

1 Introduction
Derivatives of 1,4-dihydropyridine (1,4-DHP) are one of the major categories of drugs with a wide variety of reported pharmacological activities, particularly due to their calcium channel modulating properties, which are considered as the basis for 1,4-DHPs’ cardioprotective [1,2], neuroprotective, anti-inflammatory, anti-ischemic, anti-diabetic, and many other actions [3]. The multi-faceted effects of 1,4-DHPs were explained by the privileged structure of the 1,4-DHP scaffolds with appropriate substituents, capable of interacting with diverse receptors and ion channels [4]. During the last decade, interest has arisen to neurotropic effects of different 1,4-DHPs, since experimental and clinical studies demonstrated their ability to penetrate the blood–brain barrier and reduce the risk of developing neurodegenerative diseases [5,6]. For instance, about a 27% reduction in Parkinson's disease risk was observed in patients who had taken centrally acting calcium channel blocking drugs [7]. These drugs (e.g., isradipine) are found to be beneficial also for the Alzheimer's disease treatment strategy [8–10]. 1,4-DHP derivatives of typical calcium antagonist [11] or atypical (neuronally non-antagonist) class [12] showed neuroprotective properties also in ischemic stroke models. Search for memory-improving/dementia-preventing effects of 1,4-DHP derivatives is still on the agenda in experimental laboratories and clinics [13,14]. Taking into account mitochondrial abnormalities as the essential cause of neurodegeneration, particular importance is related to 1,4-DHP mitochondria-protecting effects [15–18].
The present study is devoted to the design, synthesis and biological studies of novel 1,4-DHP derivatives by adding to their structures some pharmacophore moieties:
- • a pyridine substituent at position 4 (compounds 3 and 4) or N-alkyl pyridinium moiety at the position 4 (compounds 5–18);
- • a N-propargyl substituent at position 1 (compounds 2, 4, 6, 8, 12, 14, 16, 18).
The propargyl moiety is known to play an important role in providing neuro- and mitochondria-protecting properties of propargyl group-containing anti-depressants, selegiline and rasagiline [19–21]. The one and only literature source about the propargyl group-containing 1,4-DHP derivative – propargyl nitrendipine – demonstrated the store-operated channel blocking effect in leukemic HL-60 cells [22]. In turn, the 4-(N-dodecyl) pyridinium group was considered as a lipophilicity-giving moiety for the improvement of 1,4-DHP derivative penetration through the membranes and easier delivery to cellular targets. The dodecyl chain is found as an optimal-length alkyl substituent of different alkylphosphonium compounds for improving their mitochondria-targeted antioxidative properties [23].
The aim of the present study was to find the structure–activity relationships and to identify the importance of structure moieties as the above-mentioned 1,4-DHP derivatives for the future design of novel cytoprotective drugs. The studies were performed in vitro to assess:
- • the compounds’ calcium antagonistic properties in neuroblastoma SH-SY5Y cells and smooth muscle A7r5 cell line;
- • the compounds’ antioxidant activity with the phosphomolybdenum complex method;
- • the importance of the amphiphilic substituent in the 1,4-DHP ring. For that, two structurally similar compounds, 12 [the 4-(N-dodecyl) pyridinium group-containing derivative] and 4 (without 4-(N-dodecyl) pyridinium moiety), were tested and compared in isolated rat liver mitochondria;
- • in addition, the safety of compound 12 – in vivo subchronic toxicity test in mice.
2 Results and discussion
2.1 Chemistry
Different pharmacophore moieties were introduced into the 1,4-DHP cycle:
- • at position 4, the pyridinium substituent with alkyl moieties of different lengths at quaternised nitrogen atom;
- • at position 1, the propargyl moiety.
Previously, we had found a direct correlation between the length of the alkyl chain substituent at N-quaternised 4-β-pyridyl-l,4-dihydropyridines and their improved membranotropic effects, such as incorporation in the liposomal membranes and bilayer fluidity [24]. Furthermore, the dodecyl substituent was shown as the optimal-length moiety among different alkyl groups in alkyltriphenylphosphonium or alkylrhodamine derivatives for their mitochondria-targeted antioxidant activity [23]. In the present study, some representative types of compounds containing a N-ethyl, N-hexyl, N-octyl, N-dodecyl or N-hexadecyl pyridinium moiety at position 4 of the 1,4-DHP ring (compounds 5–14) were synthesised. The propargyl moiety was introduced in the 1,4-DHP molecule at position 1; better mitochondria-regulating properties were expected because this group was considered as essential in the antidepressant rasagiline molecule [20]. Moreover, N-alkylation of benzimidazole derivatives with propargyl halides led to more active compounds [25]. All 1,4-DHP derivatives were synthesised as N–H (N-unsubstituted) compounds (1, 3, 5, 7, 9, 11, 13, 15 and 17) and N-propargyl substituted compounds (2, 4, 6, 8, 10, 12, 14, 16 and 18). Our synthetic plan is shown in Scheme 1.
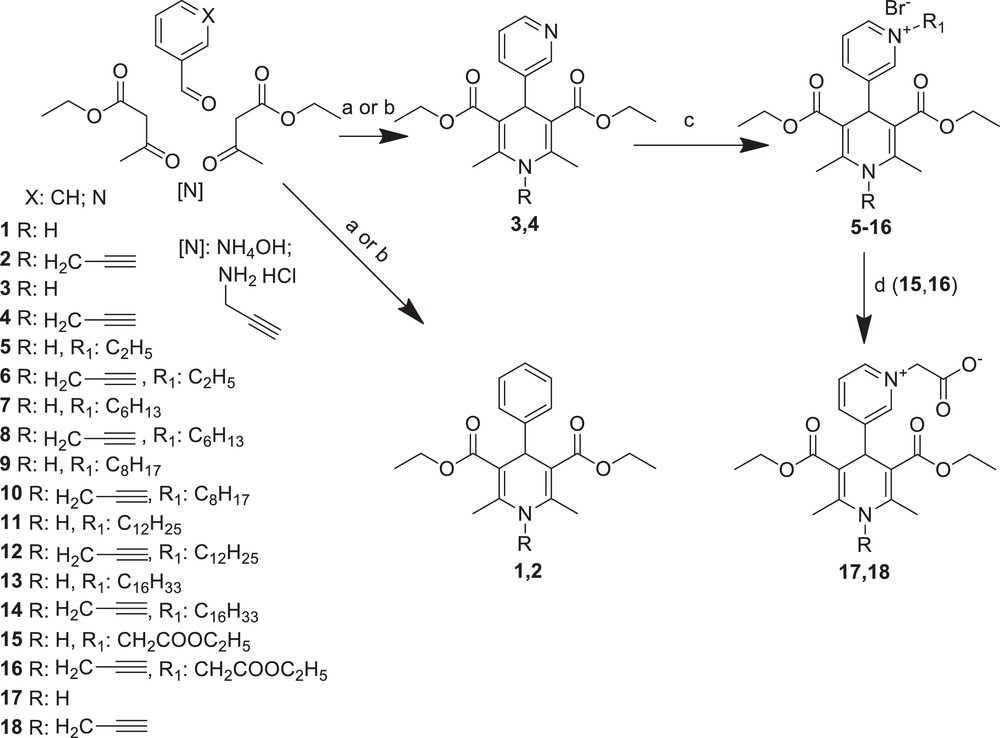
Synthesis of 1,4-DHP derivatives 1–18. Reactions conditions: (a) compounds 1 and 3: EtOH, reflux for 5–7 h; (b) compounds 2 and 4: pyridine, reflux for 5–12 h; (c) alkylbromide, acetone, reflux for 5–26 h; (d) NaOH, EtOH, water, 50 °C, 3 h.
The structures and characterisation of synthesised novel and reference compounds: N–H (N-unsubstituted) and N-propargyl-substituted 1,4-DHP derivatives are presented in Table 1.
Structures and characterisation of the studied 1,4-DHP derivatives 1–18.
| Compound | X | R | Yield (%) | Mp (°C) |
| 1 | CH | H | 92 (78[26]) | 158–160 (153[26]) |
| 2 | CH | –CH2CCH | 19 (40a) | 111–113 |
| 3 | N | H | 78 (80[27]) | 189–191 (192–194[27]) |
| 4 | N | –CH2CCH | 22 (47a) | 154–156 |
| 5 | N+–(CH2)nCH3; n = 1; Br− | H | 86 | 189–191 |
| 6 | N+–(CH2)nCH3; n = 1; Br− | –CH2CCH | 77 | 205 (decomp.) |
| 7 | N+–(CH2)nCH3; n = 5; Br− | H | 51 | 220–222 |
| 8 | N+–(CH2)nCH3; n = 5; Br− | –CH2CCH | 69 | 176–178 |
| 9 | N+–(CH2)nCH3; n = 7; Br− | H | 54 | 157–159 |
| 10 | N+–(CH2)nCH3; n = 7; Br− | –CH2CCH | 56 | 146–148 |
| 11 | N+–(CH2)nCH3; n = 11; Br− | H | 68 | 162–164 |
| 12 | N+–(CH2)nCH3; n = 11; Br− | –CH2CCH | 57 | 161–163 |
| 13 | N+–(CH2)nCH3; n = 15; Br− | H | 60 (63[28]) | 134–135 (135–136[28]) |
| 14 | N+–(CH2)nCH3; n = 15; Br− | –CH2CCH | 76 | 149–151 |
| 15 | N+–CH2COOC2H5; Br− | H | 87 (88[29]) | 210–212 (217 (decomp.)[29]) |
| 16 | N+–CH2COOC2H5; Br− | –CH2CCH | 78 | 157–159 |
| 17 | N+–CH2COO− | H | 82 (84[29]) | 128–130 (128–130[29]) |
| 18 | N+–CH2COO− | –CH2CCH | 53 | 105 (decomp.) |
a Yield of product, performing the reaction in a pressure tube as the reaction vessel.
