochromic materials, such as nickel oxide [46,47], tungsten oxide [32,48], molybdenum oxide [4,49], cerium oxide [50] and niobium oxide [51] in order to improve the electrochromic properties and charge retention of titania. They seem to be an excellent cost-effective choice for the fabrication of interdigitated electrochromic devices that have been manufactured so far using precise but expensive classical photolithography methods [24]. However, in spite of the high interest in electrochromic devices based on self-assembled titania nanotubes, there are still very few studies available on this topic and many electrochemical and structural aspects remain unclear. Given that titania nanotubes represent the starting material for all these composites, we investigated the electrochromic properties of self-assembled titania nanotubes.
2 Experimental
Mirror polished Si wafers (SSP/E-Prime grade quality, Si-Tech Inc.) doped with boron (p-type dopant, ρ = 1–10 Ωcm) cut along (100) crystallographic plane were cleaned with acetone, isopropanol and methanol, in this order, for 15 min in an ultrasonic bath. The native oxide layer was then removed by dipping the Si wafers in 1%wt HF aqueous solution for 30 s followed by rinsing with water and quick drying in a stream of Ar. The cleaned and etched Si substrates were immediately fit into the PVD (Physical Vapour Deposition) chamber which was subsequently pumped down to around 10−6 mbar.
A 2 μm thick Ti layer was DC sputtered on the as prepared Si substrate using a laboratory-made PVD device. A 99.9% Ti sputtering target was used and an ultrapure Ar atmosphere was maintained inside the deposition chamber at a pressure of 8 × 10−4 mbar during deposition.
The Ti-sputtered Si wafers were cleaved in smaller rectangular pieces using a diamond tip and then fitted into a laboratory-made special electrochemical cell described elsewhere [45]. The Ti thin-film was anodized in potentiostatic regime by applying a voltage of 60 V for 20 minutes across the two electrode cell using an EG&G Parstat 2273 potentiostat. No reference electrode was used for anodization. The working electrode (Ti thin film) was placed at 3 cm from the counter electrode (a large Pt foil). The anodization bath contained 96.7% glycerol, 1.3% NH4F and 2% H2O. The bath was stirred for 24 h before the experiment in a sealed container using a magnetic stirrer in order to ensure perfect homogeneity of the bath.
This procedure leads to the formation of a self-organized array of X-ray amorphous TiO2 nanotubes (80–100 nm in diameter, 2.2 μm long, with a wall thickness of 20 nm). The Ti thin film is not completely consumed and a continuous Ti layer, approximately 750 nm thick, is left between the Si wafer and the TiO2 nanotubes. Thus, the final configuration from top to bottom is TiO2/Ti/Si. It also has to be noted that due to a known volume expansion phenomenon during electrochemical oxidation, it is possible to grow a nanotubular oxide layer up to three times thicker than the corresponding metallic Ti film that was consumed [52].
After anodization the TiO2 nanotubes were treated with 0.1% HF for 90 s in order to remove any remainder of the initial compact layer formed during the initial stages of anodization.
The electrochemical properties of TiO2 were investigated by cyclic voltammetry and galvanostatic cycling in carefully de-aerated aqueous 1M (NH4)2SO4 solutions using a Solartron SI 1287 Electrochemical Interface coupled with a SI 1260 Impedance Analyzer and an EG&G Parstat 2273 potentiostat/galvanostat. A commercial Ag/AgCl reference electrode (Schott, Germany) was used for potential measurements. The microstructure was investigated by Scanning Electron Microscopy (SEM) using a Philips ESEM 130 Scanning Electron Microscope.
In situ diffuse reflectance measurements in the visible range (from 400 to 800 nm) were performed using a Varian 300 spectrophotometer equipped with an integrating sphere DRA-CA-30I. The sample was placed inside the spectrophotometer in a special laboratory-made two electrode electrochemical cell. The working electrode consisted of the TiO2 nanotubes while the counter electrode was a Pt wire. After filling with the electrolyte (dearerated 1M (NH4)2SO4 solution) the cell was carefully placed inside the spectrophotometer and was connected to the Solartron SI 1287 Electrochemical Interface. The transparent window of the in situ electrochemical cell originated from a commercial polymethylmetacrylate (PMMA) cuvette. For in situ optical measurements only galvanostatic techniques were used, since they do not require a reference electrode, while it is easy to account for the charge passed through the electrochemical cell. All optical measurements were performed with the nanotubular layer connected to the Solartron Electrochemical Interface thus allowing for the in situ acquisition of optical data.
