


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Molecular structure elucidation is the determination of chemical structures which can be achieved by using NMR spectroscopy techniques. The molecular environment around a nucleus determines its chemical shift, and its interpretation is important to elucidate the structure of organic molecules. Accurate predictions of NMR chemical shifts with respect to experimental values are highly valuable for structural elucidations. NMR predictions’ software primarily rely on coding and computational methods in producing NMR predictions, different programs can use different methods in their internal calculations for predictions. The focus of this study is to evaluate the accuracy of 1H NMR predictions’ software using statistical tools namely on MestReNova, ACD Workbook Suite, ChemDraw and NMRShiftDB. MestReNova and ACD Workbook suite were selected based on the similarity of what they both possess i.e., having the conventional hierarchically ordered spherical description of environment (HOSE) as one of their predictors but the latter having neural network (NN) as an additional tool that can interpolate within its trained database. These predictor’s ability was compared with ChemDraw, a predictor that only uses a single prediction method: linear additivity rules. Another predictor’s ability was compared, i.e. NMRShiftDB that only uses NNs in predicting NMR shifts.
MestReNova prediction is based on two predictors: MestreLab Predictor and Modgraph Predictor [1]. When an NMR prediction is made, each of these predictors will predict chemical shifts with a certain confidence interval based on different methods, for example with HOSE, along with substituent chemical shifts approach for Modgraph predictor, and CHARGE for MestreLab Predictor. These predicted chemical shifts will be combined using a Bayesian method to produce the final chemical shift, which has higher reliability and accuracy than the individual chemical shifts. The concept utilized is known as ensemble learning, which allows multiple machine-learning algorithms to be combined. The concept of ensemble learning is advantageous as the deficiency of one prediction method can be compensated by another prediction method, improving the overall prediction accuracy while reducing the number of outliers. For example, HOSE prediction method, which relies heavily on the size and quality of database, is highly inaccurate when predicting novel compounds, as very few database compounds possess a structure similar to the novel compound [2]. This deficiency is compensated by the substituent chemical shifts approach, which predicts by identifying substructures and substituents in a molecule and assigning base and increment values respectively based on their identity. Utilizing ensemble learning method, a database containing approximately 1000,000 shift values and 3000 available parameters, MestReNova can produce accurate 1H NMR chemical shifts by combining the strengths of each prediction method used while minimizing their respective weaknesses. ACD Workbook Suite, on the other hand utilizes multiple prediction methods which are additivity rules, HOSE, and NN [3]. HOSE can be compromised when there is error present in database values, as these errors will be reproduced in the final predicted chemical shifts [4]. NN is well-known for its ability to generalize data, it can interpolate within its trained database, consequently less dependent on database values, and thus, reducing the possibility of reproducing errors within database in the final predicted value [4, 5]. To ensure robustness, generality, and accuracy in NN, various types of descriptors, namely topological, physicochemical, and geometric descriptors (to account for stereochemistry and 3D effects) were used to better match the properties of H atoms to be predicted to those available in the database [6]. A database of approximately 1400,000 experimental 1H chemical shifts ensure robustness and accuracy of ACD Workbook Suite [7]. ChemDraw’s predictor, ChemNMR, relies on a single prediction method: linear additivity rules. Linear additivity rules are similar in concept to the substituent chemical shifts approach used in MestReNova and the additivity rules used in ACD Workbook Suite. However, it was found that linear additivity rules are inefficient in chemical shift prediction of molecules that exhibit non-linearity. Even though correction methods are implemented to improve its prediction power, it is still lacking when resonance effects are present in the molecule. Even so, ChemDraw is still reliable because it considers stereoisomerism and is backed by a large database of approximately 700 base values and 2000 increment values based on 4000 parameters [8]. NMRShiftDB utilizes deep NN in predicting NMR shifts, which achieved better precision than the HOSE code approach. When a molecule is predicted, its molecular weight, atom, and proton count will be compared to database molecules. If there are ten or more chemical shifts available, the smallest and largest chemical shift will be taken as confidence limit of the 6-sphere HOSE prediction. However, if values obtained are insufficient for 6-sphere HOSE or confidence limit estimation, the number of spheres used in HOSE will be reduced, leading to significantly decreased HOSE prediction accuracy as it relies heavily on number of spheres [9].
The diversity and variation of prediction methods among the available NMR predictor software in use are quite high and warrant further examination on the reliability and accuracy of these methods in predicting NMR chemical shifts, as well as comparisons and discussions on which method or program produces predictions that most closely matches real-world experimental data. Thirty organic compounds with 396 data points were used. Statistical comparison methods applied were mean absolute percentage error (MAPE), root mean square deviation (RMSD), one-way analysis of variance (ANOVA), Tukey’s honestly significant difference (HSD) and t-test. From the data analysis, the best NMR predictor software was determined. The predictor software was then used to predict 1H NMR of recent compounds found in literature.