The data in the literature reported that the synthesis of the well-known 4-phenyl- (1) and 4-pyridyl- (3) substituted Hantzsch-type 1,4-DHP is usually performed via the classical synthetic procedures; they can be obtained in good (78–80%) yields [26,27]. In our case, the 1,4-DHP derivatives 1 and 3 were synthesised with high yields, 92% and 78% respectively, via typical synthetic routes – the classical Hantzsch method [30], which involves the one-pot cyclocondensation of the corresponding aldehyde, ethyl acetoacetate and ammonia in ethanol under reflux for 5–7 h. The corresponding N-propargyl-substituted 1,4-DHP derivatives 2 and 4 were obtained by analogy with the synthesis of other N-substituted 1,4-DHP ones [31] – via modified Hantzsch-type cyclisation using propargyl amine hydrochloride as a nitrogen source instead of ammonia and pyridine as a solvent for the reaction mixture, under reflux for 5–12 h. The one and only literature data about the synthesis and studies of N-propargyl-substituted 1,4-DHPs, such as N-propargyl nitrendipine (N-propargyl substituted (RS)-ethyl methyl-2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate) demonstrated the alternative synthesis of N-substituted 1,4-DHP derivatives via N-alkylation of the corresponding N-unsubstituted 1,4-DHP derivative with propargyl bromide in the presence of NaH as a base with only 22% yield of product [22]. In our hands, the yields of synthesised N-propargyl-substituted 1,4-DHP derivatives – 2,6-dimethyl-4-phenyl-1-prop-2′-ynyl-1,4-dihydropyridine-3′,5′-dicarboxylic acid diethyl ester (2) and 2′,6′-dimethyl-1′-prop-2-ynyl-1′,4′-dihydro-[3,4′]bipyridinyl-3′,5′-dicarboxylic acid diethyl ester (4) – were 19% and 22%, respectively. Hantzsch-type cyclisation was carried out with a propargyl amine hydrochloride refluxing the reaction mixture for 12 h. Meanwhile, the yields of the desired products 2 and 4 were increased to 40% and 47%, respectively; these reactions, when performed in a pressure tube as the reaction vessel, were completed in just 5 h.
The synthesis of all the pyridinium moieties containing 1,4-DHP derivatives 5–14 were performed by alkylation of 4-pyridyl-1,4-DHP derivatives 3 or 4 with the corresponding alkyl bromides in acetone under reflux. The typical procedure for the quaternisation of pyridine derivatives has been reported by several research groups [32–34]. Reaction times and yields of the product are dependent on the structure of the alkyl halide. An excess of the alkylation agent was used to reduce the reaction time [29]. Compounds 15 and 16 were synthesised as synthones for the target products 17 and 18. Pyridinium betaines 17 and 18, synthesised in 82 and 53%, yields respectively, were obtained by treatment of the corresponding 4-(N-ethoxycarbonylmethyl) pyridinium-1,4-DHP bromides 15 and 16 with NaOH as a base, in analogy with the procedure described by our research group [29].
The structures of all newly synthesised derivatives 2, 4–12, 14, 16 and 18, and of those already reported compounds 1 [26], 3, [27], 13 [28], 15 [29] and 17 [29] were established and confirmed by 1H NMR, 13C NMR, MS and elemental analysis data. Molecular weights of 1,4-DHP derivatives measured by LC/MS technique were in good agreement with the calculated values for all the compounds. Details of the syntheses and full physically chemical characterisation for 1,4-DHPs 1–18 are given in Section 4. The purities of the studied compounds 1–18 were at least 97% according to high-performance liquid chromatography (HPLC) data.
2.2 Biology
2.2.1 Assessment of calcium channel blocking activity in SH-SY5Y and A7r5 cell lines
The influence of novel synthesised compounds on intracellular calcium [Ca2+]i concentration was assessed in both the neuroblastoma SH-SY5Y cells (containing L- and N-type Ca2+ channels) and the rat aorta smooth muscle A7r5 cells, which express functional L-type Ca2+ channels. The well-known calcium channel inhibitors – amlodipine and nimodipine – were used as the positive controls in this test.
As it is shown in Table 2, the most potent calcium antagonistic properties in SH-SY5Y cells demonstrated that the 1,4-DHP derivatives contained at position 4 of the 1,4-DHP ring the N-octyl pyridinium group (compound 10) and the N-dodecyl pyridinium group (compounds 11 and 12); these activities were comparable to that of amlodipine. Other compounds showed either weak calcium antagonism (compounds 4, 8 and 9), or this activity was not obtained at all. Calcium antagonistic properties were found to be higher in A7r5 cells, indicating that compounds predominantly target the L-type calcium channels in vascular smooth muscles. The highest activity comparable to that of nimodipine was revealed for compound 1, which is a well-known 4-phenyl-1,4-DHP derivative with calcium antagonistic properties [35]. In A7r5 cells, compounds 11 and 12 demonstrated at least 10-fold higher activity than that in SH-SY5Y cells, indicating their potential vasodilating activity, whereas compounds 2, 3 and 4 were about 3–4-fold less active than compounds 11 and 12; others lacked activity. Compounds 5, 7 and 13–18 did not show calcium channel blocking activity in none of the used cell lines.
Calcium overload-preventing activity (IC50) of novel 1,4-DHP derivatives in SH-SY5Y and A7r5 cells.
| Compound | Ca2+ antagonism, IC50 (μM) | |
| Cell | ||
| SH-SY5Y | A7r5 | |
| 1 | # | 0.0098 ± 0.0004* |
| 2 | > 100* | 2 ± 0.2* |
| 3 | # | 1.5 ± 0.2* |
| 4 | 53 ± 8* | 4.5 ± 1* |
| 5 | # | # |
| 6 | 100 ± 8* | > 100* |
| 7 | # | # |
| 8 | 35 ± 3 | # |
| 9 | 53 ± 8 | # |
| 10 | 12 ± 3 | # |
| 11 | 5 ± 0.8 | 0.7 ± 0.3* |
| 12 | 14 ± 5 | 0.6 ± 0.06* |
| 13 | # | # |
| 14 | # | # |
| 15 | # | # |
| 16 | # | # |
| 17 | # | # |
| 18 | # | # |
| Amlodipine | 11 ± 5 | 0.140 ± 0.012 |
| Nimodipine | 53 ± 3* | 0.008 ± 0.0014* |
* P < 0.05 vs. amlodipine; Student's paired t-test, with a two-tailed distribution.
Our results about compounds 1 and 3 are in good agreement with literature data, which demonstrated their calcium channel blocking activities [35,36].
The comparison of the Ca2+-channel blocking activity of structurally related compounds – N–H (N-unsubstituted) (compounds 1, 3, 5, 7, 9, 11, 13, 15, and 17) and N-propargyl-substituted (compounds 2, 4, 6, 8, 10, 12, 14, 16, and 18) 1,4-DHP derivatives shows either slightly increased blocking activity only for compounds 2, 4, 6 vs 1, 3, 5, and 7 in SH-SY5Y cells, or decreased activity for compound 2 vs compound 1 in A7r5 cells.
2.2.2 Assessment of total antioxidant activity (AOA)
In this study, the phosphomolybdenum complex method was used for the evaluation of the total antioxidant activity of the synthesised 1,4-DHP derivatives. Diludine (diethyl-1,4-dihydro-2,6-dimethyl-3,5-pyridinecarboxylate), a well-known 1,4-DHP derivative with antioxidant properties [37,38], was used as a positive control. The obtained results (Fig. 1) show that all synthesised 1,4-DHP derivatives possessed antioxidant properties. The highest activity (about 0.5 absorbance units) was evidenced for the N-unsubstituted derivatives – compounds 1 and 17, containing the 4-phenyl or N-carboxymethyl pyridinium moiety, respectively. The AOA of compounds 3, 6 and 15 was comparable to that of diludine. Other 1,4-DHP derivatives showed a lower AOA than diludine and even compounds that demonstrated a high calcium channel blocking activity (compounds 11 and 12), indicating a lack of correlation between their antioxidant and calcium antagonistic properties. We suggested that the AOA of the tested 1,4-DHP derivatives is mainly related to the ability of their N–H group to protonate/deprotonate at the phosphomolybdenum complex test system.
