

1 Introduction
Transition-metal-catalyzed cross-coupling reaction is one of the most powerful tools in molecular synthesis [1], and plays an important role in the synthesis of many drugs, natural products, optical devices, and industrially important starting materials [2]. In this area, nowadays, palladium-catalyzed cross-coupling reactions represent over 60% of the carbon-carbon bond formation reactions used in medicinal chemistry [3]. However, the use of transition metals such as palladium raises the issue of the presence of residual contamination in the synthetic drugs or intermediates that may affect either subsequent chemical transformations or that meet government requirements about maximum ppm level of residual metal in medicinal compounds [4]. In this context, the investigation of new cross-coupling reactions catalyzed by more benign metals is highly required. In this perspective, iron is a valuable alternative as it is a cheap, abundant and ecofriendly metal. Furthermore, during the last decade, iron was intensively studied in catalytic transformations [5]. In particular, iron complexes were used as catalysts in synthetic chemistry for carbon–carbon bond formation reactions [6], oxidation [7], and reduction reactions [8–10], which offer attractive industrial possibilities in terms of sustainable chemistry.
In the field of the extensively investigated transition-metal-catalyzed cross-coupling carbon–carbon bond formation [11], the Kumada–Corriu cross-coupling reaction of aryl Grignard reagents with primary or secondary alkyl halides was mainly dominated by palladium or nickel-based catalysts [12]. In 1971, Kochi et al. first reported Fe-catalyzed cross-coupling reactions of vinyl bromides with alkyl Grignard reagents [13]. Since this pioneering contribution, numerous reports on iron-catalyzed cross-coupling have appeared in the literature [14], and it became an obvious alternative to palladium-catalyzed processes, even in total synthesis.
On another hand, N-heterocyclic carbenes (NHCs) are certainly the class of ligands that has attracted the most attention since Arduengo isolation of the first stable NHC species in 1991 [15], following the pioneering report of Wanzlick in 1968 [16]. They have become a unique class of ligands due to their electronic and steric features: NHC ligands are generally known as strong σ-donors and weak π-acceptors and have tunable steric properties. They are widely used in many areas from organocatalysis [17], biochemistry and medicine [18] to transition-metal coordination and catalysis [19]. Despite the first iron–NHC complexes reported in the 1970s [20], the organometallic chemistry of such complexes was less extensively studied than that of the other noble transition metals, but has been a growing field over the past decade [21,22]. Thus, some NHC–iron catalytic systems have shown high activities in homogeneous catalysis [23], and in particular in cross-coupling reactions [24]. In the area of NHC–ferrate complexes, imidazolium and benzimidazolium complexes have been already described and were successfully used in cross-coupling reactions [25].
In a continuation of our research on the organometallic chemistry of iron with N-heterocyclic carbenes, we report herein the synthesis and molecular structures of imidazolidinium ferrate complexes and the preliminary results on catalytic cross-coupling Kochi reactions of alkyl halides in the presence of Grignard reagents.
2 Results and discussion
2.1 Synthesis and characterization of ferrate NHC complexes
First, 1,3-disubstituted imidazolidinium salts 1a–c were prepared according to the reported procedure by reaction of the N-substituted imidazoline with an alkyl halide for 10–12 h at 80 °C in DMF [26]. Then, the reaction of FeBr2 with one equivalent of imidazolidinium salts 1a and 1b in ethanol was performed at 70 °C for 72 h. After removal of the solvent under vacuum, recrystallization from a CHCl3/Et2O mixture led to the ferrate complexes 2a and 2b, which were isolated in 39 and 42% yields, respectively, as red–orange crystals, which were air stable both in the solid state and in solution (Scheme 1). Notably, they can be dissolved in CH2Cl2, CHCl3, and DMSO, leading to a red orange solution, but they are insoluble in diethyl ether and n-hexane.