2. Methods and analysis
The 1H NMR chemical shifts were obtained by drawing and inserting the molecules into the NMR predictor software. The 30 organic compounds are shown in the supporting documents. The predictor’s accuracy was evaluated in aliphatic, aromatic, cyclic and polycylic molecules. For aliphatic molecules, additional functional groups in the molecules add to the complexity in the prediction e.g. ester (7), ester and dicarbonyl (9), diols and ether (10) and ester, diols, alkene (11, 12 and 13). Molecules 1, 2, 4 and 13 were selected due to their cyclic conformations. It would be interesting to know whether the selected predictors are able to predict the chemical shifts of the hydrogens on the ring in these selected conformations. Molecules 1, 2 and 4 have ester functionality in the ring or near the ring, which acts as an electron withdrawing group. The ring in molecule 13 has additional complexity as it is linked to a nitrogen atom adjacent to a sulfone. Molecules 22 and 24 have cyclic moieties but whether it is in chair or boat conformation is unknown. It would be interesting to evaluate the predictor’s capability for these kinds of molecules. Again, we are curious to know the accuracy of the predictor when the cyclic compound is bonded to ester group in 22 and aromatics functionality in molecule 24. We had also selected a range of other aromatic compounds as seen in molecules 5, 6, 8, 14–21, 25, 26, 29 and 30. Various other functional groups are linked to the aromatic groups from carbonyl and thiol (5), ketone and alcohol (6), ethers and alcohol (8), sulfone (14), phosphate (15), phosphate where one oxygen atom is replaced by sulfur (16), fluorine (17–19), amide (20 and 21), thiol, carboxylic acid and amide (25), polycyclic (26) and heteropolycyclic with imine (29 and 30). The predictor’s capability was further evaluated, in the case of fused aromatic rings. The complexity was further heightened with presence of double bonds (3), polycyclic and dicarbonyl (27) and heteropolycyclic and esters (28).
The details of each software and its link are provided in the supporting document. The variation of the experimental proton shifts with predicted values for a molecule (Compound 1) is shown in the graphs in the supporting documents. The comparison of the calculated and experimental 1H NMR chemical shift values for all the compounds are provided in the SI.
MAPE was calculated by using the equations in (1) and (2), while RMSD was calculated by using formula (3).
| (1) |
| (2) |
| (3) |
MAPE was chosen as a parameter because it normalizes the absolute error between predicted and experimental values over a range of 0% to 100%. This allows comparison between datasets of different scales, as the absolute error of each predicted chemical shift is normalized by the experimental value, and an average is obtained [10]. RMSD, on the other hand, approximates the spread of predicted data from experimental data without restricting its values to any range.
2.1. Tests of statistical significance between the software
Three methods were selected to test for the significant difference between collected data, which are one-way ANOVA with a post hoc test, Tukey’s HSD, and t-test. The four sets of significance tests are shown in Table 1.
Datasets and the corresponding significance tests used
| No | Dataset | Significance test |
|---|---|---|
| 1 | MAPE of 30 organic molecules from SDBS | ANOVA + Tukey’s HSD |
| 2 | RMSD of 30 organic molecules from SDBS | ANOVA + Tukey’s HSD |
| 3 | MAPE of 3 organic compounds from literature | t-test |
| 4 | RMSD of 3 organic compounds from literature | t-test |
Both t-test and ANOVA determine the significant difference between populations by observing the mean and variance. T-test analyses two populations, whereas ANOVA is more suited for analysis of three or more populations. Thus, ANOVA was used for the first two datasets to compare the four NMR predictor programs. t-test was not repeatedly carried out as it can contribute to significant Type I error (false positive) which causes a true null hypothesis to be rejected. As ANOVA simultaneously compares all four NMR predictors, the rate of Type I error is maintained at the significance level of the hypothesis test, 𝛼 = 0.05. One-way ANOVA was used because the analysis had one independent variable: the type of NMR prediction program. Tukey’s HSD (post hoc analysis) was conducted alongside ANOVA to determine which specific NMR predictor was statistically different. Similar to t-test, Tukey’s HSD utilizes pairwise comparisons as well, however its data is presented in adjusted p values and simultaneous confidence intervals. These representations maintain the rate of occurrence of Type I errors by limiting the family-wise error rate to the significance level used, 𝛼 = 0.05 [11].
T-test was used in the third and fourth dataset where two NMR prediction software were compared, namely MestReNova and ACD Workbook Suite. The interpretation of data was based on two-tailed prediction, as the deviation of predicted chemical shift from experimental chemical shift is not unidirectional.
2.2. ANOVA and Tukey’s HSD
In performing the following ANOVA and Tukey’s HSD, the null hypothesis (H0) was that all predictor programs are equal in performance. The alternative hypothesis (H1) was that at least one of the predictor programs is significantly different in performance than the others.
The process of analysis is as follows: ANOVA (omnibus test) was separately performed on the datasets of MAPE and RMSD obtained from observing the deviations between predicted and experimental values. The MAPE and RMSD data of 30 molecules was analysed. ANOVA produces the F value (F), F-critical value (Fcrit), and P value (P). If both conditions of F > Fcrit and P < 0.05 are achieved, it indicates that at least one of the programs is significantly different in performance from the others, affirming H1 and rejecting H0.
To verify the conclusion from ANOVA and to identify which specific predictor software is significantly different, Tukey’s HSD (post hoc analysis) was performed by calculating and comparing the Q-stat value (Qstat) and Q-critical value (Qcrit) for each pair combination of predictor programs (pairwise comparisons). For pair combinations where Qstat > Qcrit, it suggests that one program is significantly different from the other in the pair. The performance of which predictor is better or worse can be determined based on their average MAPE or RMSD. Additionally, simultaneous confidence interval (CI) of each pair combination of predictor programs was calculated and a graph was plotted. The difference in performance between the programs is significant if CI does not encompass zero in the graph.
3. Statistical analysis on the NMR prediction
3.1. RMSD
The chemical structure of the 30 molecules is provided in the supporting information. The comparison of the calculated and experimental 1H NMR chemical shift values is shown in Appendix B in the supporting information. RMSD was calculated from the difference between the 1H NMR of the experimental value and predicted value as outlined in (3). The variation of the experimental proton shifts with the corresponding predicted values using the four software was carried out for all the molecules. Figure 1 shows the results of the variation for molecule 1.