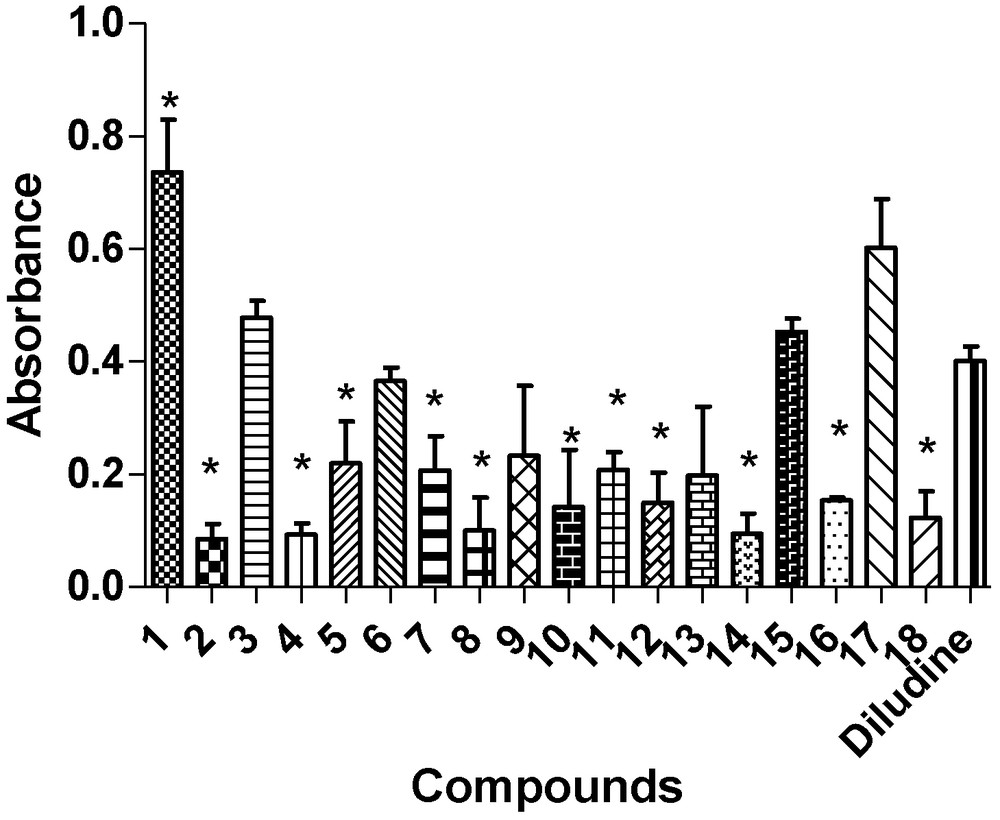
Total antioxidant activity of 1,4-DHP derivatives 1–18 (at concentration 100 μM) evaluated by the phosphomolybdenum complex method. Results are expressed as absorbance unit (at 695 nm) of sample against absorbance of a blank solution. Data are presented as a mean ± SD. *P < 0.05 vs diludine (taken as 100%), one-way ANOVA followed by Bonferroni's multiple-comparison test.
2.2.3 Influence on mitochondrial functions of rat liver isolated mitochondria
Now it is known that 1,4-DHP compounds may penetrate the blood–brain barrier [5,6]. Moreover, they are capable of penetrating cell membranes, even those of organelles, reaching mitochondria and nucleus [39]. Suggesting that an amphiphilic (N-dodecyl pyridinium) group may dramatically increase the lipophilicity of compound 12 and enhance its mitochondria-penetrating ability, we tested it in rat liver isolated mitochondria and compared their action with that of compound 4, which lacks this moiety. Both compounds also contain the N-propargyl group. We used a concentration close to IC50 obtained in the A7r5 cell line.
2.2.3.1 Effects on mitochondrial bioenergetics
Uncoupled respiration was initiated by the addition of 1 μM of p-trifluoromethoxyphenylhydrazone (FCCP). The respiratory control ratio (RCR) and ADP-to-oxygen ratio (ADP/O) were calculated. The results, presented in Table 3, show that compound 12 at concentration 1 μM did not affect mitochondrial bioenergetics; however, at higher concentration of 10 μM (about 15 times higher than IC50 in A7r5 cells), it perturbed the respiration and phosphorylation efficiency of mitochondria by affecting the permeability (integrity) of the inner mitochondrial membrane to cations, as reflected by a stimulation of states-2 and 4 respiration rate, with a parallel decrease of state-3 one (but not FCCP state) and RCR and ADP/O ratio, when glutamate/malate (Table 3) or succinate (not shown) were used as the respiratory substrates. These data indicate that the N-dodecyl group plays some role in the compound's ability to influence mitochondrial bioenergetics at higher concentrations, while compound 4 (without this structural moiety) did not show any influence on mitochondrial bioenergetics.
Effects of compounds 12 and 4 on the respiratory parameters (states 2, 3, 4, FCCP), and respiratory indices RCR and ADP/O ratio using a glutamate/malate respiratory substrate.
| Compound | Concentration (μM) | Oxygen consumption (% of control) | RCR | ADP/O | |||
| State 2 | State 3 | State 4 | State FCCP | ||||
| 12 | 0 (control) | 100.0 ± 0.0 | 100.0 ± 0.0 | 100.0 ± 0.0 | 100.0 ± 0.0 | 4.9 ± 0.5 | 3.2 ± 0.2 |
| 1 | 110.8 ± 2.9 | 93.8 ± 5.5 | 110.6 ± 3.4 | 95.2 ± 2.3 | 4.5 ± 0.6 | 3.0 ± 0.2 | |
| 10 | 157.5 ± 25** | 76.7 ± 2.7** | 149.2 ± 18.4* | 81.07 ± 4.9 | 2.5 ± 0.2** | 2.4 ± 0.1* | |
| 4 | 0 (control) | 100.0 ± 0.0 | 100.0 ± 0.0 | 100.0 ± 0.0 | 100.0 ± 0.0 | 7.9 ± 1.0 | 3.0 ± 0.05 |
| 10 | 98.3 ± 2.4 | 93.8 ± 3.9 | 115.0 ± 10.0 | 100.1 ± 7.7 | 7.2 ± 1.1 | 2.9 ± 0.13 |
* P < 0.05.
** P < 0.01.
2.2.3.2 Influence on Ca2+-induced mitochondrial pore transition (MPT)
The influences of compounds 12 and 4 were also studied on Ca2+-induced MPT to evaluate their abilities to increase the susceptibility of mitochondria to MPT induction by measuring the increase in oxygen consumption, which is a typical phenomenon that follows the induction of MPT. Mitochondria can tolerate a certain amount of Ca2+, but ultimately, their capacity of Ca2+-loading is overwhelmed and mitochondria are completely uncoupled due to a change in the inner membrane permeability. This effect is prevented by cyclosporin A (CsA), a specific inhibitor of MPT. Assays in the absence and presence of Ca2+ (100 nmol/mg protein) as well as assays in the presence of Ca2+ (100 nmol/mg protein) plus CsA (0.75 nmol/mg protein) were used as the controls for MPT induction (Fig. 2A).

A. Effects of compound 12 on MPT induction evaluated by measuring succinate (Suc)-supported mitochondrial respiration (oxygen consumption). Control assays show MPT induction by Ca2+ at a concentration of 100 nmol/mg protein and its inhibition by CsA (0.75 nmol/mg protein). B. Influence of compound 12 at concentrations 1, 5 and 10 μM (1, 5, 10, respectively) on MPT induction at Ca2+ concentration of 60 nmol/mg protein. Assay conditions: +Ca2+: presence of calcium; – Ca2+: absence of calcium. The traces in experiments A and B are representative of assays performed with three different mitochondrial preparations.
When the influence of compounds on MPT induction was tested, we used a Ca2+ concentration of 60 nmol/mg of protein that did not induce MPT by itself. The obtained results (Fig. 2B) showed that in the absence of calcium, compound 12, even at the higher concentration of 10 μM, did not induce MPT, while in the presence of calcium, this compound at concentrations 1, 5 and 10 μM significantly potentiated calcium toxic effects. For instance, at concentration 10 μM, the potentiating effect reached the control level when a concentration of 100 nmol Ca2+/mg protein was used (Fig. 2A). CsA totally blocked the potentiating effect of compound 12 (Fig. 2B, +Ca2+ + CsA + 10 μM), indicating that compound 12 has an ability to potentiate MPT induction. In contrast, compound 4 neither protected MPT nor potentiated its induction (not shown).
