


 CC-BY 4.0
CC-BY 4.0
1. Introduction
The great interest in heterocyclic core thiazole in the research of new drugs stems from the fact that there are already a variety of drugs with a thiazole ring, such as antibacterial drugs, antihistamines, vitamin B1 (thiamine), antifungal drugs, gout drugs, and drugs for the inhibition of cholinesterases [1, 2, 3]. Acotiamide (Figure 1, structure I) [4, 5, 6, 7], the drug used to treat functional dyspepsia, acts as a selective acetylcholinesterase inhibitor and is a 1,3-thiazole derivative that has stimulated the development of biologically active compounds with a thiazole nucleus with the aim of inhibiting cholinesterases. From the structure of Acotiamide (I), it can be seen that at position 2 on the thiazole, there is an amide group which was the basis for the synthesis of new compounds in this paper, which are translated into amides, amines, and imines. The need for new drugs that could be used to inhibit cholinesterases in patients with Alzheimer’s disease is high due to the various side effects of currently available drugs and the fact that as the disease progresses, more cells fail and less acetylcholine is available; hence the effectiveness of available inhibitors decreases over time. Among the active compounds containing a thiazole nucleus are also the 1,3-thiazolylimine (Figure 1, structure II) [6], 1,3-thiazolylhydrazones (Figure 1, structure III) [4], and the 1,3-thiazole-piperazine derivatives (Figure 1, structure IV) [7].
Examples of thiazole-bearing cholinesterase inhibitors.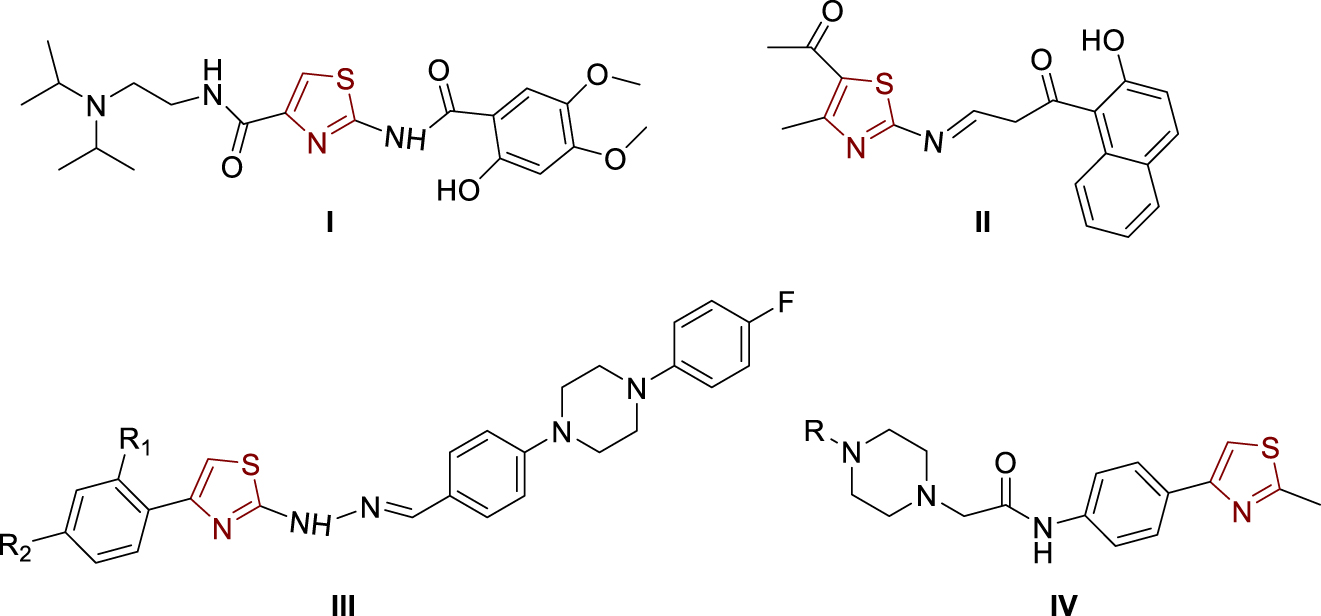
Some of the mentioned compounds show promising activity and selectivity to acetylcholinesterase, while a small number of them showed some binding and inhibition of butyrylcholinesterase. Thus, 1,3-thiazolylhydrazones (Figure 1, structure III) show very selective activity against acetylcholinesterase, but only two of them show any activity against butyrylcholinesterase. As it is known, acetylcholinesterase has much higher catalytic activity towards the hydrolysis of acetylcholCine and is present in much larger quantities than butyrylcholinesterase, which is why the results of such compounds are promising. Hydroxynaphthalenyl thiazole II (Figure 1) achieves powerful inhibition of acetylcholinesterase (IC50 1.78 ± 0.11 μM), and also, it can inhibit butyrylcholinesterase in a certain extent (IC50 11.9 ± 0.43 μM) [6]. Thiazolylhydrazones III (Figure 1) showed significant acetylcholinesterase inhibition but slight inhibition of butyrylcholinesterase [4]. The best inhibition of acetylcholinesterase was achieved with the following substituents at positions R1 and R2: R1, R2 = H, IC50 0.0496 ± 0.002 μM; R1 = H, R2 = OCH3, IC50 0.0317 ± 0.001 μM and R1 = H, R2 = CF3, IC50 = 0.2158 ± 0.010 μM. As a reference, Donepezil was used as an acetylcholinesterase inhibitor (IC50 = 0.0287 ± 0.005 μM) [4]. The results on thiazolylhydrazones III showed that the structure with the methoxy substituent (R2) at the para-position is the best acetylcholinesterase inhibitor with the inhibitory activity very close to the values of donepezil.
In order to ensure the diversity of new possible acetylcholinesterase inhibitors, the approaches of different papers are based on different types of molecules and include the introduction of different substituents to the thiazole scaffold. Thus, some 1,3-thiazole-piperazine derivatives (Figure 1, structure IV) [7] proved to be very effective acetylcholinesterase inhibitors almost without any butyrylcholinesterase inhibition activity (R = 2-pyridyl, IC50 0.051 ± 0.002 μM; R = benzyl, IC50 0.011 ± 0.001 μM; R = 2-furoyl, IC50 0.27 ± 0.001 μM). Donepezil was used again as a standard (IC50 0.054 ± 0.002 μM) [7]. Those results showed that inhibition by 2-pyridyl and benzyl derivatives was even better than by donepezil, which gives them great importance in further development and research.
In our previous study concerning the newly synthesized 1,3-thiazoles 1–17 (Figure 2) and their anti-inflammatory characteristics [8], amide 7 with quinoxaline system showed the best activity. Additionally, some other screened compounds were found to be potent in the series by inhibiting LPS-induced TNFα and IL-8 release with IC50 values in the μM range. Amide 2 with para-methoxyphenyl substituent was also beneficial for TNFα inhibition. On the other hand, amide 3, which was inactive, by methylating gave amide 17 with higher activity toward the inhibition of TNFα. Benzyl amine analogues 11–14, with variously substituted benzyl units, were also active, indicating the importance of rotatable bonds for activity. What is very important is that the evaluation of compounds 1–17 by screening in vitro cell viability assays did not show a cytotoxic effect [8].