3 Results
This anodization procedure leads to the formation of a self-organized array of highly parallel TiO2 nanotubes which are 80–100 nm in diameter, 2.2 μm long and have a wall thickness of around 20 nm (Fig. 1). It is important to notice that the pristine titania nanotubes are amorphous. It was not possible to obtain any useful diffraction pattern for these nanotube layers even after prolonged X-ray diffraction experiments.

Top view and cross-section of titania nanotubes. The tilt angle of the cross-sectional observations was 30° and the calculated tube length is 2.2 μm. From the SEM micrographs, the porosity of the titania nanotubes is estimated at approximately 75% of the total layer volume.
The titania nanostructured electrodes studied here are recognized in the literature to belong to the second generation of self-organized anodic titania nanotubes. The relatively fast fabrication process does not allow for the clearing of the incipient compact layer that covers the titania nanotubes after fabrication. Nevertheless, after treatment with HF 0.1% very little of the initial oxide compact layer is left and most of the tubes are open, as it can be seen in Fig. 1. It should be noted that the cross-section image was obtained under a tilt angle of 30°. After perspective corrections the nanotubes length was determined to be 2.2 μm.
Cyclic voltammetry experiments performed in 1 M (NH4)2SO4 solution at sweep rates of 2, 50 and 200 mV/s (Fig. 2), revealed that the nanostructured electrode undergoes a noticeable electrochemical reaction below a potential of −0.5 V vs Ag/AgCl. The influence of the substrate is considered to be minimal and can be ignored according to the blank experiment performed on a pristine, naked Ti thin film. The ammonium sulphate solution was preferred since it can act as a buffer solution having a slightly acidic pH of 4.5. Also, the ammonium cation is considered sufficiently large to prevent any unwanted co-insertion together with the protons within the titania structure. It is known that the effective ionic radius of NH4+ is between 1.50–1.61 Å [53] while for the H+ the effective ionic radius is between 0.18–0.38 Å [54] and depends strongly on the coordination number. It is also known that some transition metal oxides may only insert protons from a solution containing Na+ and K+ cations [55]. In fact, it is this selective proton insertion that enables the good performance of the positive electrodes of commercial Leclanché primary cells. In both classical and alkaline battery systems neither the ammonia ions from the NH4Cl electrolyte nor the K+ from the KOH electrolyte are inserted in the γ-MnO2 (pirolusite) phase due to their significantly larger size [56].
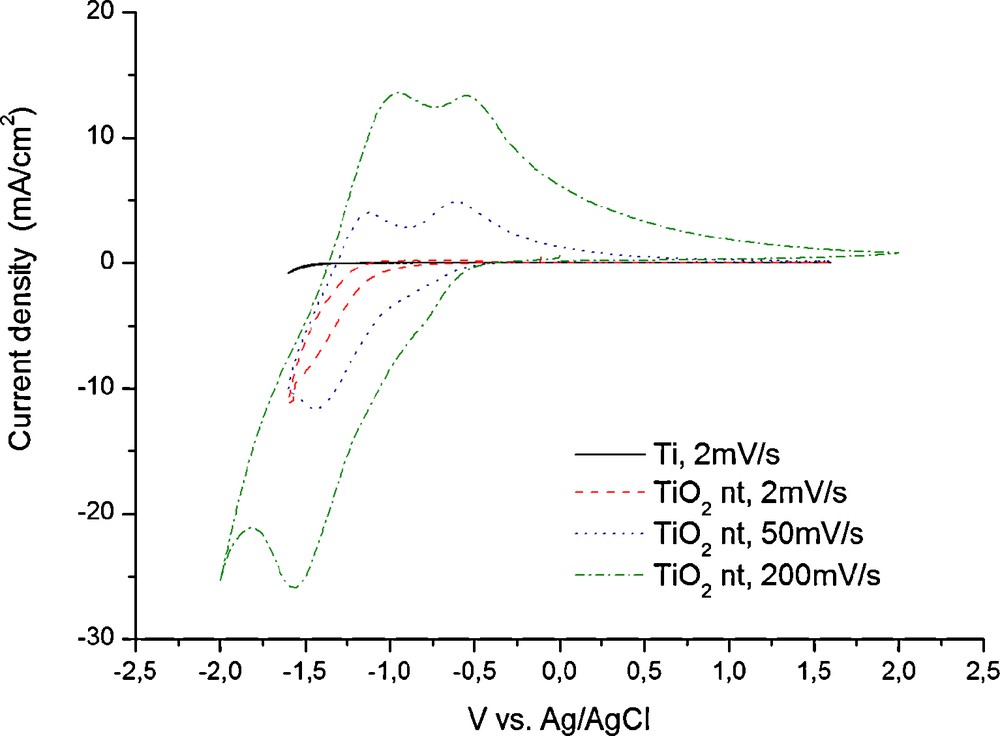
Cyclic voltammograms of titania nanotubes at different sweep rates in 1M (NH4)2SO4 aqueous solution.