Therefore, mitochondrial transition pore opening is not induced by compound 12 per se, indicating the mitochondria safety. Under certain pathological conditions (in our case, the calcium ion overload), it may potentiate calcium entry into mitochondria.
2.2.3.3 Influence on mitochondrial oxidative stress
The effects of compounds 12 and 4 on mitochondrial oxidative damage were assessed by detecting mitochondrial membrane lipid peroxidation induced by the pro-oxidant pair ADP/Fe2+ by evaluating the formation of thiobarbituric acid reactive substances (TBARs). Both compounds 12 and 4 did not affect TBARs formation, indicating that they do not possess mitochondrial antioxidant properties (Table 4).
Effect of compounds 12 and 4 on membrane lipid peroxidation of rat liver mitochondria induced by the pro-oxidant pair ADP/Fe2+ by evaluating TBARs formation.
| Compound | Concentration (μM) | TBARs (nmol/mg protein,10 min) |
| 12 | 0 | 11.3 ± 0.5 |
| 1 | 11.0 ± 0.2 | |
| 10 | 11.7 ± 1.0 | |
| 20 | 10.9 ± 1.3 | |
| 50 | 11.5 ± 1.6 | |
| 100 | 12.0 ± 1.1 | |
| 4 | 0 | 11.3 ± 0.5 |
| 60 | 10.4 ± 0.5 | |
| -ADP/Fe2+ | 0.44 ± 0.25 |
2.3 Subchronic toxicity in mice for compound 12
To clarify whether the changes in mitochondrial bioenergetics caused by compound 12 may play a crucial role in cell survival, we tested this compound in vivo for its subchronic toxicity. Compound 12 was administered for 7 days as a single daily intraperitoneal dose of 100 mg/kg. Mice were observed during 10 days. No toxic symptom was observed. Therefore, it can be considered that some toxic influences found in vitro at higher concentrations are not essential for cell-integrative in vivo processes, and the compound can be considered as a cell-safe agent.
3 Conclusions
The obtained data have demonstrated that among the tested N-quaternised 4-β-pyridyl-l,4-dihydropyridines 5–14, the highest calcium channel blocking activity was shown by compounds comprising the amphiphilic group – the N-dodecyl pyridinium moiety at position 4 of the 1,4-DHP cycle (compounds 11 and 12). This activity, in smooth muscle cell line A7r5, was considerably higher than that in neuroblastoma cell line SH-SY5Y, suggesting that these compounds are predominantly targeting the L-type calcium channels. These compounds possessed low antioxidant activity if compared to 1,4-DHP derivatives 1, 3 and 17. Compound 12 at concentrations close to those of others demonstrating L-type calcium channel blocking activity did not affect mitochondrial functioning. This compound can be considered as a safe agent also in vivo (no toxicity up to 100 mg/kg). The propargyl group at position 1 of 1,4-DHP ring in general did not influence essentially the biological activity of the tested 1,4-DHP derivatives assessed in the used test systems. The compounds containing the N-dodecyl pyridinium moiety at position 4 of the 1,4-DHP cycle can be regarded as the prototype molecules for further chemical modifications and studies in animal models of cardiovascular or neurological diseases.
The structure–activity relationships of synthesised 4-pyridinium and/or N-propargyl substituted 1,4-DHP derivatives are summarised in Fig. 3.
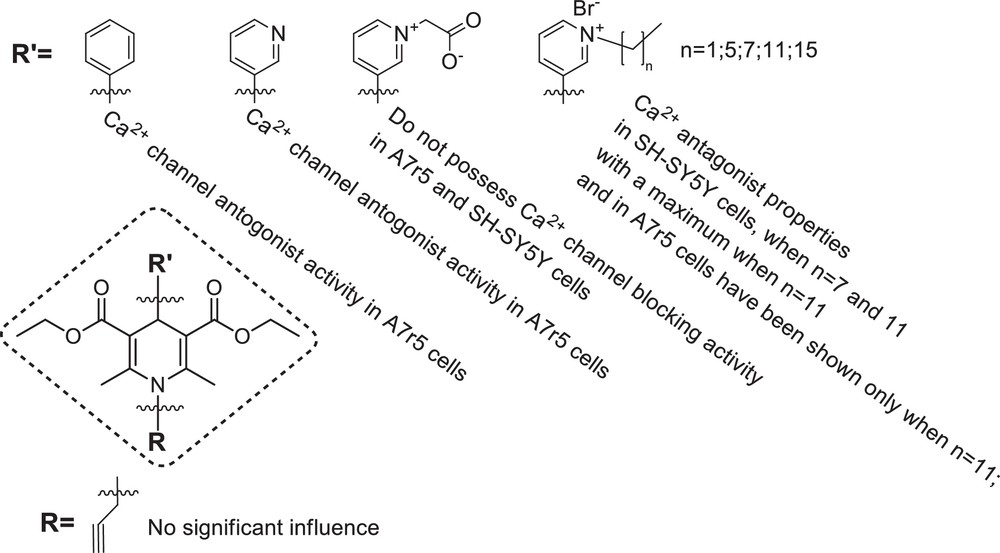
Structure–activity relationships of the studied 4-pyridinium and/or N-propargyl-substituted 1,4-DHP derivatives.
4 Experimental
4.1 Chemistry
4.1.1 General
All the reagents and solvents were purchased from commercial suppliers: Acros, Sigma–Aldrich, Alfa Aesar or Merck and used without further purification. TLC was performed on Silica gel 60 F254 Aluminium sheets 20 × 20 cm (Merck). The purities of compounds were determined by HPLC on a Waters Alliance 2695 system and Waters 2489 UV–vis detector equipped with an Alltima C18 column, 5 μm, 4.6 × 150 mm, using a gradient elution with acetonitrile/H3PO4 (0.1%) in water, at a flow rate of 1 mL/min. Peak areas were determined electronically with a Waters Empower 2 chromatography data system. 1H NMR spectra were recorded on a Varian-Mercury BB (200 MHz or 400 MHz) spectrometer. 13C NMR spectra were recorded on a Varian-Mercury BB (100 MHz) spectrometer using CDCl3 or DMSO-d6 as the solvents. The chemical shifts of the atoms are reported in parts per million (ppm) relatively to the residual signals of the solvent: CDCl3 (δ: 7.26) or DMSO-d6 (δ: 2.50) for 1H NMR spectra and CDCl3 (δ: 77.16) or DMSO-d6 (δ: 39.52) for 13C NMR. Multiplicities are abbreviated as: s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; m, multiplet; dq, double quartet; dd, double doublet; dq, double quartet; bs, broad singlet. The coupling constants are expressed in Hz. Mass spectral data were determined on a Waters Alliance 2695 Separation module connected with a Micromass 3100 mass detector (Waters) operating in the ESI positive or negative ion mode on a Xbridge C18 column (5 μm, 2.1 mm × 50 mm; Waters) using a gradient elution with acetonitrile/formic acid (0.1%) in water, at a flow rate of 0.6 mL/min. Melting points were determined on an OptiMelt (SRS Stanford Research Systems) equipment. Elemental analyses were determined on an Elemental Combustion System ECS 4010 (Costech Instruments) or on an EA 1106 (Carlo Erba Instruments).
4.1.2 Synthesis
Compounds 1, 3, 13, 15 and 17 were synthesised according to the literature. Briefly, the 2′,6′-dimethyl-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylic acid diethyl ester (1) [26,40] and 2′,6′-dimethyl-1′,4′-dihydro-[3,4′]bipyridinyl-3′,5′-dicarboxylic acid diethyl ester (3) [27] were obtained from the corresponding aldehyde, ethyl acetoacetate and ammonia in the classical Hantzsch synthesis; 3′,5′-bisethoxycarbonyl-1-hexadecyl-2′,6′-dimethyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (13) and 3′,5′-bisethoxycarbonyl-1-ethoxycarbonylmethyl-2′,6′-dimethyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (15) from 2′,6′-dimethyl-1′,4′-dihydro-[3,4′]bipyridinyl-3′,5′-dicarboxylic acid diethyl ester (3) and hexadecyl bromide [28] or ethyl bromoacetate [29], according to procedures described from Makarova et al., respectively; 1-carboxymethyl-2′,6′-dimethyl-3′,5′-bisetoxycarbonyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium (17) from equimolar amounts of 3′,5′-bisethoxycarbonyl-1-ethoxycarbonylmethyl-2′,6′-dimethyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (15) and metallic sodium in absolute ethanol, according to the procedure described by Makarova et al. [29]. 1H NMR spectra data and elemental analysis of compounds 1, 3 13, 15 and 17 were in agreement with those reported in the corresponding literature.
