

1 Introduction
The π-conjugated molecules have been drawing broad research attention because of their practical applications in device technology such as solar cells [1,2]. In the field of organic solar cells, conjugated molecules are a subject of an increasing interest in recent years because of their advantages of light weight, low cost, and potential to make flexible photovoltaic devices in comparison with the traditional silicon-based solar cells.
Dye-sensitized solar cells (DSSCs) reported by O'Regan and Grätzel in 1991 attracted attention of the scientific community as new devices for solar cell application. These solar cells are composed of wide gap semiconductors (such as TiO2, ZnO, and so on) having a high surface area on which is adsorbed a dye (such as a ruthenium complex or an organic dye). The cell also contains a redox couple electrolyte (typically I−/I3−) [1,3], which is in contact with an electrode (usually platinum). In the excited state (D∗), after the absorption of light, the dye injects an electron into the conduction band of the semiconductor. The oxidized dye is reduced by a reducing agent (I−), the latter is regenerated against the electrode [4]. In the end, no species are consumed or formed during operation of the cell. So, there is appearance of a photocurrent.
The semiconductor used must have a wide gap to have no overlap between the absorption and the solar spectrum. The most widely used semiconductor is titanium oxide (TiO2) in the anatase form. For a high power conversion efficiency, the DSSCs must have the following characteristics: the HOMO (highest occupied molecular orbital) level of dyes must be down than the HOMO level of electrolyte for making easy the reception of the electron from a redox electrolyte pair (I−/I3−), the LUMO (lowest unoccupied molecular orbital) must be located on the conduction band of TiO2 for facilitating the injection phenomenon into its conduction band.
The dyes having a donor–π–acceptor (D–π–A) structure are the most studied currently, which exhibit several advantages such as low cost of production and high molar extinction coefficients [5]. In this structure, the process photoexcitation has been done by the intramolecular charge transfer (ICT) between D (donor) and A (acceptor) via π-spacer. The electron injects into the conduction band of the semiconductor (TiO2) at the anchoring unit, usually a 2-cyanoacrilic acid function (acceptor unit). Modification of the donor group affects the HOMO and LUMO energy levels [6]. However, many researchers have carried out with the aim to built new π-conjugated materials having a D–π–A structure. Recently, special interest was expressed for compounds containing thienopyrazine unit as a donor [7,8].
The compounds containing thienopyrazine were proved to be excellent precursors to produce organic materials having a low band gap and facilitating the ICT. This effect could be explained by the existence of the thiophene unit, which shares a portion of its electron density to the neighboring pyrazine unit [9–11]. Recently, photovoltaic devices consisting of thienopyrazine-based low band gap compounds [12–15] as hole-transporting materials have shown improved efficiencies. In this context, the aim of this work was to study theoretically a new family of organic dyes based on thienopyrazine (D1, D2, D3, D4, D5, and D6), to determine their capacity to generate a photocurrent in the field of DSSC (Fig. 1).
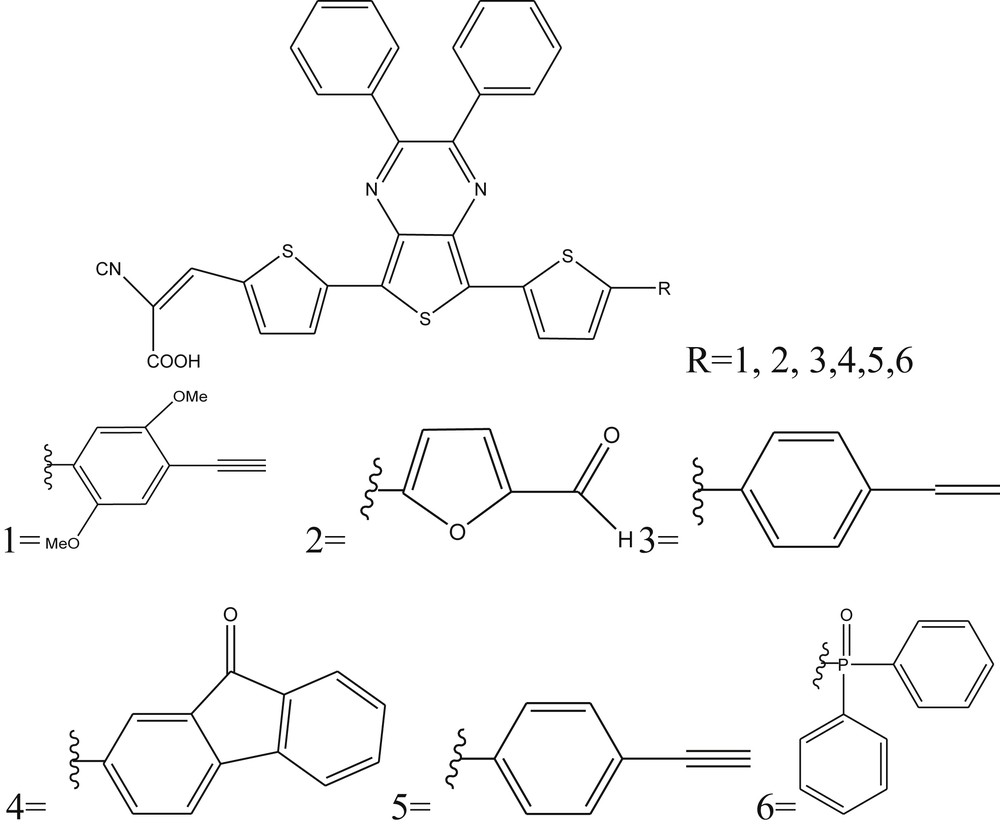
Chemical structure of studied compounds Di (i = 1–6).