As it can be seen in Fig. 2 at high sweep rates, it is possible to evidence the current peaks corresponding to electrochemical reactions at the electrode. In the cathodic direction only one peak is visible while in the anodic direction a double peak is observed. The cathodic processes can be attributed to proton insertion into the TiO2 nanotubes and the hydrogen evolution side reaction. The anodic waves correspond to the de-insertion of the protons from the structure and probably the re-oxidation of some adsorbed hydrogen that was generated during the cathodic scan. The H+ insertion reaction can be written:
| (1) |
The titania nanotube layer was submitted to electrochemical galvanostatic cycling at 0.5 mA/cm2 in order to evaluate the stability of the electrode with respect to the coloring (H+ insertion) and bleaching (H+ de-insertion) reactions. A typical potential-time profile obtained during galvanostatic cycling is shown in Fig. 3 while the coloring/bleaching electrochemical capacities are shown in Fig. 4. The potential limits were set to −1 V vs. Ag/AgCl for coloring and to +1 V vs. Ag/AgCl for the bleaching process. This relatively wide potential range used for galvanostatic cycling represents a compromise between the cycling rate, the charge injected into the material and the hydrogen evolution side reaction. These limits allow for cycling at higher currents and thus faster color switching, an important property of an electrochromic device. Within the potential limits used for galvanostatic cycling the hydrogen evolution is kept at levels that can be easily determined.
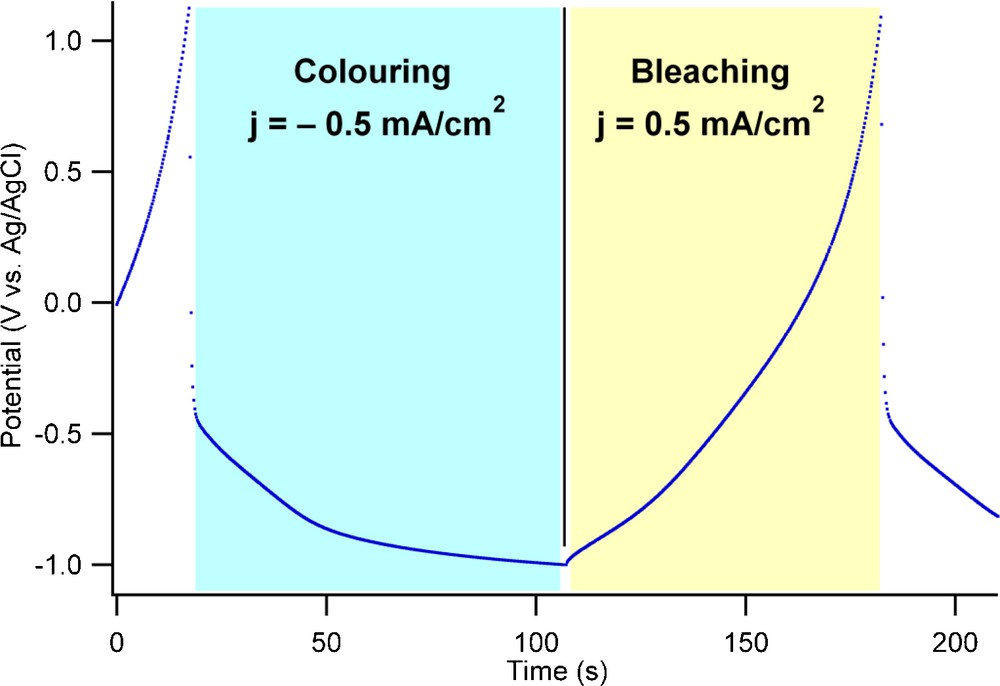
Typical potential-time profiles recorded during galvanostatic cycling of titania nanotubes corresponding to the proton insertion process (coloring) and the proton de-insertion process (bleaching) at a current density of ±0.5 mA/cm2.