4.1.2.1 2′,6′-Dimethyl-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylic acid diethyl ester (1)
Yield: 92%. Mp: 158–160 °C (78%; M: 153 °C [26]. 1H NMR (400 MHz, CDCl3) δ: 1.20 (t, 6H, J = 7.4); 2.29 (s, 6H); 4.01–4.12 (m, 4H); 4.97 (s, 1H); 5.85 (bs, 1H); 7.08–7.12 (m, 1H); 7.16–7.20 (m, 2H); 7.24–7.28 (m, 2H). MS (+ESI) m/z (relative intensity) 330 ([M]+, 60%). Anal. calcd for C19H23NO4: C, 69.28; H, 7.04; N, 4.25; found: C, 69.35; H, 7.04; N, 4.19.
4.1.2.2 2′,6′-Dimethyl-1′,4′-dihydro-[3,4′]bipyridinyl-3′,5′-dicarboxylic acid diethyl ester (3)
Yield: 78%. Mp: 189–191 °C (80%; M: 192–194 °C [27]. 1H NMR (200 MHz, CDCl3) δ: 1.21 (t, 6H, J = 7.3); 2.33 (s, 6H); 4.08 (q, 4H, J = 7.3); 4.97 (s, 1H); 6.03 (bs, 1H); 7.12–7.18 (m, 1H); 7.57–7.61 (m, 1H); 8.36 (d, 1H, J = 5.1); 8.52 (s, 1H). 13C NMR (CDCl3) δ: 14.24; 19.18; 37.87 (4-C–DHP); 59.80 (3,5-OCH2); 103.13 (3,5-C–DHP); 123.20; 135.95; 143.76; 145.05; 147.00; 149.40; 167.53 (CO). MS (+ESI) m/z (relative intensity) 331 ([M]+, 100%). Anal. calcd for C18H22N2O4: C, 65.44; H, 6.71; N, 8.48; found: C, 65.40; H, 6.73; N, 8.56.
4.1.2.3 3′,5′-Bisethoxycarbonyl-1-hexadecyl-2′,6′-dimethyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (13)
Yield: 60%. Mp: 134–135 °C (63%; M: 135–136 °C [28]. 1H NMR (400 MHz, DMSO-d6) δ: 0.84 (t, 3H, J = 7.0); 1.12 (t, 6H, J = 7.0); 1.18–1.26 (m, 26H); 1.85 (quint, 2H, J = 7.0); 2.30 (s, 6H); 4.00 (dq, 4H, J = 7.0 and J = 3.5); 4.64 (t, 2H, J = 7.0); 4.98 (s, 1H); 8.05 (dd, 1H, J = 8.2 and J = 6.0); 8.35 (d, 1H, J = 8.2); 8.84 (bs, 1H); 8.92 (d, 1H, J = 6.0); 9.20 (s, 1H). 13C NMR (DMSO-d6) δ: 14.54; 14.76; 18.95; 22.70; 25.80; 28.98; 29.30; 29.36; 29.46; 29.56; 29.58; 29.61; 29.63; 29.64; 29.66; 31.50; 31.89; 38.71 (4-C-DHP); 60.03 (N+-CH2); 61.09 (3,5-OCH2); 100.14 (3,5-C-DHP); 128.68; 143.25; 143.39; 144.82; 148.20; 148.99; 166.66 (C=O). MS (+ESI) m/z (relative intensity) 555 (79Br) ([M–Br]+, 100%). Anal. calcd for C34H55N2O4Br: C, 64.24; H, 8.72; N, 4.41; found: C, 64.01; H, 8.78; N, 4.29.
4.1.2.4 3′,5′-Bisethoxycarbonyl-1-ethoxycarbonylmethyl-2′,6′-dimethyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (15)
Yield: 87%. Mp: 210–212 °C. (88%; Mp: 217 °C (decomp.) [29]. 1H NMR (400 MHz, DMSO-d6) δ: 1.10 (t, 6H, J = 7.0); 1.23 (t, 3H, J = 7.0); 2.30 (s, 6H); 3.99 (dq, 4H, J = 7.0 and J = 3.5); 4.22 (q, 2H, J = 7.0); 5.00 (s, 1H); 5.70 (s, 2H); 8.13 (dd, 1H, J = 7.8 and J = 5.9); 8.45 (d, 1H, J = 7.8); 8.84 (bs, 1H); 8.87 (d, 1H, J = 5.9); 9.21 (s, 1H). 13C NMR (DMSO-d6) δ: 13.67; 13.88; 18.12; 37.93 (4-C–DHP); 59.25 (3,5-OCH2); 60.05 (N+-…OCH2); 62.12 (N+-CH2); 99.25 (3,5-C–DHP); 127.30; 143.91; 144.31; 145.15; 147.44; 148.00; 165.78 (3,5-CO); 166.16 (N+-…C=O). MS (+ESI) m/z (relative intensity) 417 (79Br) ([M–Br]+, 100%). Anal. calcd for C22H29N2O6Br: C, 53.13; H, 5.88; N, 5.63; found: C, 52.73; H, 5.81; N, 5.47.
4.1.2.5 1-Carboxymethyl-2′,6′-dimethyl-3′,5′-bisethoxycarbonyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium (17)
Yield: 82%. Mp: 128–130 °C (84%; M: 128–130 °C [29]). 1H NMR (200 MHz, CDCl3) δ: 1.22 (t, 6H, J = 7.0); 2.28 (s, 6H); 4.06 (q, 4H, J = 7.0); 5.04 (s, 1H); 5.16 (s, 2H); 7.67 (dd, 1H, J = 8.2 and J = 5.9); 8.29 (d, 1H, J = 8.2); 8.37 (d, 1H, J = 5.9); 8.77 (s, 1H); 9.39 (bs, 1H). MS (+ESI) m/z (relative intensity) 389 ([M]+, 100%). Anal. calcd for C20H24N2O6: C, 56.69; H, 6.65; N, 6.60; found: C, 56.45; H, 6.60; N, 6.33.
4.1.2.6 General method for synthesis of compounds (2 and 4)
The solution of ethyl acetoacetate (14.2 g, 0.109 mol, 13.7 mL), propargylamine hydrochloride (5.0 g, 0.055 mol) and the corresponding aldehyde (benzaldehyde or pyridine-3-carboxaldehyde) (0.055 mol) in pyridine (15 mL) was refluxed for 5–12 h, the reaction mixture was poured in an ice/water mixture, extracted by diethyl ether (3 × 15 mL), then the organic phase was dried over Na2SO4. The solvent was evaporated in vacuo, after which the residue was recrystallized from ethyl acetate, then from ethanol, and dried in vacuo.
4.1.2.6.1 2,6-Dimethyl-4-phenyl-1-prop-2′-ynyl-1,4-dihydropyridine-3′,5′-dicarboxylic acid diethyl ester (2)
Yield: 19 or 40%. Mp: 111–113 °C. 1H NMR (200 MHz, DMSO-d6) δ: 1.14 (t, 6H, J = 7.3); 2.51 (s, 6H); 3.50 (t, 1H, J = 2.2); 4.04 (q, 4H, J = 7.3); 4.50 (d, 2H, J = 2.2); 4.93 (s, 1H); 7.10–7.20 (m, 5H). 13C NMR (CDCl3) δ: 14.22; 15.21; 36.38 (N–CH2); 38.52 (4-C–DHP); 60.21 (3,5-OCH2); 73.82 (–C); 79.48 (CH); 107.97 (3,5-C–DHP); 125.94; 127.25; 128.14; 146.11; 147.83; 167.78 (CO). MS (+ESI) m/z (relative intensity) 368 ([M]+, 30%). Anal. calcd for C22H25NO4: C, 71.91; H, 6.86; N, 3.81; found: C, 71.59; H, 6.90; N, 3.77.
4.1.2.6.2 2′,6′-Dimethyl-1′-prop-2-ynyl-1′,4′-dihydro-[3,4′]bipyridinyl-3′,5′-dicarboxylic acid diethyl ester (4)
Yield: 22 or 47%. Mp: 154–156 °C. 1H NMR (200 MHz, CDCl3) δ: 1.25 (t, 6H, J = 7.3); 2.45 (t, 1H, J = 2.2); 2.60 (s, 6H); 4.14 (q, 4H, J = 7.3); 4.32 (d, 2H, J = 2.2); 5.06 (s, 1H); 7.12 (dd, 1H, J = 8.1 and J = 5.1); 7.54 (d, 1H, J = 8.1); 8,37 (dd, 1H, J = 5.1 and J = 2.2); 8.46 (d, 1H, J = 2.2). 13C NMR (CDCl3) δ: 14.27; 16.18; 36.14 (N–CH2); 36.84 (4-C–DHP); 60.18 (3,5-OCH2); 73.93 (-C); 79.00 (CH); 106.98 (3,5-C–DHP); 123.08; 135.08; 141.37; 147.56; 148.47; 148.48; 149.30; 167.22 (CO). MS (+ESI) m/z (relative intensity) 369 ([M]+, 100%). Anal. calcd for C21H24N2O4: C, 68.46; H, 6.57; N, 7.60; found: C, 68.08; H, 6.57; N, 7.31.