Investigated thiazole derivatives 1–17.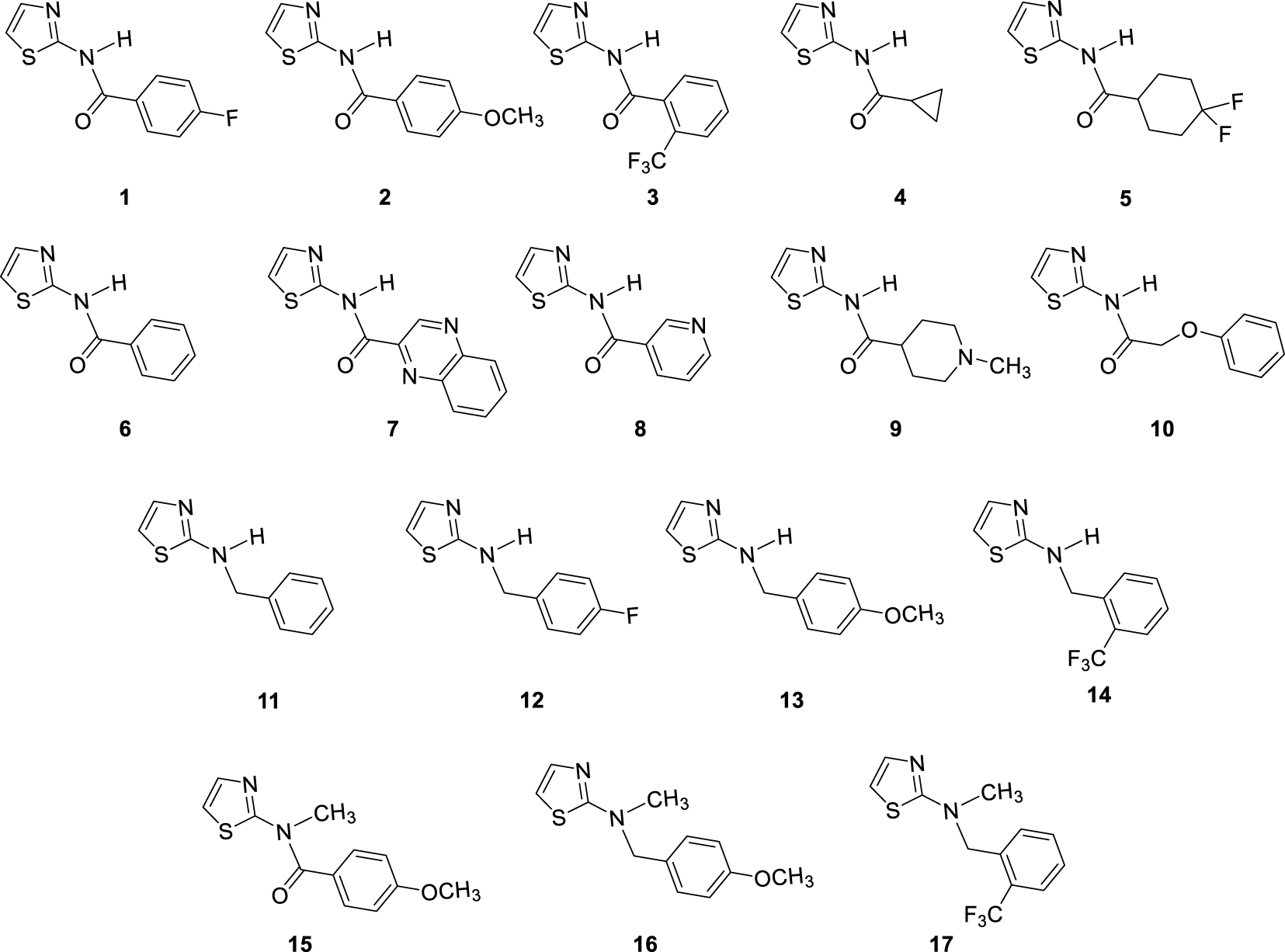
In the current study series of 1,3-thiazole fragments 1–17 (Figure 2) were further investigated as potent cholinesterase inhibitors prompted by the results on anti-inflammatory activity without cytotoxic activity. Some of these compounds (Figure 2), as well as those from the literature (Figure 1), especially with secondary amines as linkers might be cations at physiological pH. While performing in vitro assays we haven’t observed issues with solubility at the measured concentrations. Our aim in this fragments library design was to follow rule of 3, considering the MW, clogP, number of hydrogen donors and acceptors including rotatable bonds. Obtained results are just preliminary data that can lead to conclusions of developing of existing compounds to drug-like compounds. The anti-inflammatory effect of AChE inhibitors was documented previously [9, 10, 11, 12]; it appears that this neuro-immunomodulation comes as the result of the protection of ACh from being split by the AChE enzyme, enhancing the cholinergic pathway. According to the experimental results in this research, the cholinesterase inhibitory and anti-inflammatory activity of 1,3-thiazoles 1–17 will be correlated to see if the same compounds inhibited LPS-stimulated TNFα cytokine production in PBMCs and enzymes cholinesterases. Molecular docking is employed to visualize the possible conformations of thiazoles as cholinesterases inhibitors and to obtain insight into the stabilizing interactions present within the docked active site of AChE and BChE.