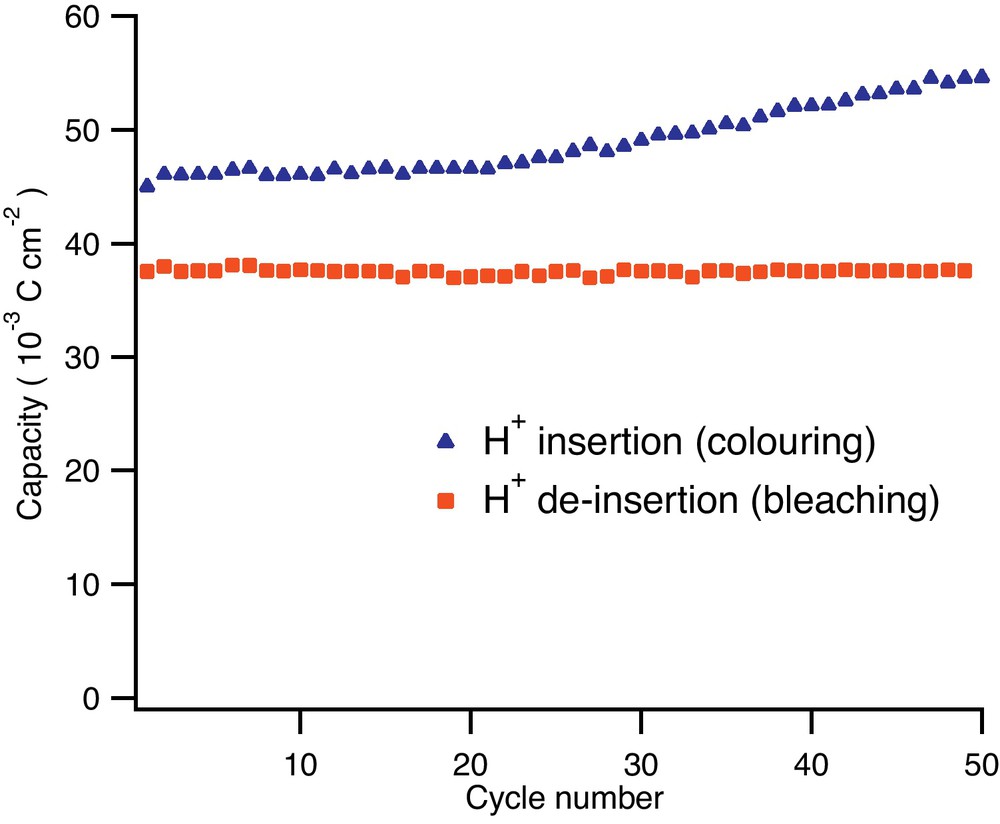
Galvanostatic cycling behaviour of titania nanotubes with respect to proton insertion/extraction reaction shows good reversibility. The difference between the forward (insertion) and reverse (de-insertion) reactions is due to H2 nucleation and evolution on the titania nanotubes. The current densities used were −0.5 mA/cm2 (insertion) and +0.5 mA/cm2 (de-insertion) while the potential limits were set to −1 V respectively +1 V vs. Ag/AgCl reference electrode.
4 Discussion
The lack of a definite plateau on the potential-time profile acquired during galvanostatic cycling is an indication that the electrochemical insertion occurs without phase change; the transition between charged and discharged states involves a state analogous to a solid solution. While protons are inserted and diffuse into the titania structure, electrons are injected into the conduction band of titania. The nanotubes become thus a degenerated semiconductor, with high electrical conductivity and consequently absorb light. From a chemical point of view this translates into having some Ti4+ ions reduced to Ti3+ ions. This behaviour is similar to the insertion of H+ into γ-MnO2 where a similar behaviour is recorded; Mn3+/Mn4+ are effectively forming a solid solution [56].