2 Theoretical methodology
In this work, all calculations are made with the Gaussian 09 program [16] using the density functional theory (DFT) with the hybrid functional B3LYP [17–19] in which this functional has shown that the calculated electronic properties of compounds based on thienopyrazine are in good agreement with those obtained experimentally [20,21]. The structure of the ground state of each dye was optimized with the base Pople 6-31G(d,p) and a frequency calculation was performed to ensure that the geometry corresponded to a minimum. The vertical electronic transitions were evaluated using the TD-DFT and coulomb-attenuating method based on B3LYP (CAM-B3LYP)/6-31G(d,p) [22]. As for the absorption, Céron-Carrasco et al. [23] and Dessì et al. [24] have shown that for a set of thienopyrazine and thiazolothiazole organic dyes, the most CAM-B3LYP functional gave accurate transition energies, which are in good agreement with the experimental results with an error of 0.1 eV. Therefore, the vertical electronic transitions and electronic absorption spectra were evaluated using TD-CAM-B3LYP in this work. The solvent effects were included in the calculation by using an approach of the polarizable continuum (PCM) and more particularly the “conductor-like-PCM” such as implemented in the Gaussian program.
3 Results
3.1 Frontier molecular orbitals
To enhance the light harvesting efficiency (LHE) of DSSCs, the choice of the appropriate donor and acceptor unit is essential. Strong electron donating groups give a high EHOMO energy level, whereas strong accepting electron groups cause a low ELUMO energy level. The calculated HOMO/LUMO energies using B3LYP/6-31G(d,p) are −5.025/−3.057 for D1, −5.276/−3.293 for D2, −5.091/−3.099 for D3, −5.139/−3.124 for D4, −5.155/−3.140 for D5, and −3.140/−3.159 eV for D6, corresponding to energy gaps of 1.968 for D1, 1.983 for D2, 1.992 for D3, 2.015 for D4, 2.015 for D5, and 2.171 eV for D6 (Table 1). The increase in the conjugation of these π-systems permits to decrease the HOMO–LUMO gap, giving a red-shifted absorption.
Estimated electrochemical parameters for all dyes.
| Dyes | EHOMO | ELUMO | Eg | λh | λe | λtotal | E00 | Edyes | Edyes∗ | ΔGinject | LHE | VOC |
| D1 | −5.025 | −3.057 | 1.968 | 0.22 | 0.27 | 0.49 | 2.0963 | 5.025 | 2.9287 | −1.0713 | 0.95 | 0.943 |
| D2 | −5.276 | −3.293 | 1.983 | 0.22 | 0.14 | 0.36 | 2.1215 | 5.276 | 3.1545 | −0.8455 | 0.94 | 0.707 |
| D3 | −5.091 | −3.099 | 1.992 | 0.22 | 0.27 | 0.49 | 2.1183 | 5.091 | 2.9727 | −1.0273 | 0.94 | 0.901 |
| D4 | −5.139 | −3.124 | 2.015 | 0.23 | 0.25 | 0.48 | 2.1334 | 5.138 | 3.0046 | −0.9954 | 0.95 | 0.876 |
| D5 | −5.155 | −3.140 | 2.015 | 0.22 | 0.27 | 0.49 | 2.1362 | 5.155 | 3.0188 | −0.9812 | 0.94 | 0.860 |
| D6 | −5.330 | −3.159 | 2.171 | 0.30 | 0.36 | 0.66 | 2.2618 | 5.330 | 3.0682 | −0.9318 | 0.91 | 0.841 |
We notice that the energy gap increases when going from D1 to D6 in the following order: D1 < D2 < D3 < D4 < D5 < D6. Finally, the energy gaps of D1, D2, and D3 are much smaller than the three other compounds. This is because of the strong electron donor property of the donor unit (R) that enhances the ICT character, which is an important factor to the red-shift in the absorption spectra.