4.1.2.7 General method for synthesis of compounds (5–12, 14, 16)
The corresponding alkyl (ethyl, hexyl, octyl, dodecyl or hexadecyl) bromide or ethyl bromoacetate (9.0 mmol) was added to a solution of the corresponding 2′,6′-dimethyl-1′,4′-dihydro-[3,4′]bipyridinyl-3′,5′-dicarboxylic acid diethyl ester (3) or 2′,6′-dimethyl-1′-prop-2-ynyl-1′,4′-dihydro-[3,4′]bipyridinyl-3′,5′-dicarboxylic acid diethyl ester (4) (3.0 mmol) in acetone (30 mL). The mixture was refluxed for 5–26 h, cooled to 0 °C, the resulting precipitate was filtered off, washed with cooled acetone, recrystallized from acetone, and dried in vacuo.
4.1.2.7.1 3′,5′-Bisethoxycarbonyl-1-ethyl-2′,6′-dimethyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (5)
Yield: 86%. Mp: 189–191 °C. 1H NMR (200 MHz, CDCl3) δ: 1.23 (t, 6H, J = 6.9); 1.71 (t, 3H, J = 7.3); 2.51 (s, 6H); 4.09 (q, 4H, J = 6.9); 4.90 (q, 2H, J = 7.3); 5.10 (s, 1H); 7.93 (dd, 1H, J = 8.0 and J = 5.9); 8.37 (bs, 1H); 8.39 (d, 1H, J = 8.0); 8.85 (s, 1H); 9.10 (d, 1H, J = 5.9). 13C NMR (CDCl3) δ: 14.29; 17.31; 19.48; 39.02 (4-C–DHP); 57.37 (N+–CH2); 60.23 (3,5-OCH2); 99.99 (3,5-C–DHP); 127.99; 142.10; 142.33; 144.68; 148.67; 150.14; 167.15 (CO). MS (+ESI) m/z (relative intensity) 359 (79Br) ([M–Br]+, 100%). Anal. calcd for C20H27N2O4Br: C, 54.68; H, 6.19; N, 6.38; found: C, 54.77; H, 6.17; N, 6.32.
4.1.2.7.2 3′,5′-Bisethoxycarbonyl-1-ethyl-2′,6′-dimethyl-1′-prop-2-ynyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (6)
Yield: 77%. Mp: 205 °C (decomp.). 1H NMR (400 MHz, CDCl3) δ: 1.24 (t, 6H, J = 7.0); 1.63 (t, 3H, J = 7.4); 2.51 (t, 1H, J = 2.4); 2.68 (s, 6H); 4.14 (q, 4H, J = 7.0); 4.67 (d, 2H, J = 2.4); 5.07 (q, 2H, J = 7.4); 5.14 (s, 1H); 7.95 (dd, 1H, J = 8.2 and J = 6.3); 8.42 (d, 1H, J = 8.2); 8.77 (s, 1H); 9.29 (d, 1H, J = 6.3). 13C NMR (CDCl3) δ: 9.63; 12.06; 12.50; 33.14 (N–CH2); 33.23 (4-C–DHP); 52.60 (N+–CH2); 55.96 (3,5-OCH2); 69.41 (–C); 74.79 (CH); 98.78 (3,5-C–DHP); 123.07; 137.48; 138.12; 140.01; 142.93; 146.58; 162.39 (CO). MS (+ESI) m/z (relative intensity) 397 (79Br) ([M–Br]+, 100%). Anal. calcd for C23H29N2O4Br: C, 57.87; H, 6.12; N, 5.87; found: C, 57,74; H, 6,08; N, 5.74.
4.1.2.7.3 3′,5′-Bisethoxycarbonyl-1-hexyl-2′,6′-dimethyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (7)
Yield: 51%. Mp: 220–222 °C. 1H NMR (400 MHz, DMSO-d6) δ: 0.83 (t, 3H, J = 7.0); 1.11 (t, 6H, J = 7.0); 1.16–1.26 (m, 6H); 1.85–1.90 (quint, 2H, J = 7.0); 2.30 (s, 6H); 4.09 (dq, 4H, J = 7.0 and J = 3.9); 4.66 (t, 2H, J = 7.0); 4.78 (s, 1H); 8.06 (dd, 1H, J = 8.2 and J = 5.9); 8.35 (d, 1H, J = 8.2); 8.45 (bs, 1H); 8.94 (d, 1H, J = 5.9); 9.22 (s, 1H). 13C NMR (DMSO-d6) δ: 13.86; 14.24; 18.43; 21.90; 24.92; 30.60; 30.93; 38.18 (4-C–DHP); 59.53 (3,5-OCH2); 60.56 (N+–CH2); 99.60 (3,5-C–DHP); 128.14; 142.66; 142.93; 144.34; 147.71; 148.17; 166.15 (CO). MS (+ESI) m/z (relative intensity) 415 (79Br) ([M–Br]+, 100%). Anal. calcd for C24H35N2O4Br: C, 58.18; H, 7.12; N, 5.65; found: C, 58.18; H, 7.14; N, 5.55.
4.1.2.7.4 3′,5′-Bisethoxycarbonyl-1-hexyl-2′,6′-dimethyl-1′-prop-2-ynyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (8)
Yield: 69%. Mp: 176–178 °C. 1H NMR (400 MHz, DMSO-d6) δ: 0.83 (t, 3H, J = 6.7); 1.18 (t, 6H, J = 7.1); 1.20–1.25 (m, 6H); 1.81–1.90 (m, 2H); 2.58 (s, 6H); 3.58 (s, 1H); 4.10 (q, 4H, J = 7.1); 4.60 (s, 2H); 4.66 (t, 2H, J = 6.7); 5.02 (s, 1H); 8.05 (dd, 1H, J = 7.8 and J = 5.1); 8.37 (d, 1H, J = 7.8); 8.73 (s, 1H); 8.97 (d, 1H, J = 5.1). 13C NMR (DMSO-d6) δ: 14.02; 14.37; 16.16; 22.07; 25.16; 30.80; 31.15; 36.55 (N–CH2); 37.09 (4-C–DHP); 60.36 (3,5-OCH2); 60.93 (N+–CH2); 76.69 (–C); 80.45 (CH); 103.80 (3,5-C–DHP); 128.46; 142.69; 143.34; 144.22; 146.42; 151.13; 163.30 (CO). MS (+ESI) m/z (relative intensity) 453 (79Br) ([M–Br]+, 100%). Anal. calcd for C27H37N2O4Br: C, 60.79; H, 6.99; N, 5.25; found: C, 60.59; H, 7.01; N, 5.12.
4.1.2.7.5 3′,5′-Bisethoxycarbonyl-2′,6′-dimethyl-1-octyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (9)
Yield: 54%. Mp: 157–159 °C. 1H NMR (400 MHz, DMSO-d6) δ: 0.83 (t, 3H, J = 7.0); 1.11 (t, 6H, J = 7.0); 1.16–1.27 (m, 10H); 1.87 (quint, 2H, J = 7.0); 2.30 (s, 6H); 4.00 (dq, 4H, J = 7.0 and J = 3.5); 4.65 (t, 2H, J = 7.0); 4.98 (s, 1H); 8.05 (dd, 1H, J = 8.2 and J = 6.0); 8.35 (d, 1H, J = 8.2); 8.85 (bs, 1H); 8.95 (d, 1H, J = 6.0); 9.23 (s, 1H). 13C NMR (DMSO-d6) δ: 13.44; 13.72; 17.90; 21.58; 24.75; 27.89; 27.97; 30.47; 30.63; 37.65 (4-C–DHP); 58.99 (3,5-OCH2); 60.03 (N+–CH2); 99.08 (3,5-C–DHP); 127.62; 142.15; 142.40; 143.81; 147.19; 147.94; 165.62 (CO). MS (+ESI) m/z (relative intensity) 443 (79Br) ([M–Br]+, 100%). Anal. calcd for C26H39N2O4Br: C, 59.65; H, 7.51; N, 5.35; found: C, 59.52; H, 7.50; N, 5.24.
4.1.2.7.6 3′,5′-Bisethoxycarbonyl-2′,6′-dimethyl-1-octyl-1′-prop-2-ynyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (10)
Yield: 56%. Mp: 146–148 °C. 1H NMR (400 MHz, DMSO-d6) δ: 0.84 (t, 3H, J = 7.0); 1.18 (t, 6H, J = 7.0); 1.19–1.27 (m, 10H); 1.85 (quint, 2H, J = 7.0); 2.58 (s, 6H); 3.58 (t, 1H, J = 2.0); 4.05 (dq, 4H, J = 7.0 and J = 3.0); 4.95 (d, 2H, J = 2.0); 4.65 (t, 2H, J = 7.0); 5.02 (s, 1H); 8.05 (dd, 1H, J = 8.0 and J = 6.0); 8.36 (d, 1H, J = 8.0); 8.73 (s, 1H); 8.96 (d, 1H, J = 6.0). 13C NMR (DMSO-d6) δ: 14.09; 14.30; 16.08; 22.22; 25.45; 28.54; 28.60; 31.12; 31.27; 36.46 (N–CH2); 37.02 (4-C–DHP); 60.28 (3,5-OCH2); 60.85 (N+–CH2); 76.60 (–C); 80.36 (CH); 103.75 (3,5-C–DHP); 128.39; 142.62; 143.22; 144.15; 146.39; 150.75; 166.22 (CO). MS (+ESI) m/z (relative intensity) 481 (79Br) ([M–Br]+, 100%). Anal. calcd for C29H41N2O4Br: C, 62.03; H, 7.36; N, 4.99; found: C, 61.74; H, 7.41; N, 4.77.