Variation of the experimental proton shifts with the corresponding predicted values for molecule 1.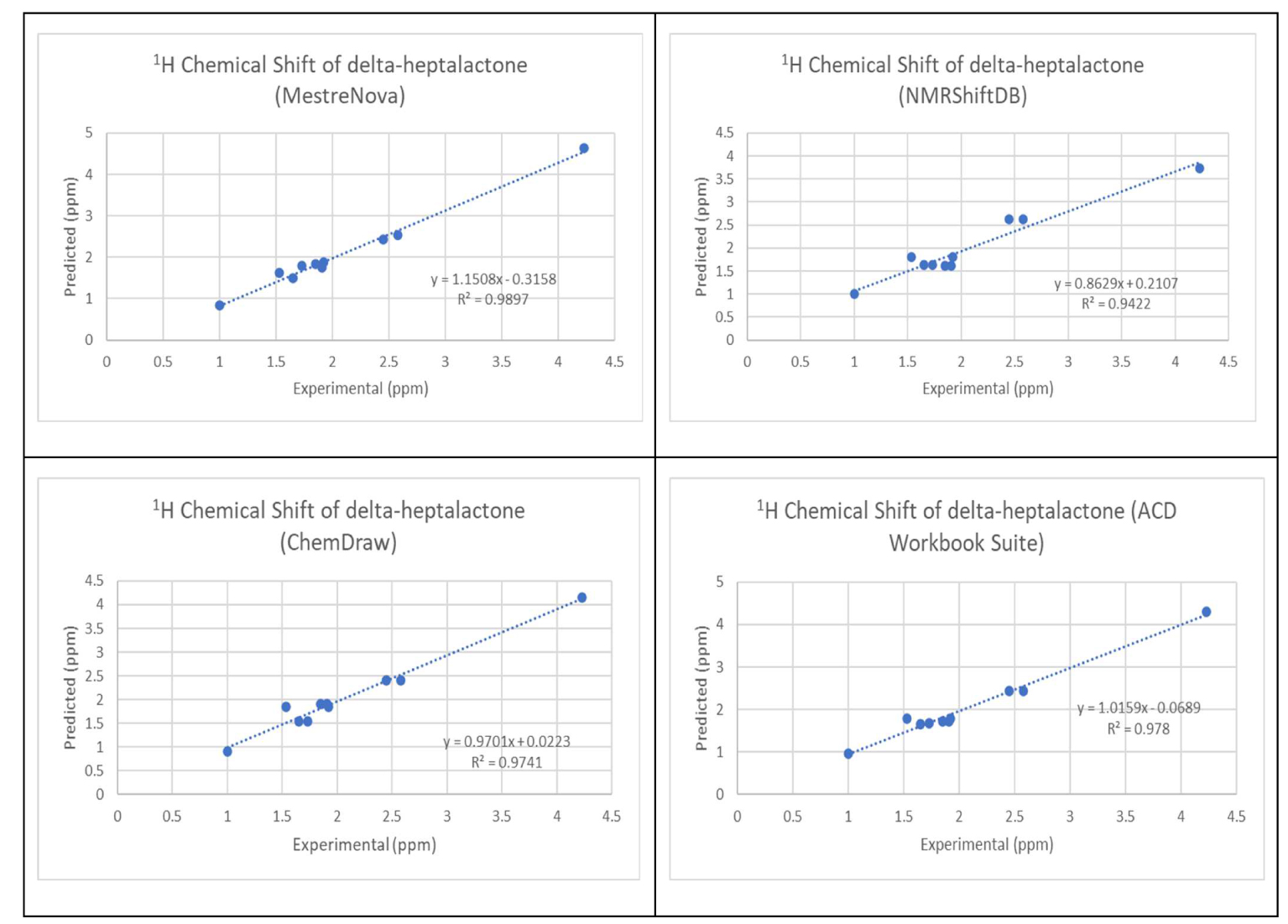
3.1.1. Average RMSD
RMSD is calculated as per (3) and the average RMSD was calculated from the RMSD of the 30 molecules and shown in the table below.
RMSD of the 30 organic molecules
| Molecule | RMSD of the 30 organic molecules | |||
|---|---|---|---|---|
| MestReNova | NMRShiftDB | ChemDraw | ACD Workbook Suite | |
| 1 | 0.158199558 | 0.223282556 | 0.139216019 | 0.131732304 |
| 2 | 0.132205018 | 0.507366156 | 0.429319714 | 0.231302399 |
| 3 | 0.250353084 | 0.412088178 | 0.46808653 | 0.248822226 |
| 4 | 0.250149132 | 0.313776616 | 0.275316716 | 0.28554893 |
| 5 | 0.153337714 | 0.398996127 | 0.221822411 | 0.134359423 |
| 6 | 0.097377429 | 0.101795696 | 0.117088156 | 0.244548564 |
| 7 | 0.157633637 | 0.408167969 | 0.200715085 | 0.254124164 |
| 8 | 0.991556128 | 0.991556128 | 0.092443376 | 0.023242681 |
| 9 | 0.184746313 | 0.254360374 | 0.10793146 | 0.363292444 |
| 10 | 0.064758292 | 0.326990409 | 0.099995454 | 0.283289925 |
| 11 | 0.169342355 | 0.290319077 | 0.110152167 | 0.46665628 |
| 12 | 0.164378167 | 0.415193493 | 0.182995777 | 0.1007084 |
| 13 | 0.153414797 | 1.611406249 | 0.219695471 | 0.192520908 |
| 14 | 0.231103061 | 0.455596998 | 0.436298207 | 0.186641367 |
| 15 | 0.042973247 | 0.291949824 | 0.029132456 | 0.121849087 |
| 16 | 0.075511589 | 0.335740972 | 0.260335797 | 0.157330573 |
| 17 | 0.116416398 | 0.392535207 | 0.43611097 | 0.116511325 |
| 18 | 0.348320398 | 0.440394255 | 0.244824631 | 0.44057576 |
| 19 | 0.329876837 | 0.996771425 | 0.285785361 | 0.382548036 |
| 20 | 0.093588272 | 0.500206957 | 0.34992764 | 0.281360978 |
| 21 | 0.275964529 | 0.553884464 | 0.426578308 | 0.378532554 |
| 22 | 0.209280016 | 0.295367356 | 0.331437211 | 0.299683062 |
| 23 | 0.090381414 | 0.394404361 | 0.15513478 | 0.332540373 |
| 24 | 0.617479738 | 0.883528232 | 0.656619684 | 0.683054603 |
| 25 | 0.058695187 | 0.316056957 | 0.078370913 | 0.051740941 |
| 26 | 0.459865431 | 0.548149289 | 0.468122923 | 0.40571084 |
| 27 | 0.624529823 | 1.068597211 | 0.513298159 | 0 |
| 28 | 0.480409201 | 1.732503785 | 0.229454491 | 0.260868654 |
| 29 | 0.181191762 | 0.407916547 | 0.979724943 | 0.127477984 |
| 30 | 0.17631676 | 0.418365391 | 0.502282391 | 0.317947165 |
| Average | 0.244645176 | 0.542908942 | 0.30160724 | 0.250150732 |
The results show the following trend in order of increasing RMSD, MestReNova < ACD Workbook Suite < ChemDraw ≪ NMRShiftDB.