The narrow band gap Eg in D1 and D2 is attributed to an increase in the conjugation caused by their donor units, which possess a heteroatom (oxygen in the dyes D1 and D2). On the contrary, the result indicates that a phenylene group is not appropriate to extend the conjugation [25].
The frontier orbitals in the ground state of all compounds are presented in Fig. 2. The graphical representation of orbitals shows that the HOMO of D1, D2, and D3 is almost located on the whole molecule, but that from D4, D5, and D6 is mainly located on the π-bridge (thiophene–thienopyrazine–thiophene) and only a little part of it is located on the acceptor unit (carboxyl groups), whereas the LUMO for the six compounds is located on the π-bridge groups and near the carboxyl groups (acceptor of electrons). These results show that electrons are moving from the donor unit to acceptor unit via thiophene–thienopyrazine–thiophene and then to TiO2 during the excitation process.

The contour plots of HOMOs and LUMOs of the studied compounds Di (i 1–6).
3.2 Photovoltaic properties
The analysis of the frontier orbitals (HOMO and LUMO) of the six dyes allows finding their donor behavior in the face of semiconductor TiO2. Comparing the LUMO levels from these six dyes with the conduction band of TiO2 (−4.00 eV) [26] (Table 1), we deduce that all compounds are likely to inject an electron in the conduction band of the semiconductor, in the excited state. This suggests that these compounds can be useful in photovoltaic devices.
The power conversion efficiency (η) was calculated according to Eq. 1:
| (1) |
| (2) |
3.2.1 Theoretical background
Generally, in DSSCs, the short-circuit current density Jsc is determined as
| (3) |
| (4) |
On the basis of Eq. 3, the large Φinject leads to a high Jsc, in which Φinject is related to the driving force (ΔGinject) of the electron injection from the photoinduced excited states of organic dyes to the semiconductor surface. Generally, the larger the ΔGinject, the larger will be Φinject, and it can be expressed (in eV) as [28]
| (5) |
| (6) |
As can be seen in Eq. 3, the high value of Jsc needs the weak value of total reorganization energy (λtotal), in which λtotal is defined as the sum of reorganization energy of the hole and those of the electron. Hence, we computed the hole and the electron reorganization energy (λh and λe) according to the following formula [29]:
| (7) |
On the basis of Eq. 3, we notice that LHE and the electronic injection free energy (ΔGinject) are the two main influencing factors on Jsc and consequently on the efficiency of the organic dyes. The LHE is considered as a very important factor for any dyes, in which we could appreciate the role of these dyes in the DSSCs, that is, absorbing photons and injecting photoexcited electrons to the conduction band of TiO2. To give an impression as how the donor unit (D) influences the LHE in our compounds, we simulated the UV–vis absorption spectra of the six dyes. We found that when we change the donor unit, the oscillator strengths were changed slightly. As shown in Table 1, the LHE of all dyes ranged from 0.91 to 0.95. The LHE values of the dyes are in narrow ranges. However, this means that all of the dyes will give similar photocurrent.
To enhance the value of Jsc, another factor was introduced which is the electronic injection free energy ΔGinject. The details of the calculation of ΔGinject have been described in Eq. 5. The ground state oxidation potential energy is related to ionization potential energy according to the Koopmans theorem [29]. Furthermore, Edye can be estimated as negative EHOMO and Edye∗ is calculated based on Eq. 6. Through the results shown in Table 1, we observe that the electron donor significantly influences Edye∗. We also notice that for all studied dyes, ΔGinject is negative; this reveals that the electron injection process is spontaneous, and the calculated ΔGinject for these six dyes is decreased in the following order: D2 > D6 > D5 > D4 > D3 > D1. Among these six dyes, we observe that the dyes D2, D6, D5, and D4 have the largest ΔGinject, this may be because of the influence of the donor spacer on our dyes. As a result and on the basis of LHE and ΔGinject related to Jsc, we could conclude that the cell containing the dyes D2, D6, D5, and D6 should have the highest Jsc because of its high value of LHE compared with the dyes D1, D3.