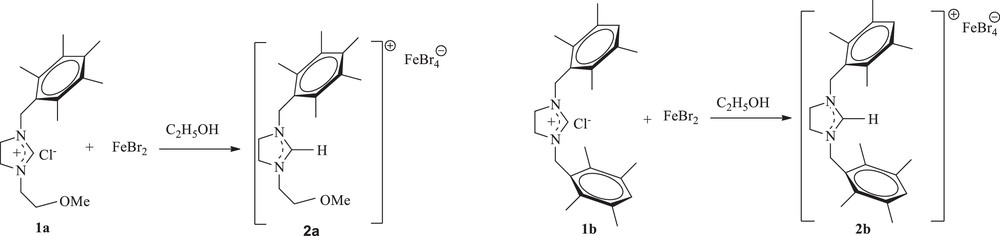
Preparation of ferrate imidazolidinium complexes.
When the reaction was performed starting from Fe(OAc)2 (2 equiv) as the iron precursor, with 1 equiv of imidazolidinium salts 1b and 1c in acetonitrile at 60 °C for 48 h, the corresponding complexes (imidazolidinium)2(Fe2OCl6) 2c and 2d were isolated in 36 and 40% yield, respectively, as white solids that are air stable both in the solid state and in solution (Scheme 2).
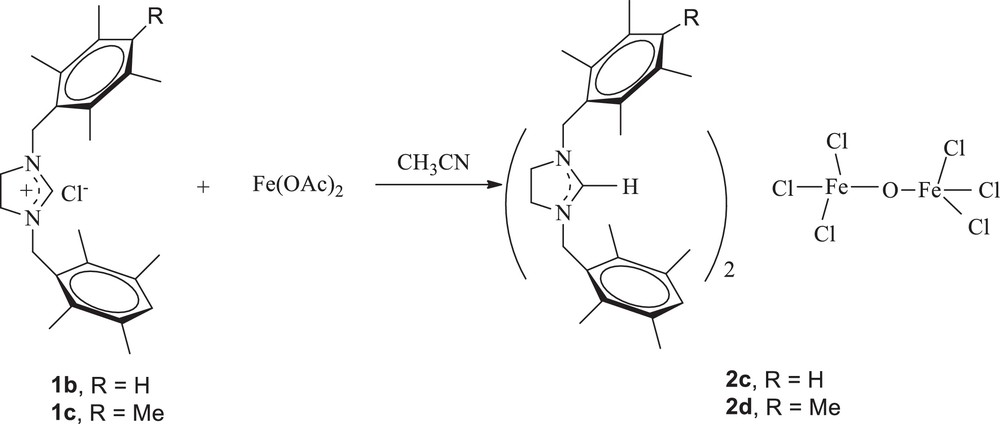
Preparation of (imidazolidinium)2(Fe2OCl6) complexes.
All obtained complexes are paramagnetic and were then characterized by elemental analysis and by crystallographic analysis. The crystals of 2a, 2c and 2d, obtained from a chloroform solution layered with diethyl ether under air atmosphere, were suitable for X-ray single-crystal diffraction analysis. The crystallography and measurement data are shown in Figs. 1 and 2. The molecular structures of the complexes, with the atom numbering scheme, are shown in Figs. 1 and 2 (Tables 1 and 2).