3.1.2. ANOVA, Tukey’s HSD and CI from RMSD of the 30 organic molecules
To test for the significant difference in RMSD from Table 2, one-way ANOVA with a post hoc test and Tukey’s HSD were carried out. The results are shown in Table 3a, 3b and 3c and Figure 2.
Single factor ANOVA from RMSD of 30 organic molecules
| Source of variation | SS | df | MS | F | P value | F crit |
|---|---|---|---|---|---|---|
| Between groups | 1.791134363 | 3 | 0.597044788 | 9.38209254 | 1.33255E-05 | 2.682809407 |
| Within groups | 7.38184953 | 116 | 0.063636634 | |||
| Total | 9.172983893 | 119 |
Tukey’s HSD from RMSD of 30 organic molecules
| Pair | Difference | Q stat | Significant statistical difference |
|---|---|---|---|
| MN | 0.298263766 | 6.476010207 | Yes |
| MC | 0.056962064 | 1.236780827 | No |
| MA | 0.005505555 | 0.119538602 | No |
| NC | 0.241301702 | 5.23922938 | Yes |
| NA | 0.29275821 | 6.356471605 | Yes |
| AC | 0.051456508 | 1.117242225 | No |
(M = MestReNova, N = NMRShiftDB, C = ChemDraw, A = ACD Workbook Suite).
Confidence interval (CI) from RMSD of 30 organic molecules
| Pair | Difference | Upper CI limit | Lower CI limit |
|---|---|---|---|
| MN | 0.298263766 | 0.468056457 | 0.128471075 |
| MC | 0.056962064 | 0.226754755 | −0.112830627 |
| MA | 0.005505555 | 0.175298246 | −0.164287135 |
| NC | 0.241301702 | 0.411094393 | 0.071509011 |
| NA | 0.29275821 | 0.462550901 | 0.122965519 |
| AC | 0.051456508 | 0.221249199 | −0.118336182 |
(M = MestReNova, N = NMRShiftDB, C = ChemDraw, A = ACD Workbook Suite).
Tukey simultaneous 95% CI for predictor program pair combinations (M = MestReNova, N = NMRShiftDB, C = ChemDraw, A = ACD Workbook Suite).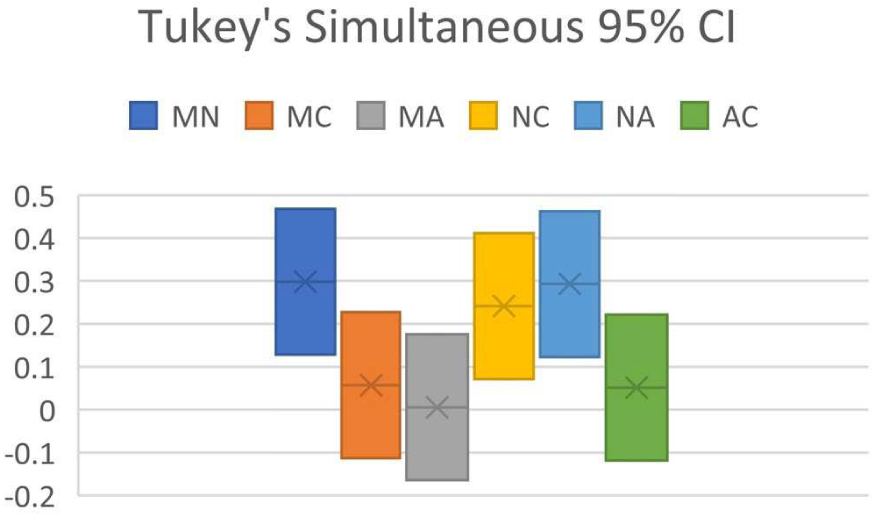
ANOVA of the dataset showed that F = 9.38, Fcrit = 2.68, and P = 0.0000133. As P < 0.05 and F > Fcrit, it suggests that H0 is false, whereas H1 is true: at least one of the predictor programs is significantly different in performance than the others. Tukey’s HSD indicates that Qstat > Qcrit between NMRShiftDB paired with MestReNova, with ChemDraw, and with ACD Workbook Suite, affirming the conclusion reached by ANOVA. Results from Figure 2 shows the CI analysis where the CI between NMRShiftDB and all other programs does not intersect zero, indicating that there is significant difference between NMRShiftDB and all other programs, verifying the conclusion of Tukey’s HSD.