4.1.2.7.7 3′,5′-Bisethoxycarbonyl-2′,6′-dimethyl-1-dodecyl-1′,4′-dihydro[3,4′]bipyridinyl-1-ium bromide
Yield: 68%. Mp: 162–164 °C. 1H NMR (200 MHz, CDCl3) δ: 0.85 (t, 3H, J = 7.4); 1.21 (t, 6H, J = 7.0); 1.22–1.25 (m, 18H) overlap; 1.98 (quint, 2H, J = 7.4); 2.50 (s, 6H); 4.07 (q, 4H, J = 7.0); 4.78 (t, 2H, J = 7.4); 5.07 (s, 1H); 7.91 (dd, 1H, J = 8.2 and J = 6.3); 8.22 (d, 1H, J = 8.2); 8.48 (bs, 1H); 8.76 (s, 1H); 9.02 (d, 1H, J = 6.3). 13C NMR (CDCl3) δ: 14.07; 14.36; 19.44; 22.65; 26.03; 28.98; 29.00; 29.28; 29.31; 29.44; 29.51; 29.54; 31.84 (N+–CH2–CH2); 31.92 (4-C–DHP); 39.01 (N+–CH2); 60.21 (3,5-OCH2); 100.02 (3,5-C–DHP); 127.97; 141.81; 142.44; 144.62; 148.67; 149.91; 167.36 (CO). MS (+ESI) m/z (relative intensity) 499 (79Br) ([M–Br]+, 100%). Anal. calcd for C30H47N2O4Br: C, 62.34; H, 8.17; N, 4.83; found: C, 62.34; H, 8.22; N, 4.72.
4.1.2.7.8 3′,5′-Bisethoxycarbonyl-2′,6′-dimethyl-1-dodecyl-1′-prop-2-ynyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (12)
Yield: 57%. Mp: 161–163 °C. 1H NMR (200 MHz, CDCl3) δ: 0.90 (t, 3H, J = 6.6); 1.22–1.39 (m, 22H); 1.50–1.65 (m, 2H); 1.99–2.04 (m, 2H); 2.51 (t, 1H, J = 2.2); 2.74 (s, 6H); 4.21 (q, 4H, J = 7.3); 4.70 (d, 2H, J = 2.2); 5.03 (t, 2H, J = 6.6); 5.20 (s, 1H); 8.01 (dd, 1H, J = 8.1 and J = 5.9); 8.33 (d, 1H, J = 8.1); 8.69 (s, 1H); 8.91 (d, 1H, J = 5.9). 13C NMR (CDCl3) δ: 14.07; 14.30; 16.79; 22.63; 25.98; 29.05; 29.28; 29.32; 29.44; 29.46; 29.52; 29.54; 31.84 (N+–CH2–CH2); 32.00 (N–CH2); 37.88 (4-C–DHP); 60.70 (N+–CH2); 62.04 (3,5-OCH2); 74.12 (–C); 79.54 (CH); 103.34 (3,5-C–DHP); 127.87; 142,36; 143.24; 144.78; 147.46; 151.39; 167.13 (CO). MS (+ESI) m/z (relative intensity) 537 (79Br) ([M–Br]+, 100%). Anal. calcd for C33H49N2O4Br: C, 64.17; H, 8.00; N, 4.50; found: C, 64.31; H, 8.04; N, 4.50.
4.1.2.7.9 3′,5′-Bisethoxycarbonyl-1-hexadecyl-2′,6′-dimethyl-1′-prop-2-ynyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (14)
Yield: 76%. Mp: 149–151 °C. 1H NMR (400 MHz, DMSO-d6) δ: 0.85 (t, 3H, J = 6.6); 1.18 (t, 6H, J = 6.8); 1.19–1.30 (m, 26H); 1.85 (quint, 2H, J = 6.9); 2.58 (s, 6H); 3.57 (s, 1H); 4.09 (dq, 4H, J = 6.6 and J = 3.0); 4.59 (s, 2H); 4.64 (t, 2H, J = 6.9); 5.02 (s, 1H); 8.05 (dd, 1H, J = 7.8 and J = 5.7); 8.36 (d, 1H, J = 7.8); 8.72 (s, 1H); 8.97 (d, 1H, J = 5.7). 13C NMR (DMSO-d6) δ: 13.80; 13.96; 15.74; 21.94; 25.11; 28.25; 28.55; 28.62; 28.73; 28.82; 28.84; 28.86; 28.88; 28.89; 28.91; 30.79; 31.14; 36.12; 36.68 (4-C–DHP); 59.93 (3,5-OCH2); 60.50 (N+–CH2); 76.29 (–C); 80.12 (CH); 103.41 (3,5-C–DHP); 128.06; 142.23; 142.91; 143.76; 146.05; 150.41; 165.88 (CO). MS (+ESI) m/z (relative intensity) 593 (79Br) ([M–Br]+, 100%). Anal. calcd for C37H57N2O4Br: C, 65.96; H, 8.53; N, 4.16; found: C, 65.62; H, 8.57; N, 4.03.
4.1.2.7.10 3′,5′-Bisethoxycarbonyl-1-ethoxycarbonylmethyl-2′,6′-dimethyl-1′-prop-2-ynyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (16)
Yield: 78%. Mp: 157–159 °C. 1H NMR (400 MHz, CDCl3) δ: 1.28 (t, 6H, J = 7.0); 1.29 (t, 3H, J = 7.0); 2.46 (t, 1H, J = 1.9); 2.71 (s, 6H); 4.14 (q, 4H, J = 7.0); 4.25 (q, 2H, J = 7.0); 4.65 (d, 2H, J = 1.9); 5.20 (s, 1H); 6.35 (s, 2H); 7.88 (dd, 1H, J = 8.2 and J = 5.5); 8.48 (d, 1H, J = 8.2); 8.77 (s, 1H); 9.09 (d, 1H, J = 5.5). 13C NMR (CDCl3) δ: 14.49; 14.73; 17.18; 38.25; 38.41 (4-C-DHP); 61.14 (3,5-OCH2); 61.64 (N+–…OCH2); 63.63 (N+–CH2); 74.46 (–C); 80.25 (CH); 103.60 (3,5-C–DHP); 127.18; 144.39; 145.50; 146.07; 147.70; 152.18; 166.36 (N+–…CO); 167.40 (3,5-CO). Anal. calcd for C25H31N2O6Br: C, 56.08; H, 5.84; N, 5.23; found: C, 56.00; H, 5.78; N, 5.09.
4.1.2.7.11 1-Carboxymethyl-2′,6′-dimethyl-3′,5′-bisethoxycarbonyl-1′prop-2-ynyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium (18)
3′,5′-Bisethoxycarbonyl-1-ethoxycarbonylmethyl-2′,6′-dimethyl-1′-prop-2-ynyl-1′,4′-dihydro-[3,4′]bipyridinyl-1-ium bromide (16) (0.5 g, 0.93 mmol) was dissolved in ethanol (5 mL) and a solution of NaOH (37.2 mg, 0.93 mmol) in water (2.5 mL) was added, then, the reaction mixture was heated to 50 °C for 3 h. The reaction mixture was concentrated in vacuo till approximately half of solvent was evaporated off, after which the residue was mixed with water. The resulting precipitate was filtered off, recrystallized from water and dried in vacuo. Yield: 53% Mp: 105 °C (decomp.). 1H NMR (200 MHz, CDCl3) δ: 1.28 (t, 6H, J = 7.3); 2.49 (t, 1H, J = 2.3); 2.64 (s, 6H); 4.19 (q, 4H, J = 7.3); 4.48 (d, 2H, J = 2.3); 5.05 (s, 2H); 5.22 (s, 1H); 7.71 (dd, 1H, J = 8.0 and J = 5.9); 8.33 (d, 1H, J = 8.0); 8.42 (s, 1H); 8.56 (d, 1H J = 5.9). MS (+ESI) m/z (relative intensity) 427 ([M]+, 100%). Anal. calcd for C23H26N2O6: C, 64.78; H, 6.14; N, 6.35; found: C, 64.39; H, 6.13; N, 6.38.