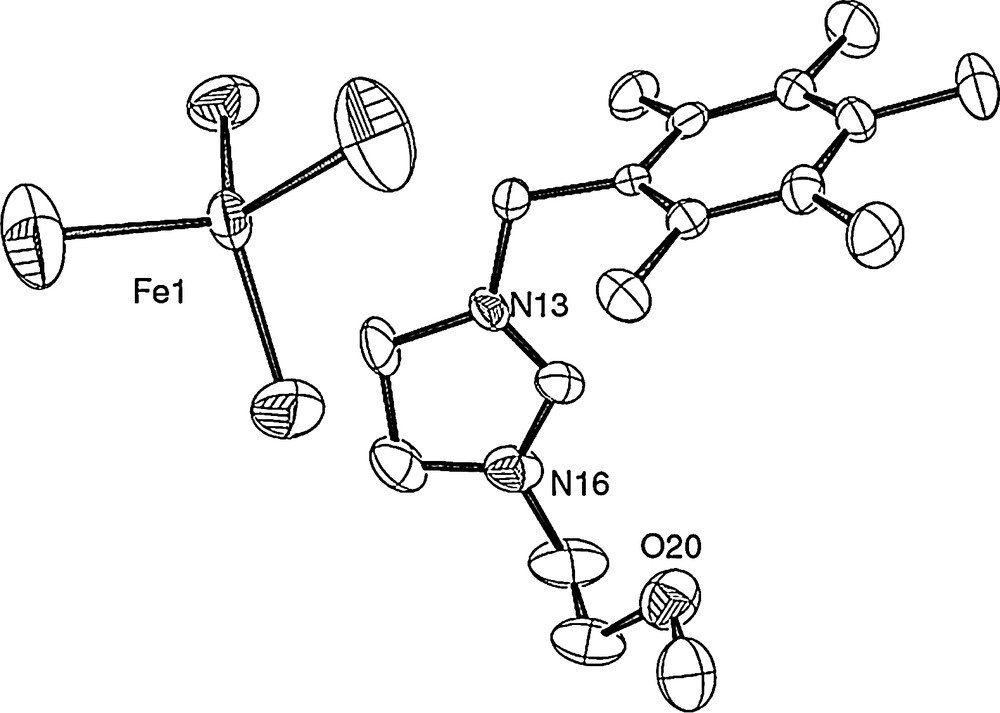
Molecular structure of 2a. Selected bond lengths (Ǻ) and angles (deg): Fe(1)–Br(1) = 2.3192(18), Fe(1)–Br(2) = 2.2960(18), Br(4)–Fe(1) = 2.3079(16), Br(3)–Fe(1) = 2.2760(19); Br(3)–Fe(1)–Br(2) = 109.14(8), Br(3)–Fe(1)–Br(4) = 107.90(7), Br(2)–Fe(1)–Br(4) = 112.84(7), Br(3)–Fe(1)–Br(1) = 111.39(8), Br(2)–Fe(1)–Br(1) = 108.08(7), Br(4)–Fe(1)–Br(1) = 107.53(7).
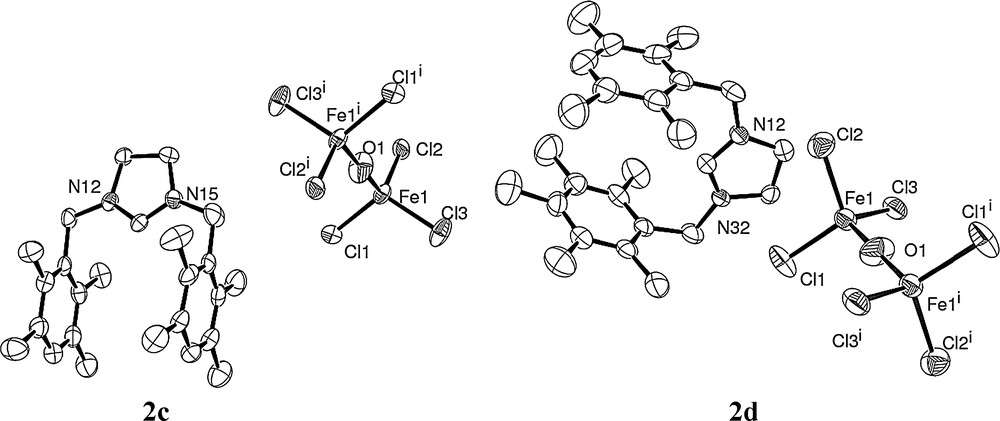
Molecular structures of 2c and 2d.