The results obtained from RMSD lead to the conclusion that NMRShiftDB (with the highest average RMSD) is the weakest in 1H NMR prediction whereas the remaining three predictors are equally good in performance.
3.2. MAPE (%)
MAPE (%) was calculated from the total percentage error as outlined in (2).
3.2.1. Average MAPE (%)
The average MAPE (%) was calculated from the MAPE (%) of the 30 molecules and is shown in Table 4.
MAPE of the 30 organic molecules
| Molecule | MAPE (%) of the 30 organic molecules | |||
|---|---|---|---|---|
| MestReNova | NMRShiftDB | ChemDraw | ACD Workbook Suite | |
| 1 | 5.717016795 | 7.757044772 | 6.374206787 | 5.705317882 |
| 2 | 3.457274581 | 14.90233757 | 15.12577338 | 7.372038802 |
| 3 | 2.754474665 | 4.060824356 | 4.75206928 | 3.015019914 |
| 4 | 15.63547599 | 18.69213871 | 16.15365045 | 14.68724293 |
| 5 | 4.134137032 | 7.480177279 | 5.017721695 | 3.509145976 |
| 6 | 1.804341875 | 3.473166633 | 2.645913839 | 7.528264736 |
| 7 | 1.58826661 | 7.374456369 | 3.521567751 | 7.299849584 |
| 8 | 8.584075843 | 7.179467635 | 1.53870372 | 0.493973606 |
| 9 | 3.654524363 | 6.098809222 | 2.134442416 | 13.57000892 |
| 10 | 1.384165294 | 8.092697791 | 3.249486222 | 9.203666198 |
| 11 | 3.128689071 | 6.653747635 | 2.516097881 | 15.15575568 |
| 12 | 1.951321346 | 7.3119789 | 2.52315348 | 3.550421853 |
| 13 | 7.753393404 | 40.0219553 | 6.563769996 | 9.194718558 |
| 14 | 2.979856941 | 5.289498235 | 5.780252463 | 2.960532135 |
| 15 | 1.420874825 | 7.109018275 | 1.044595353 | 3.749748505 |
| 16 | 1.321297802 | 8.26591674 | 7.02811375 | 2.461663019 |
| 17 | 2.546452891 | 5.690348764 | 5.488150679 | 3.175524462 |
| 18 | 5.10986636 | 10.06512895 | 3.039594832 | 5.356112132 |
| 19 | 5.342243544 | 14.86655201 | 3.271496429 | 5.810154517 |
| 20 | 3.952488954 | 13.91247433 | 15.2768481 | 7.191562469 |
| 21 | 5.288046347 | 13.0807625 | 15.36188933 | 8.205957251 |
| 22 | 12.87252284 | 17.02719402 | 19.34607702 | 14.74142521 |
| 23 | 2.836695567 | 7.768817027 | 4.26845463 | 7.62937135 |
| 24 | 15.28376466 | 21.82579088 | 16.67810021 | 19.63752167 |
| 25 | 0.970251774 | 5.573267219 | 1.279891869 | 0.884969588 |
| 26 | 11.32680729 | 11.99120799 | 10.9485264 | 8.640007363 |
| 27 | 7.233767583 | 18.62754286 | 17.64618799 | 0 |
| 28 | 6.135162462 | 12.26033072 | 4.988802026 | 4.26720172 |
| 29 | 2.196676326 | 4.903645145 | 2.937826634 | 1.526134792 |
| 30 | 2.549187235 | 5.02799848 | 6.227786948 | 4.33685596 |
| Average | 5.030437342 | 10.74614321 | 7.090971719 | 6.695338893 |
The results show the following trend in order of increasing MAPE (%), MestReNova < ACD Workbook Suite < ChemDraw ≪ NMRShiftDB. Molecules such as compound 4 showed higher MAPE (%). The 1H NMR of the protons in the chair conformation of its cyclohexane contributed to the high error. For molecule 22, the highest error was shown by the hydrogen in the cyclopentane. The 1H NMR assumed a flat cyclopentane, however it could be either in envelope or half-chair conformation. Molecule 24 had various five- and six-membered cyclic moiety and its exact conformation was not considered in the 1H NMR predictions. The same case was encountered with compound 26, where the six-membered ring protons gave the highest error.
3.2.2. ANOVA, Tukey’s HSD and CI from MAPE (%) of the 30 organic molecules
To test for the significant difference in MAPE (%) from Table 4, one-way ANOVA with a post hoc test and Tukey’s HSD were carried out. The results are shown in Table 5a, 5b and 5c and Figure 3.
Single factor ANOVA of RMSD of 30 organic molecules
| Source of variation | SS | df | MS | F | P value | F crit |
|---|---|---|---|---|---|---|
| Between groups | 522.096092 | 3 | 174.0320307 | 5.492488409 | 0.001454083 | 2.682809407 |
| Within groups | 3675.513547 | 116 | 31.68546162 | |||
| Total | 4197.609639 | 119 |
Tukey’s HSD for MAPE (%) of the 30 organic molecules
| Pair | Difference | Q stat | Significant statistical difference |
|---|---|---|---|
| MN | 5.715705869 | 5.561609341 | YES |
| MC | 2.060534377 | 2.004981974 | NO |
| MA | 1.66490155 | 1.620015485 | NO |
| NC | 3.655171492 | 3.556627367 | NO |
| NA | 4.050804318 | 3.941593856 | YES |
| CA | 0.395632826 | 0.384966489 | NO |
(M = MestReNova, N = NMRShiftDB, C = ChemDraw, A = ACD Workbook Suite).