On the other hand, the reorganization energy λtotal is also an important influencing factor in the kinetics of electron injection. Therefore, to analyze the relationship between the electronic structure and the Jsc it is necessary to calculate the reorganization energy (λtotal). As mentioned previously, the small λtotal increase the Jsc. We notice that the calculated λtotal of all compounds proposed for use as dyes in a solar cell is increased in the following order: D2 < D4 < D1 = D3 = D5 < D6 (Table 1). These results show that the compounds D2 and D4 have the smallest λtotal, whereas the compound D6 has the largest λtotal. Furthermore, for high efficient DSSCs, the balanced transport of both holes and electrons is important, that is, the more the balanced reorganization energy between the hole and electron is obtained, the higher will be the luminous efficiency will obtained [30]. From Table 1, we also notice that the differences between λh and λe of six compounds are 0.05, 0.08, 0.05, 0.02, 0.05, and 0.06 eV successively. As a result, combined with discussion of the electron injection driving force, LHE, and the reorganization energies of the studied organic dyes, we could predict that these six dyes are favorable candidates for DSSCs and can exhibit a favorable Jsc
3.3 Absorption and emission properties
TD-DFT/CAM-B3LYP/6-31G(d,p) level was performed to simulate the optical property of the six compounds used as dyes in the solar cell. The maximum (λmax) wavelengths for UV–vis absorption spectra of all dyes are tabulated in Table 2. The absorption wavelengths increase progressively with the increase in conjugation lengths. As shown in Table 2 and Fig. 3, the absorption wavelengths of D1, D2, D3, D4, D5, and D6 are 625.38, 618.01, 620.04, 615.49, 613.46, and 574.33 nm respectively, indicating that all molecules have only one band in the visible region (λabs > 400 nm) and D1 has the maximum absorbent wavelength.
Absorption and emission spectra data for all dyes obtained with PCM-CAM-B3LYP/6-31G(d,p).
| Compounds | D1 | D2 | D3 | D4 | D5 | D6 |
| λmax emi (nm) | 805.02 | 794.65 | 801.53 | 793.82 | 790.72 | 727.01 |
| λmax abs (nm) | 625.38 | 618.01 | 620.04 | 615.49 | 613.46 | 574.33 |
| SS (nm) | 179.64 | 176.64 | 181.49 | 178.33 | 177.26 | 152.68 |

Simulated UV–vis optical absorption spectra of the title compounds with the calculated data at the TD-DFT/CAM-B3LYP/6-31G(d,p) level in chloroform solvent.
The emission spectra data obtained at the TD-DFT/CAM-B3LYP/6-31G(d,p) level of all compounds recorded in chloroform are presented in Table 2 and depicted in Fig. 3. The normalized photoluminescence spectrum of the studied compounds shows a maximum at 805.02 for D1, 794.65 for D2, 801.53 for D3, 793.82 for D4, 790.72 for D5, and 727.01 nm for D6. The red-shifted emission of the photoluminescence spectra observed is in reasonable agreement with the obtained results of absorption. We can also note that relatively high values of Stokes shift (SS) are obtained from all dyes D1 (179.64), D2 (176.64), D3 (181.49), D4 (178.33), D5 (177.26), and D6 (152.68 nm) (Table 2), indicating that the compounds having a weak SS present a minimal conformational reorganization between the ground state and excited state. Indeed, this stops the intermolecular transfer charge and delays the injection phenomenon from LUMO of the dye to the conduction band of TiO2 [31,32].
The SS is the difference between the vertical absorption energy (EVA) and the vertical emission (EVE) of the same electronic transition, which is usually related to the bandwidths of both, the absorption and emission bands [33,34].
4 Conclusions
We have used the DFT/B3LYP and TD-DFT/CAM-B3LYP methods to investigate theoretically the optoelectronic properties of the six dyes based on thienopyrazine. The band gaps obtained using DFT-B3LYP with 6-31G(d,p) as a basis set range from 1.968 to 2.171 eV, indicating that these compounds have a high order of conjugation. The LUMO levels of all dyes are much higher than the conduction band of TiO2, suggesting that the photoexcited electron escapes easily from Di to TiO2. The Voc range from 0.706 to 0.942 eV, indicating that these values are sufficient for an efficient electron injection. The absorption wavelengths of D1, D2, D3, D4, D5, and D6 are 625.38, 618.01, 620.04, 615.49, 613.46, and 574.33 nm, respectively, indicating that spectra of these dyes cover the spectra emitted from sunlight. Finally, this study has been carried out to propose the synthesis of novel materials with specific electronic properties.
Acknowledgments
This work was supported by Volubilis Program (No. MA/11/248) and the convention CNRST/CNRS (Project chime 1009).