X-ray crystallographic data for complexes 2a, 2c, and 2d.
| 2a | 2c | 2d | |
| Empirical formula | C18H29Br4FeN2O | C50H66Cl6Fe2N4O | C52H74Cl6Fe2N4 O |
| fw | 664.92 | 1063.47 | 1095.55 |
| Temperature (K) | 150(2) | 150(2) | 150(2) |
| λ(Mo Ka) (Å) | 0.71073 | 0.71073 | 0.71073 |
| Cryst syst | Orthorhombic | Monoclinic | Monoclinic |
| Space group | Pcab | P21/a | P21/a |
| Unit cell dimension | |||
| a (Å) | 9.7617(3) | 13.9025(13) | 13.9317(8) |
| b (Å) | 21.8705(7) | 11.0340(12) | 10.8833(6) |
| c (Å) | 22.5179(8) | 19.1209(19) | 18.6985(12) |
| β (deg) | 90 | 110.711 | 92.533(2) |
| V (Å3) | 4807.4(3) | 2743.6(5) | 2832.4(3) |
| Z | 8 | 2 | 2 |
| Dcalc (g·cm−3) | 1.837 | 1.287 | 1.285 |
| μ (mm−1) | 7.287 | 0.858 | 0.833 |
| F(000) | 2600 | 1112 | 1152 |
| Crystal size (mm) | 0.3 × 0.25 × 0.03 | 0.16 × 0.14 × 0.07 | 0.25 × 0.24 × 0.02 |
| θ range (deg) | 3.55–27.48 | 2.92–27.48 | 2.93–27.48 |
| Number of reflns collected | 32,053 | 18,783 | 23,902 |
| Number of reflns unique, Rint | 5297, 0.0651 | 6076, 0.0768 | 6475, 0.0783 |
| Goodness-of-fit on F2 | 1.04 | 1.018 | 0.986 |
| R1, wR2 [I > 2σ(I)] | 0.0819, 0.2418 | 0.064, 0.1531 | 0.0553, 0.1244 |
| R1, wR2 (all data) | 0.1499, 0.2926 | 0.1293, 0.1924 | 0.126, 0.1554 |
Selected bond lengths (Ǻ) and angles (°) for 2c and 2d.
| 2c | 2d | |
| Fe(1)–O(1) | 1.7444(6) | 1.7411(5) |
| Fe(1)–Cl(1) | 2.2217(13) | 2.2133(11) |
| Fe(1)–Cl(2) | 2.2326(14) | 2.2165(11) |
| Fe(1)–Cl(3) | 2.2211(14) | 2.2279(11) |
| O(1)–Fe(1a) | 1.7444(6) | 1.7411(5) |
| O(1)–Fe(1)–Cl(1) | 107.08(5) | 111.01(4) |
| O(1)–Fe(1)–Cl(2) | 111.15(5) | 107.35(4) |
| Cl(1)–Fe(1)–Cl(2) | 108.91(5) | 111.08(5) |
| O(1)–Fe(1)–Cl(3) | 110.90(5) | 110.81(4) |
| Cl(1)–Fe(1)–Cl(3) | 111.04(6) | 107.49(4) |
| Cl(2)–Fe(1)–Cl(3) | 107.76(5) | 109.11(4) |
| Fe(1a)–O(1)–Fe(1) | 180 | 180.00(3) |
As shown in Fig. 1, the iron center is coordinated by four bromide atoms in a distorted tetrahedral geometry with angles at iron in the range 107.53(7)–112.84(7)°, with a mean value of 109.48° close to the ideal angle in tetrahedral structure. The Fe–Br distances are between 2.2760(19) and 2.3192(18) Å, with a mean value of 2.300 Å, in accordance with those already reported [25d–f,27]. As already described, for example, with palladium [28], or nickel [29], complex 2a results in the combination of two ion pairs, the imidazolidinium one and FeBr4−.
When using Fe(OAc)2 as the iron salt source, the reaction led also to the complexes resulting in the combination of ion pairs, two imidazolidinium moieties, and one (FeCl3)2O ion.