It can be observed that during galvanostatic cycling these materials show a relatively high charge imbalance of about 17% on average between the cathodic coloring and the anodic bleaching. This is due to the hydrogen evolution side reaction. However, it has to be noted that the bleaching reaction corresponding to anodic H+ de-insertion shows highly constant capacities from cycle to cycle with no capacity decay after 50 cycles. It is known that in every electrochemical insertion reaction the active materials undergo a phenomenon known as “active material breathing” which is mainly represented by volume modifications during the ionic species insertion. Such volume modification of a regular microcrystalline material will induce stress in the materials’ structure. Eventually, cracks will develop, leading to electric contact loss and hence capacity fading. In the present case, the titania nanotube layers are able to buffer the volume modification stresses and remain in contact with the current collector. This effect is quite remarkable since the cycling was performed at a rather harsh current density of 0.5 mA/cm2.
Given that hydrogen evolution seems the source of an important loss of cycling efficiency, a more detailed analysis was carried out by evaluating the derivative of the capacity (the charge spent during the coloring/bleaching processes) with respect to the electrode potential. A plot of the differential capacity against the electrode potential acquired in galvanostatic mode at −0.5 mA/cm2 is shown in Fig. 5. It can be noticed that in spite of plotting the derivative of the charge inserted in the structure the peaks corresponding to the proton insertion/de-insertion reactions are still very broad. This is another indication of a reaction taking place through the occurrence of a solid solution. It can be seen that the insertion reaction starts at around −0.4 V vs. Ag/AgCl and its rate increases steadily until around −0.8 V vs. Ag/AgCl where the hydrogen evolution reaction takes over.

Derivative of the charged/discharged capacity with respect to the insertion potential showing the particularities of the proton insertion reaction into the titania nanotubes. This data was acquired in galvanostatic mode at a current density of −0.5 mA/cm2.
The H+ de-insertion following the reversal of the current flow through the material presents an even broader peak that stretches throughout the entire potential domain. It is reasonable to assume that the capacity recorded during the bleaching process corresponds mostly to the proton de-insertion reaction. The same very broad peak is recorded during proton extraction pointing to the previously described solid solution mechanism. This mechanism is not commonly recorded for crystalline titania in equivalent conditions. For instance, Li+ insertion into anatase leads to sharp peaks; Li+ insertion exhibits a voltage plateau corresponding to equilibrium between distinct lithiated and non-lithiated phases. However, the titania nanotubes are fully amorphous; they do not exhibit any crystallinity when investigated by common diffraction techniques using either X-rays or electrons. It is then highly probable that in such disordered phases Ti3+ and T4+ ions form a solid solution when ionic species are inserted into titania. Similar observations were previously made for Li+ insertion and de-insertion from amorphous titania nanotubes used as negative electrodes in Li-ion batteries [39]. Since during proton insertion titania undergoes a transition from semiconductor to degenerated (pseudo-metallic) behaviour, it is also very likely that a very small fraction of the protons will be impossible to remove by the anodic (bleaching) process. This situation is somehow similar to the semiconductor-metallic transition of LiCoO2 when first cycled in a Li-ion battery that is never returning to its original semiconducting state once Li+ ions have been displaced from the structure [57–59].
Photographs of the samples in bleached and colored form are shown in Fig. 6; they were taken in the miniature electrochemical cell that was fit into the spectrophotometer for optical measurements. The colored sample is the result of 60 mC of charge injected into the nanotubular electrode. The photographs were taken at an angle of approximately 10° from the normal to the sample in order to avoid unwanted reflections from the Ti thin film which acts as a mirror. The red wire is connected to the titanium thin film via a protected silver paste contact visible at the bottom of the sample while the other connection is the counter electrode constituted of a Pt wire. A video showing the several coloration/bleaching cycles is available in the support section of this publication (Supporting video).

Photographs of the bleached (left) and colored (right) states. The width of the rectangular cuvette is 1 cm while the diameter of the nanotube layer switching color is 7 mm.
Reflectance data of bleached and colored titania nanotubes are shown in Fig. 7. It can be noticed that the sample gets very easily colored at the beginning of the insertion process while the electrochromic process is less efficient afterwards. The parameter that is usually used to evaluate the efficiency of an electrochromic material is the color efficiency which is defined:
| (2) |
- • is the color efficiency at a given wavelength;
- • is the variation of optical density between the colored and bleached states at the given wavelength;
- • Q is the specific area burst of charge that was used to obtain the variation of optical density.