4.2 Biology
4.2.1 Intracellular Ca2+ measurements
Fluo-4 NW Calcium Assay Kit was purchased from Invitrogen (Sweden), all the others from Sigma–Aldrich (St Louis, MO, USA). A7r5 (rat aorta smooth muscle cells) were obtained from the ATTC cell collection (USA), SH-SY5Y (human neuroblastoma cell line) from the European Collection of Animal Cell Cultures (ECACC, UK). The cells were grown at 37 °C in a humidified atmosphere with 5% CO2/95% air in Dulbecco's modified Eagle medium (DMEM), containing 2 mM of glutamine and supplemented with 10% fetal bovine serum. Cells were passaged once a week using 0.25% trypsin, 0.53 mM EDTA solution and grown in 75 mm2 plastic culture flasks until confluence.
For the experiment, SH-SY5Y cells were plated into 96-well plates at a density of 30,000 cells per well and incubated for 24 h. A7r5 cells were seeded into 96-well plates at a density of 10,000 cells per well and incubated for 72 h. Changes in intracellular [Ca2+]i concentration were studied using the Fluo-4 NW Calcium Assay Kit, accordingly to the manufacturer's instructions. The SH-SY5Y and A7r5 cells were loaded with Fura-4NW for 45 min. Then, the cells were pre-incubated in the dark for 15 min with the tested compounds at concentrations from 0.8 to 100 μM. The compounds were dissolved in DMSO.
Application of carbachol (100 nM) to Fura-4 loaded SH-SY5Y cells was used to stimulate changes in the intracellular [Ca2+]i concentration, whereas A7r5 cells were treated with 1.5 mM CaCl2 and KCl (50 mM) for 5 min to induce an increase in the [Ca2+]i concentration. The intracellular [Ca2+]i concentration was measured using the fluorescence spectrophotometer's (Thermo Ascient, Finland) settings appropriate for an excitation at 494 nm and an emission at 516 nm. Amlodipine, nimodipine were used as the positive control. IC50 values as mean ± SD were calculated using GraphPad Prism 3.0 software.
4.2.2 Evaluation of the total antioxidant activity
The total antioxidant activity of the synthesised 1,4-DHP derivatives was studied using a spectrophotometric quantitation of the antioxidant capacity through the formation of the phosphomolybdenum complex [41]. Stock solutions of the tested compounds and of reference diludine were prepared in ethanol with concentration 1 × 10−3 M just before use.
The reagent solution contains 0.6 M sulphuric acid, 28 mM sodium phosphate, and 4 mM ammonium molybdate solutions in water.
An aliquot of 0.3 mL of the sample solution was combined in an Eppendorf tube with 3 mL of the reagent solution. The tubes were capped and incubated in a thermal block at 95 °C for 90 min. After the samples had been cooled to room temperature, the absorbance of the sample solution of each was measured at 695 nm against a blank sample. A typical blank solution contained 3 mL of the reagent solution and the appropriate volume of ethanol as the solvent, and was incubated under the same conditions as the rest of the samples.
4.2.3 Assays in rat liver isolated mitochondria
Male Wistar rats (250–350 g) were obtained from Central Vivarium, Faculty of Medicine, University of Coimbra, Coimbra, Portugal). All the experimental procedures were performed in accordance with the guidelines of Directive86/609/EEC. “European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes” (1986) and were approved by the National Ethics Committee. All the chemicals were obtained from Sigma (St Louis, MO, USA).
Liver mitochondria were isolated by differential centrifugation according to the conventional methods. The experimental procedures were performed in accordance to those described previously [15].
Briefly, after washing, the pellet was gently re-suspended in the washing medium at a protein concentration of about 50 mg/mL. The protein content was determined by the biuret method, using bovine serum albumin as a standard.
1,4-DHP derivatives were dissolved in DMSO; concentration did no exceed 0.1% in the medium.
4.2.4 Measurement of respiratory activities
Oxygen consumption was monitored polarographically with a Clark-type electrode at 30 °C in a closed glass chamber equipped with a magnetic stirring device. Mitochondria (1 mg/mL) were incubated in a respiratory medium containing 130 mM of sucrose, 5 mM of HEPES (pH 7.2), 50 mM of KCl, 2.5 mM of K2HPO4, and 2.5 mM of MgCl2 (in the presence and absence of compounds 4 or 12) for 3 min before energisation with 10 mM glutamate/5 mM malate. State-2 respiration (consumption of oxygen in the presence of substrate); state-3 respiration (consumption of oxygen in the presence of substrate and ADP) was induced by the addition of adenosine diphosphate (ADP, 150 μM) and state-4 respiration (consumption of oxygen after ADP phosphorylation). FCCP (p-trifluoromethoxyphenylhydrazone)-stimulated respiration was initiated by the addition of 1 μM of FCCP. The respiratory control ratio (RCR), which is calculated by the ratio between states 3 and 4, is an indicator of the mitochondrial membrane integrity. The ADP/O ratio, which is expressed by the ratio between the amounts of ADP and the oxygen consumed during state-3 respiration, is an index of oxidative phosphorylation efficiency. Respiration rates were calculated assuming that the saturation of oxygen concentration was 250 μM at 30 °C, and the values are expressed in percentage of control (% of control).
4.2.5 Ca2+-induced mitochondrial membrane transition pore
Ca2+–induced MPT was evaluated by measuring changes in oxygen consumption using a Clark-type electrode. The reactions were conducted in a medium containing 200 mM of sucrose, 10 mM of MOPS–Tris (pH 7.4), 1 mM of KH2PO4, and 10 μM of EGTA, supplemented with 2 μM of rotenone, as previously described [42,43]. Mitochondria (1 mg/mL), previously incubated at 30 °C for 3 min (in the absence and presence of compounds 4 or 12), were energised with 10 mM succinate, and a single addition of Ca2+ (100 mol/mg protein) was used to induce MPT. Control assays, in both the absence and presence of Ca2+, and in the presence of Ca2+ plus 0.75 nmol/mg of protein cyclosporin A (CsA) and the compound (when necessary), were also performed. To evaluate the ability of the compounds to increase the susceptibility of mitochondria to MPT induction, we used a Ca2+ concentration (60 nmol/mg of protein) that did not induce MPT by itself.
4.2.6 Lipid peroxidation
Lipid peroxidation was determined by measuring thiobarbituric acid reactive substances (TBARs), using the routine thiobarbituric acid assay. Aliquots of mitochondrial suspensions (0.5 mL each), removed 10 min after the addition of ADP/Fe2+, were added to 0.5 mL of ice cold 40% trichloroacetic acid. Then, 2 mL of 0.67% of aqueous thiobarbituric acid containing 0.01% of 2,6-di-t-butyl-p-cresol was added. The mixtures were heated at 90 °C for 15 min, then cooled on ice for 10 min, and centrifuged at 850 g for 10 min. Controls, in the absence of ADP/Fe2+, were performed under the same conditions. The supernatant fractions were collected and lipid peroxidation was estimated spectrophotometrically at 530 nm. As blanks, we used control reactions performed in the absence of mitochondria and ADP/Fe2+. The amount of TBARs formed was calculated using a molar extinction coefficient of 1.56 × 105 mol−1 cm−1 and expressed as nmol TBARs/mg of protein.
4.2.7 Statistical analysis
The mitochondrial experiments were performed using three independent experiments with different mitochondrial preparations. The values are expressed as means ± SE. Means were compared using one-way ANOVA for multiple-comparisons, followed by Tukey's test. Statistical significance was set at P < 0.05.
4.2.8 Subchronic toxicity in mice
The test was performed in male ICR mice weighing 20 ± 2 g (obtained from the Laboratory of Experimental Animals, Riga Stradins University, Riga, Latvia).
Compound 12 was administered as a single daily intraperitoneal dose of 100 mg/kg for 7 days. Number of animals n = 10. Control group (n = 10) received a saline solution. The behaviour of the mice was observed during 10 days.
All experimental procedures were performed in accordance with the EU Directive 2010/63/EU for animal experiments and were approved by the Animal Ethics Committee of the Food and Veterinary Service (Riga, Latvia).
Acknowledgements
This study was supported by ESF project No. 2009/0217/1DP/1.1.1.2.0/09/APIA/VIAA/031; the EuroNanoMed project “CheTherDel”; Portuguese Research Council (FCT), Faculty of Medicine, Centre for Neuroscience and Cell Biology (CNC) and Marine and Environmental Research Centre (IMAR–CMA) of the University of Coimbra, Portugal.
We thank colleagues from the University of Coimbra, particularly Catarina R. Oliveira, António J. M. Moreno and Maria S. Santos for the assistance to perform mitochondrial studies. We obliged also the students from the University of Latvia: Nelda Lencberga, Marta Pavasare, Evija Sidorova, Katrina Hauka for their participation in mitochondrial assays at University of Coimbra, in a framework of ERASMUS programme, as well as Elga Poppela for technical assistance to carry out in vivo test. Also thanks to Dr. Gunars Tirzitis for fruitful discussions, Dr. Brigita Cekavicus for synthesis of reference compounds 1 and 3.