In both structures 2c and 2d, the iron center of the (OFe2Cl6)2−counter-ion is in a distorted tetrahedral geometry with angles at iron with mean values of 109.473° and 109.475°, respectively, close to the ideal angle in tetrahedral structure. Furthermore, bridging oxide O atom displays a linear Fe–O–Fe with an angle of 180° [30]. The Fe–Cl bond lengths range from 2.2211(14) Å to 2.2326(14) Å (mean value of 2.225 Å) for 2c and from 2.2133(11) Å to 2.2279(11) Å (mean value of 2.219 Å) for 2d. The Fe–O distances are 1.7444(6) Å and 1.7411(5) Å for 2c and 2d, respectively. Such values are in accordance with the described (OFe2Cl6)2−-type complexes. Notably, for these two complex structures, the two aromatic rings for the benzyl arm of the imidazolinium moiety are parallel, which differs from the corresponding [bis(1,3-bibenzylimidazolium)][(FeCl3)2O] [31].
2.2 Imidazolidinium ferrate-catalyzed cross-coupling reaction with Grignard reagents
The catalytic activity of anionic iron complexes 2a–2d was then examined for the coupling reaction of a simple aryl/alkyl chloride and bromide with Grignard reagents (Table 3).
Iron-catalyzed cross-coupling of aryl or alkyl halides and arylmagnesium bromidea.
| Entry | R-X | [Fe] (mol%) | Conditions | Yields (%)b | |
| 3 | 4 | ||||
| 1 | 2a (2.5) | THF, RT, 10 min | 66 | 34 | |
| 2 | 2a (2.5) | Et2O | 69 | 31 | |
| 3 | 2a (1.0) | Et2O | 82 | 18 | |
| 4 | 2a (0.5) | Et2O | 42 | 58 | |
| 5 | 2a (1.0) | Et2O | 85 | 15 | |
| 6 | 2b (1.0) | Et2O | 73 | 27 | |
| 7 | 2c (1.0) | Et2O | — | — | |
| 8 | 2d (1.0) | Et2O | — | — | |
| 9 | 2ac (1.0) | Et2O | 65 | 35 | |
| 10 | 2bc (1.0) | Et2O | 62 | 38 | |
| 11 | 2cc (1.0) | Et2O | — | 80 | |
| 12 | 2dc (1.0) | Et2O | — | 71 | |
| 13 | 2a (1.0) | Et2O, RT, 15 h | 67 | 33 | |
| 14 | 2b (1.0) | Et2O | 63 | 37 | |
| 15 | 2c (1.0) | Et2O | — | 94 | |
| 16 | 2a (1.0) | Et2O, RT, 15 h | 5 | 95 | |
| 17 | 2c (1.0) | Et2O, RT, 15 h | — | 13 | |
| 18 | 2a (1.0) | Et2O, RT, 3 h | 41 | 59 | |
| 19 | 2b (1.0) | Et2O | 37 | 63 | |
| 20 | 2c (1.0) | Et2O | — | 9 | |
| 21 | 2a (1.0) | Et2O, RT, 15 h | 32 | 68 | |
| 22 | 2b (1.0) | Et2O, RT, 15 h | 26 | 74 |
a Reaction conditions: all reactions were carried out in THF or Et2O at room temperature with 1 mmol of aryl or alkyl halides, 1.5 mmol of PhMgBr (3 M in Et2O) and iron catalyst (0.5–2.5 mol %).
b Yields were determined by GC–MS.
c Reaction with 1.5 mmol (1.5 equivalent) of p-tolylmagnesium bromide.
The cross-coupling reaction of phenyl magnesium bromide with bromocyclohexane was chosen as a test reaction because it is a common benchmark example of an aryl Grignard with a secondary alkyl halide bearing a hydrogen atom in β-position.