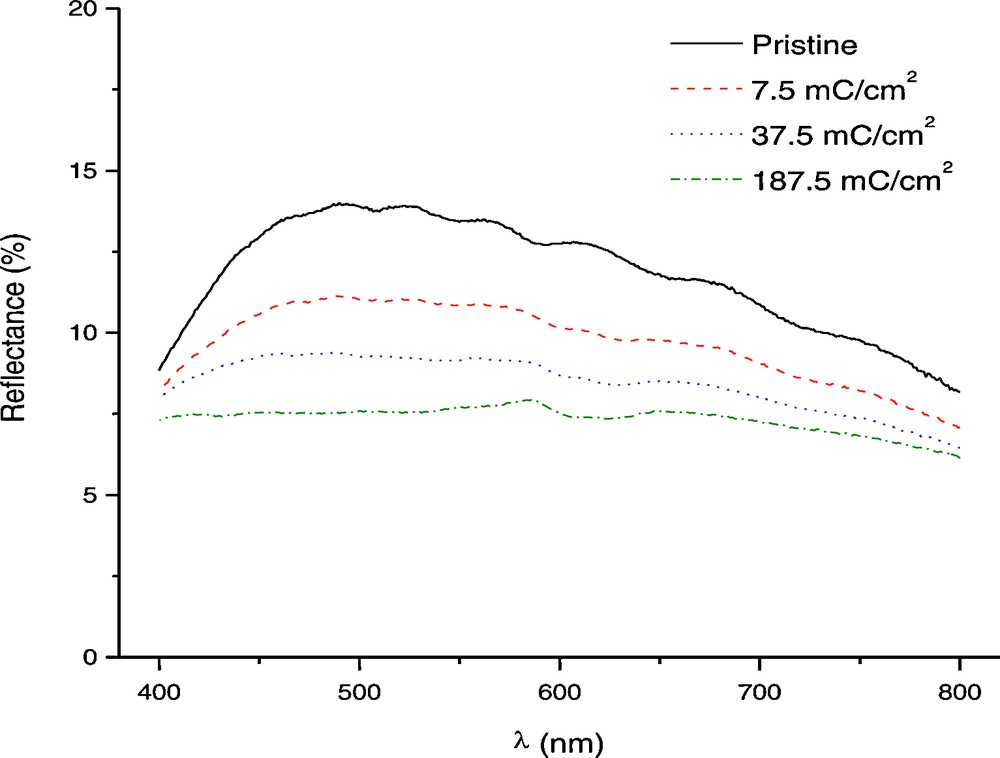
In situ reflectance measurements of colored titania nanotubes at different charge values. The colouration was obtained by applying current densities of −0.5 mA/cm2 for 15 s, 60 s and 300 s which lead to 7.5, 37.5 and 187.5 mC/cm2 respectively.
The specific burst of charge was simply calculated by multiplying the applied current by the time and then by dividing by the area of the sample. The optical data was recorded immediately after the application of the burst of charge and the variation of the optical density was calculated with respect to the bleached state. The color efficiency expresses the number of area units that can be colored with 1 coulomb of charge passed through the electrochromic material.
The maximum color efficiency has a value around 13 cm2/C (Fig. 8) when calculated for the first burst of charge of 7.5 mC/cm2. In comparison with some literature data for TiO2, this can be considered a reasonable value and comparable with published data for some other inexpensive electrochromic materials, such as tungsten oxide, and TiO2-V2O5 thin films [8]. Further calculations for higher specific area charges show that the color efficiency is decreasing due to the hydrogen evolution until they reach minimum values around 1.5 cm2/C. Practically, after the initial coloration, most of the charge is used for proton reduction into gaseous hydrogen that escapes the system. Nevertheless, titania is known to exhibit relatively low color efficiency and due to this the contrast of electrochromic materials based on titania is relatively poor.