Confidence interval (CI) for MAPE (%) of 30 organic molecules
| Pair | Difference | Upper CI limit | Lower CI limit |
|---|---|---|---|
| MN | 5.715705869 | 9.504451169 | 1.926960568 |
| MC | 2.060534377 | 5.849279677 | −1.728210924 |
| MA | 1.66490155 | 5.453646851 | −2.12384375 |
| NC | 3.655171492 | 7.443916793 | −0.133573809 |
| NA | 4.050804318 | 7.839549619 | 0.262059018 |
| CA | 0.395632826 | 4.184378127 | −3.393112474 |
(M = MestReNova, N = NMRShiftDB, C = ChemDraw, A = ACD Workbook Suite).
Tukey Simultaneous 95% CI for predictor program pair combinations (M = MestReNova, N = NMRShiftDB, C = ChemDraw, A = ACD Workbook Suite).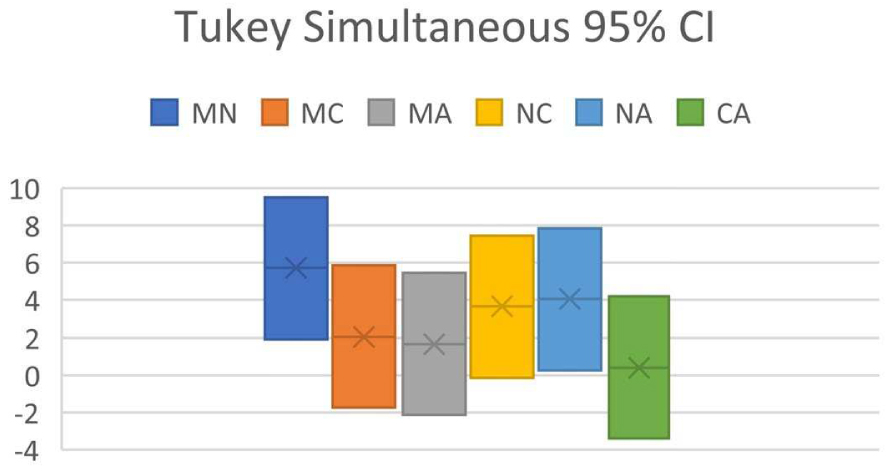
ANOVA of dataset shows that F = 5.49, Fcrit = 2.68, and P = 0.00145. As P < 0.05 and F > Fcrit, it indicates that H0 is false, whereas H1 is true: at least one of the predictor programs is significantly different in performance from the others. In Tukey’s HSD, it is found that Qstat > Qcrit between MestReNova and NMRShiftDB, and between NMRShiftDB and ACD Workbook Suite, in line with the conclusion reached by ANOVA.
Results in Figure 3 show the CI analysis where the CI between MestReNova and NMRShiftDB, and between NMRShiftDB and ACD Workbook Suite do not encompass zero, indicating that there exists significant difference between MestReNova and NMRShiftDB, and between NMRShiftDB and ACD, affirming the conclusion obtained by Tukey’s HSD. The results obtained from MAPE lead to conclude that MestReNova and ACD Workbook Suite are equally good in performance, followed by ChemDraw and NMRShiftDB. This conclusion differs slightly from that obtained using RMSD.
3.3. Conclusion from RMSD and MAPE statistical studies
The conclusion based on MAPE is that MestReNova and ACD Workbook Suite are equally good in performance, followed by ChemDraw and NMRShiftDB, because MAPE is more reliable as it normalizes our data over the experimental value to a range of 0% to 100%, as compared to RMSD which is unscaled. Based on Table 4, although MestReNova exhibits the lowest MAPE, it could be due to chance, as ANOVA and Tukey’s HSD indicate that the difference in its mean is not significant as compared to ACD Workbook Suite.
3.4. Using NMR software to predict chemical shifts
Using the best NMR predictors, the 1H NMR chemical shifts of three published compounds, with 60 data points namely epitetrathiodiketopiperazine (referred to as Compound 31), bis(p-fluorobenzyl)disulfide (Compound 32), and tosylate (Compound 33) [12], were predicted and compared with literature. The structural formulae of the compounds are shown in Figure 4.
The predicted 1H NMR chemical shifts and experimental data are shown in Table 6.