In THF, the reaction proceeded in 10 minutes using 2.5 mol% of complex 2a at room temperature and provided the cross-coupling product 3a in 66% yield, with 34% of biphenyl 4a resulting from the homocoupling of the Grignard reagent (entry 1). When the reaction was performed in diethyl ether under similar conditions, the cross-coupling product 3a was obtained in 69% in association with 31% of biphenyl 4a (entry 2). When decreasing the catalytic loading to 1 mol%, 82% of cyclohexylbenzene 3a can be obtained within 10 min at r.t. and 85% within 10 min at 0 °C (entries 3 and 5). The decrease in the loading to 0.5 mol had a deleterious effect as only 42% of 3a is obtained with 58% of 4a. Under the optimized conditions (1 mol% of complex, in diethyl ether at room temperature for 10 min), complex 2b led to 73% of 3a and 27% of 4a, whereas complexes 2c and 2d gave no reaction (entries 6–8). With p-tolylmagnesium bromide, to reach full conversions, the reactions were performed at room temperature for 3 h; with complexes 2a and 2b, the cross-coupling compound 3b was obtained in 65 and 62% yields, respectively, in mixture with the homocoupling bis-p-tolyl derivative 4b (entries 9 and 10). By contrast, complexes 2c and 2d led exclusively to the homocoupling product 4b in 80 and 70% yields, respectively, which shows that μ-oxido-bis[trichloroferrate(III)] complexes have a different chemoselectivity (entries 11 and 12). Cahiez has shown that alkyl Grignard reagents react with alkenyl halides in the presence of [Fe(acac)3] to give excellent yields of the cross-coupling product when NMP is used as a co-solvent [32]. Also, Fürstner has described and demonstrated the generality of the iron-catalyzed coupling reactions with alkyl Grignard reagents with the use of NMP as a co-solvent [33]. In this work, we described the cross-coupling reaction between halides (alkyl or aryl) and aryl Grignard reagents. We also tried the use of NMP as a co-solvent and observed 41% cross-coupling product and 59% homocoupling product yields with p-tolylmagnesium bromide and cyclohexylbromide with 2a catalyst. We obtained better results without NMP as a co-solvent under the same reaction conditions (entry 9).
The reaction was then done with different alkyl and aryl halides and phenylmagnesium bromide. With 2-cyclohexylmethyl bromide, the cross-coupling compound 2-phenyl-1-cyclohexylmethane 3b was obtained in 67 and 63% yields using complexes 2a and 2b, respectively, but in mixture with the biphenyl 4a, whereas complex 2c led specifically to the homocoupling derivative 3a (entries 13–15). By contrast, using aromatic halides such as p-anisyl bromide, p-anisyl iodide and p-tolyl bromide led to the homocoupling compound 4a as the major product (entries 16–22). The most efficient catalyst for the cross-coupling reaction is complex 2a, whereas complex 2c pertains to the formation of the homocoupling product 4.
3 Conclusions
In summary, two tetrabromoferrate and two μ-oxido-bis[trichloroferrate] imidazolidinium complexes have been synthesized, starting from the corresponding imidazolium bromides and iron salts. Notably, they have been structurally characterized by X-ray analysis. The two tetrabromoferrate imidazolidinium complexes 2a and 2b were pre-catalysts for the cross-coupling reaction of alkyl halides with aromatic Grignard reagents under mild conditions at room temperature, whereas complexes 2c and 2d led to homocoupling compounds.
Further investigations of these ferrate complexes containing N-heterocyclic carbene ligands are currently evaluated in our laboratories.
4 Experimental
All reactions were performed under an argon atmosphere using standard Schlenk tubes and vacuum line techniques. Solvents were distilled under argon over sodium/benzophenone (THF and Et2O) and degassed prior to use. Chemicals and solvents were purchased from Sigma Aldrich Co. (Dorset, UK). Imidazolidinium salts were synthesized in our laboratory according to the literature [26]. Elemental analyses were performed using a LECO CHNS-932 elemental analyzer. Melting points were measured in open capillary tubes with an Electrothermal-9200 melting point apparatus. All catalytic reactions were monitored on a Agilent 6890 N GC system by GC-FID with a HP-5 column of length 30 m, diameter 0.32 mm, and film thickness 0.25 μm. Column chromatography was performed using silica gel 60 (70–230 mesh). Solvent ratios are given as v/v.