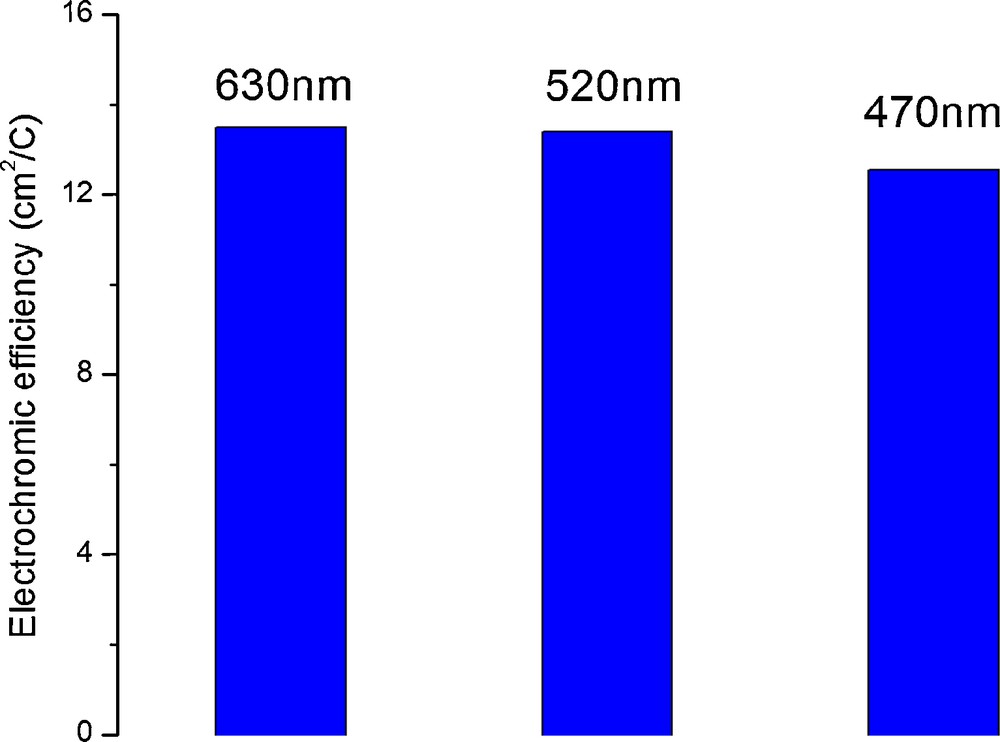
Maximum electrochromic (color) efficiencies determined for three different wavelengths corresponding to red, green and blue light. The coloration was achieved after a burst of charge of 7.5 mC/cm2 delivered at a current density of −0.5 mA/cm2.
It can also be noticed in Fig. 8 that the color efficiency is somehow higher for absorption at wavelengths in the red region of the spectrum than for light absorption in the blue region of the spectrum. In other words, the material absorbs more in the red than in the blue region. This relatively small imbalance is nevertheless enough to impress a slight bluish tint in the colored state. However, this effect is barely visible with the naked eye as it can be noticed from the photographs in Fig. 6 and from the supporting video. A non-negligible factor during reflectance measurements was the angle of incidence of the spectrophotometer beam on the sample surface. For reproducibility reasons the spectrophotometer beam was kept normal to the sample. However, it was observed that small variations from this geometry led to somehow lower reflectance values, although it was impossible with the instrumentation available to establish a correlation between the reflectance and the angle of incidence. Since the beam of light travels a longer length in the optical media when the incidence is different from normal it is expected that more light will be absorbed in this case. Some other effects may contribute as well to this apparent angle enhanced selective light absorption, for instance light scattering and other optical effects [60] onto the array of parallel nanotubes that may increase further the effective optical path travelled by photons within the material. This effect is even more pronounced as the absorbing layer is relatively thick (2.2 μm) as compared to usual electrochromic layers (20–200 nm). This effect may lead to the design of new electrochromic devices such as electrochromic static diaphragms for photographic lens and other optical systems.
5 Conclusions
The electrochromic properties of self-assembled titania nanotubes have been investigated using an in situ technique. Reflectance measurements were conducted while the coloration reaction took place inside the spectrophotometers’ cuvette.
Titania exhibits good color efficiency at the beginning of the proton insertion but the efficiency decays afterwards because of the hydrogen evolution. The materials show excellent cycling behaviour; no perceptible decay of the capacity was detected after 50 cycles. The capacity measured during bleaching is very stable, while the capacity during coloration steadily increases due to evolution of hydrogen at the nanotubular electrode. The proton insertion reaction starts around −0.5 V vs. Ag/AgCl, determined by analysis of the derivative capacity.
The observation of the dependence of the absorbance on the incidence angle is qualitative and requires further study. In spite of a somehow lower contrast, these materials may serve very well as a low-cost substrate for new devices such as interdigitated multicolor electrochromic devices.