The predicted 1H NMR chemical shifts and experimental data
| Hydrogen position | Experimental (ppm) | Predicted (ppm) | ||
|---|---|---|---|---|
| Literature | MestReNova | ACD Workbook Suite | ||
| 1H NMR chemical shift of compound 31 | ||||
| SO2Ph-o-H | 7.96 | 7.95 | 7.859 | |
| SO2Ph-p-H | 7.54 | 7.71 | 7.495 | |
| SO2Ph-m-H | 7.435 | 7.58 | 7.455 | |
| 8 | 7.435 | 7.17 | 6.997 | |
| 7 | 7.235 | 7.36 | 7.207 | |
| 6 | 7.05 | 7.04 | 6.848 | |
| 5 | 6.99 | 7.2 | 7.71 | |
| 2 | 6.89 | 6.03 | 6.492 | |
| 2′ | 6.84 | 7.12 | 7.095 | |
| 3′ | 6.7 | 6.83 | 6.64 | |
| 5′ | 3.76 | 3.8 | 3.767 | |
| 12a | 3.27 | 3.15 | 3.716 | |
| 12b | 3.1 | 2.97 | 3.587 | |
| 17 | 3.05 | 3.14 | 2.845 | |
| 18 | 1.99 | 1.87 | 1.873 | |
| 1H NMR chemical shift of compound 32 | ||||
| 8 | 7.64 | 7.17 | 6.992 | |
| SO2Ph-o-H | 7.59 | 7.71 | 7.859 | |
| 3′′ | 7.365 | 7.34 | 7.317 | |
| SO2Ph-p-H | 7.365 | 7.95 | 7.495 | |
| 7 | 7.365 | 7.36 | 7.207 | |
| SO2Ph-m-H | 7.16 | 7.58 | 7.455 | |
| 5 | 7.16 | 7.2 | 7.72 | |
| 6 | 7.16 | 7.04 | 6.848 | |
| 3′′′ | 7.08 | 7.34 | 7.317 | |
| 4′′ | 7.04 | 7.09 | 7.049 | |
| 4′′′ | 6.91 | 7.09 | 7.044 | |
| 2′ | 6.74 | 7.12 | 7.105 | |
| 3′ | 6.63 | 6.83 | 6.64 | |
| 2 | 6.59 | 5.98 | 6.352 | |
| 15 | 4.91 | 5.6 | 5.192 | |
| 1′′a | 4.21 | 3.99 | 4.073 | |
| 1′′′a | 4.01 | 3.96 | 4.035 | |
| 1′′b | 3.83 | 3.96 | 4.073 | |
| 1′′′b | 3.79 | 3.94 | 4.035 | |
| 5′ | 3.77 | 3.8 | 3.767 | |
| 12a | 3.55 | 2.96 | 3.693 | |
| 17 | 3.12 | 3.1 | 3.056 | |
| 12b | 3.06 | 2.95 | 2.571 | |
| 1H NMR chemical shift of compound 33 | ||||
| 9′ | 7.77 | 7.62 | 7.7 | |
| 8 | 7.61 | 7.19 | 6.997 | |
| SO2Ph-o-H | 7.47 | 7.71 | 7.753 | |
| SO2Ph-p-H | 7.34 | 7.95 | 7.495 | |
| 7 | 7.275 | 7.36 | 7.217 | |
| 10′ | 7.275 | 7.46 | 7.34 | |
| 5 | 7.105 | 7.23 | 7.389 | |
| 6 | 7.105 | 7.04 | 6.856 | |
| SO2Ph-m-H | 7.105 | 7.58 | 7.455 | |
| 2′ | 6.66 | 7.11 | 7.115 | |
| 3′ | 6.54 | 6.83 | 6.809 | |
| 2 | 6.15 | 5.86 | 6.052 | |
| 11 | 4.39 | 4.55 | 4.256 | |
| 5′ | 4.25 | 4.11 | 4.007 | |
| 15 | 4.04 | 4.43 | 3.66 | |
| 7′ | 3.95 | 4.17 | 4.125 | |
| 12a | 3.13 | 2.54 | 3.035 | |
| 12b | 2.88 | 2.46 | 2.572 | |
| 17 | 2.88 | 2.91 | 2.87 | |
| 12′ | 2.4 | 2.39 | 2.447 | |
| 6′ | 2.13 | 2.16 | 2.159 | |
| 18 | 1.58 | 1.31 | 0.822 | |
The structural formula of epitetrathiodiketopiperazine (Compound 31), bis(p-fluorobenzyl)disulfide (Compound 32), and tosylate (Compound 3), with their atoms labelled.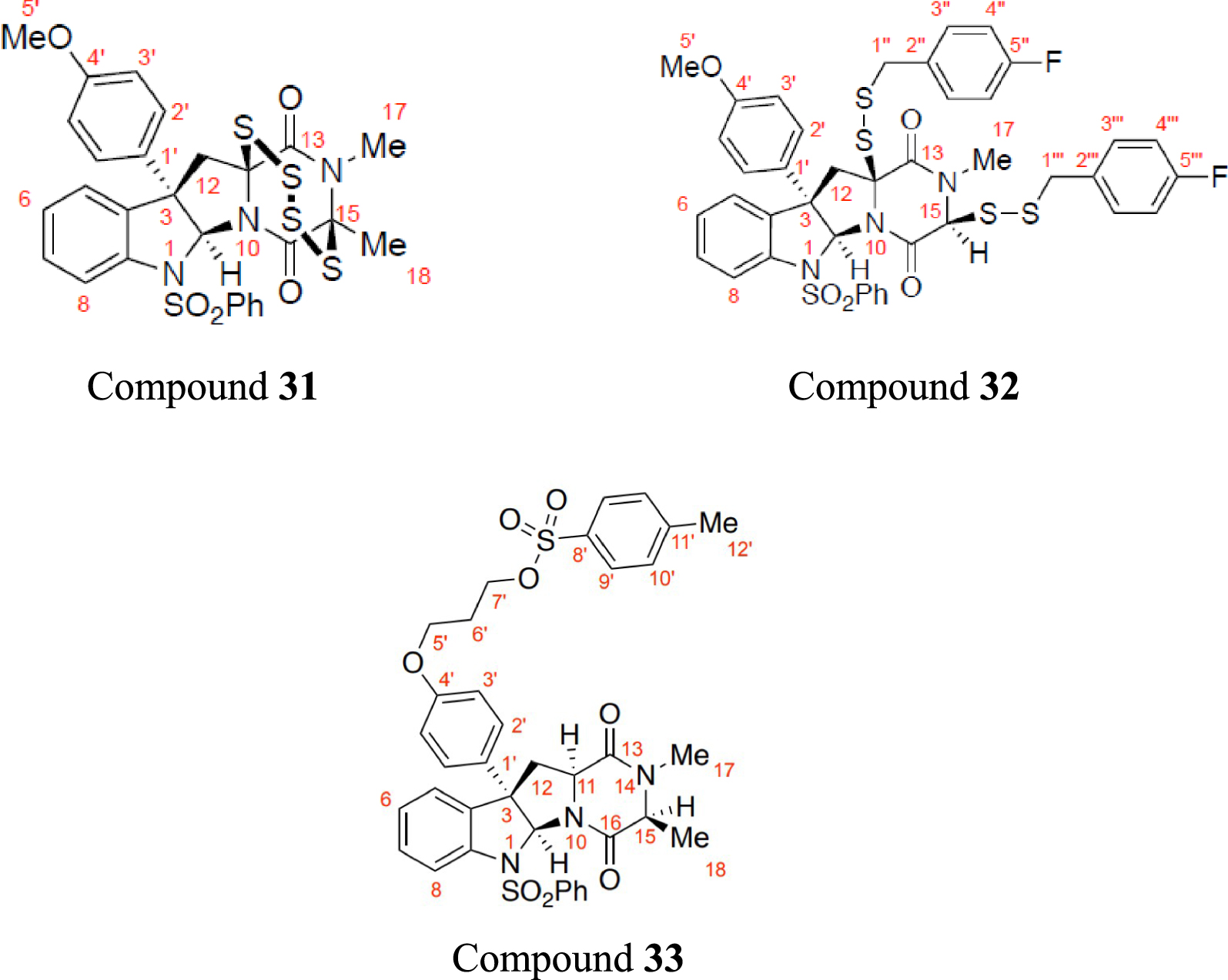
3.4.1. RMSD and MAPE (%) of three organic compounds from literature [12]
Table 7 shows the average RMSD and MAPE (%) for MestReNova and ACD Workbook Suite calculated from the findings in Table 6.