4.1 General experimental procedure for the preparation of the anionic iron complexes
4.1.1 Synthesis of 2a and 2b complexes
A mixture of imidazolidinium salts (1 mmol) and FeBr2 (1 mmol) in ethanol (25 mL) was stirred at 70 °C for three days. The solvent was removed under vacuum and the crude product was crystallized from CHCl3/Et2O and yielded reddish–purple crystals suitable for elemental analyses and X-ray diffraction determination. NMR spectra could not be recorded as it is a paramagnetic compound.
4.1.1.1 Complex 2a
Yield after recrystallization: 259 mg (39%), m.p.: 123–124 °C, m/z 289.18 [M+] calculated for C18H29N2O. Anal. calcd for C18H29N2OBr4Fe: C, 32.52; H, 4.40; N, 4.21, found: C, 32.60; H, 4.51; N, 4.25%.
4.1.1.2 Complex 2b
Yield after recrystallization: 310 mg (42%), m.p.: 171–172 °C, m/z 363.4 [M+] calculated for C25H35N2. Anal. calcd for C25H35N2Br4Fe: C, 40.63; H, 4.77; N, 3.79, found: C, 40.72; H, 4.71; N, 3.84%.
4.1.2 Synthesis of 2c and 2d complexes
A mixture of imidazolidinium salts (2 mmol) and Fe(OAc)2 (1 mmol) in dry acetonitrile (25 mL) was stirred at 60 °C for 2 days. The solvent was removed under vacuum and the crude product was crystallized in the air from CHCl3/Et2O and yielded yellow crystals suitable for elemental analyses and X-ray diffraction determination. NMR spectra could not be recorded as it is a paramagnetic compound.
4.1.2.1 Complex 2c
Yield after recrystallization: 384 mg (36%), m.p.: 298–299 °C, m/z 363.3 [M+] calculated for C25H35N2. Anal. calcd for C50H70N4Cl6Fe2O: C, 56.25; H, 6.61; N, 5.25, found: C, 56.31; H, 6.67; N, 5.18%.
4.1.2.2 Complex 2d
Yield after recrystallization: 438 mg (40%), m.p.: 285–286 °C, m/z 377.5 [M+] calculated for C26H37N2. Anal. calcd for C52H74N4Cl6Fe2O: C, 57.01; H, 6.81; N, 5.11, found: C, 57.13; H, 6.77; N, 5.20%.
4.2 General method for Grignard reagents cross-coupling with aryl/alkyl halides
Under a nitrogen atmosphere and at room temperature, 1.0 mmol of aryl/alkyl halides, iron catalyst (0.5–2.5 mol %) and a solution of 1.5 mmol of phenylmagnesium bromide (3 M solution in Et2O) or p-tolylmagnesium bromide (1 M in THF) were added in THF or Et2O (3 mL) as the solvent. The mixture was stirred at room temperature or 60 °C for 10 min–48 h. The reaction mixture was quenched with H2O (5 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic layer was dried over anhydrous MgSO4 and evaporated under reduced pressure. The crude material was purified by column chromatography to afford the desired coupling product in high purity. 1H NMR and GC–MS were consistent with the known material.
Acknowledgments
This work was supported financially by the Technological and Scientific Research Council of Turkey (TUBİTAK), and the CNRS (France) [project 210T051] and the İnönü University Research Fund (2013/170). CD and JBS are grateful to the “Université de Rennes-1”, “Rennes Métropole” and the French “Ministère de l’Enseignement supérieur et de la Recherche” for support.
Vous devez vous connecter pour continuer.
S'authentifier