Average RMSD and MAPE (%)
| Molecule | MestReNova | ACD Workbook Suite |
|---|---|---|
| RMSD | ||
| Compound 1 | 0.267129806 | 0.314466214 |
| Compound 2 | 0.320154514 | 0.278211993 |
| Compound 3 | 0.311245664 | 0.300011515 |
| Average | 0.299509995 | 0.297563241 |
| MAPE (%) | ||
| Compound 1 | 3.279491594 | 4.940945337 |
| Compound 2 | 4.287435252 | 4.002110785 |
| Compound 3 | 5.613675005 | 5.885956871 |
| Average | 4.39353395 | 4.943004331 |
Table 7 shows that MestReNova has a lower average MAPE and the same RMSD as compared to ACD Workbook Suite.
3.4.2. T-test of RMSD and MAPE
T-test was performed to determine the statistical difference between the average values obtained from MestReNova and ACD Workbook Suite. Statistical difference is significant when both following conditions are met: (a) TSTAT value is lesser than TCRIT value, (b) P (T⩽t) two-tail value is more than 0.05. When it is the case, the NMR prediction software with either the lower average RMSD or the lower average MAPE (%) has performed significantly better than the other. Based on the values obtained in both Table 8a and 8b, there was no instance in which both conditions proved that statistical difference was simultaneously reached.
t-test: RMSD of 3 organic compounds from literature
| MestReNova | ACD Workbook Suite | |
|---|---|---|
| Mean | 0.299509995 | 0.297563241 |
| Variance | 0.000806199 | 0.000333088 |
| Observations | 3 | 3 |
| Pearson correlation | −0.885835578 | |
| Hypothesized mean difference | 0 | |
| df | 2 | |
| t Stat | 0.074338735 | |
| P(T ≦ t) one tail | 0.473753524 | |
| t Critical one tail | 2.91998558 | |
| P(T ≦ t) two tail | 0.947507049 | |
| t Critical two tail | 4.30265273 |
t-test: MAPE (%) of three organic compounds from literature
| MestReNova | ACD Workbook Suite | |
|---|---|---|
| Mean | 4.39353395 | 4.943004331 |
| Variance | 1.370545749 | 0.887222199 |
| Observations | 3 | 3 |
| Pearson correlation | 0.567987829 | |
| Hypothesized mean difference | 0 | |
| df | 2 | |
| t Stat | −0.949287589 | |
| P(T ≦ t) one tail | 0.221334666 | |
| t Critical one tail | 2.91998558 | |
| P(T ≦ t) two tail | 0.442669333 | |
| t Critical two tail | 4.30265273 |
The t-test showed that the difference between the average MAPE and RMSD of MestReNova and ACD Workbook Suite is not significant. Based on this result, we concluded that MestReNova and ACD Workbook Suite are equally good in terms of 1H NMR prediction, even though the average MAPE of MestReNova is lower than that of ACD Workbook Suite, as the statistical tests have shown that there is no significant difference between their average MAPEs.
4. Conclusion
In contrast to NMR spectrometers, NMR prediction programs are certainly more cost-effective, versatile, and time-saving, as they are easily accessible and acquirable through the internet. In addition, NMR software allows different types of NMR predictions based on a variety of interchangeable solvents and frequencies. The ability to employ high frequencies in NMR prediction allows the chemical shift data to be more pronounced while saving cost on acquiring high-end spectroscopic instruments. NMR prediction programs can rapidly generate chemical shift data with high accuracy using well-studied and reliable computational methods and include other functions such as peak processor, multiplet analysis, structure elucidation programs, and embedded chemical shift databases. The ability to rapidly generate highly accurate chemical shift data without using any spectroscopic instruments makes NMR prediction programs an invaluable tool for scientists.
The data obtained from this project suggest that among the four 1H NMR software, MestReNova and ACD Workbook Suite provide equally accurate 1H NMR predictions, with NMRShiftDB and ChemDraw predictions having equally less accuracy than the previous two programs. The equal level of accuracy between MestReNova and ACD Workbook Suite, and between ChemDraw and NMRShiftDB is due to the inability of the data collected to prove statistical significance between the two programs within the respective pairings, and thus we cannot definitively confirm that one program is better or worse than the other within each pairing. The results of this study can prove extremely useful in deciding which software to be prioritized in order to obtain more accurate 1H NMR predictions, encouraging the use of NMR software that provide accurate predictions, and discouraging the use of programs that do not.
Conflicts of interest
Authors have no conflict of interest to declare.
Acknowledgements
The authors are grateful to ACD/Labs and IT Tech Research (M) Sdn. Bhd. for providing free access to ACD software for 30 days.