2. Results and discussion
2.1. Chemistry
Thiazole derivatives 1–17 (Figure 2) synthesized earlier have been investigated for cholinesterase inhibitory activity due to their evidenced potential as anti-inflammatory agents [8].
Breafly, the amides 1–10 were synthesized from 2-aminothiazole and various carboxylic acids (Figure 2) activated with 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxide-hexa-fluorophosphate (HATU), along with diisopropylethylamine (DIPEA). All amines 11–14 (Figure 2) were synthesized by a reductive amination reaction starting from 2-amino thiazole [13]. Reductive amination was carried out at 80 °C on a stirrer with a heating element overnight. Reductive amination is a reaction in two steps; in the first step, imines are formed, also isolated and characterized in the previous study of the anti-inflammatory activity of 1,3-thiazoles 1–17. The corresponding amines 11–14 were obtained with NaBH4 as a reducing agent. To assess the influence of the NH group as an H-bond donor on cholinesterase inhibitory activity, amide 2 and amine 14 are N-methylated and gave amide 15 and amine 17, respectively. For this purpose, iodomethane with NaH was used as the base.
2.2. Cholinesterase inhibitory activity
Structures of tested 1,3-thiazole derivatives 1–17 (Figure 2) can be grouped into four different sets of compounds. Compounds 1–10 are amides with various substituents at acyl part. The second set of tested 1,3-thiazoles were four amines 11–14, reduced analogs of amides with the same substituent. One amide (2) and two amines (13, 14) were methylated in order to examine the influence of the –NH group on biological activity, as mentioned above.
Presented compounds were screened for acetylcholinesterase and butyrylcholinesterase inhibition activity (for details see Section 3.2). The results are given in Table 1 and reported as maximal inhibition of AChE and BChE at the maximal concentration tested and, where achieved, the IC50 value was calculated. Final concentrations of compounds in tested solutions are expressed in μM values. Maximal tested concentrations were not the same for all compounds due to the problems with solubility in the applied system. Compounds were tested in a wide range of concentrations and adjusted to the solubility of the individual compound.
Inhibition of AChE and BChE by 1,3-thiazoles 1–17 and calculated IC50 values
| Compound | AChE IC50 (μM) | AChE % Inhibition∗ | BChE IC50 (μM) | BChE % Inhibition∗ |
|---|---|---|---|---|
| 1 | — | 31.8 ± 3.6 (500) | — | 39.1 ± 3.6 (500) |
| 2 | — | 28.6 ± 0.8 (500) | — | 30.0 ± 3.3 (500) |
| 3 | 621 | 56.7 ± 3.8 (800) | — | 45.2 ± 3.9 (1000) |
| 4 | — | 47.4 ± 1.2 (1000) | — | 53.1 ± 3.7 (1000) |
| 5 | — | 35.0 ± 3.1 (1000) | 304 | 71.5 ± 3.0 (1000) |
| 6 | — | 40.0 ± 2.1 (1000) | 195 | 75.3 ± 2.9 (1000) |
| 7 | 91 | 56.3 ± 2.1 (100) | — | 19.5 ± 4.1 (100) |
| 8 | 765 | 54.8 ± 1.1 (1000) | — | 26.1 ± 2.1 (1000) |
| 9 | 502 | 57.4 ± 4.4 (1000) | — | 47.0 ± 4.1 (800) |
| 10 | — | 18.9 ± 6.9 (1000) | — | 33.7 ± 2.1 (1000) |
| 11 | — | 32.1 ± 2.3 (1000) | — | 47.4 ± 3.6 (1000) |
| 12 | — | 36.4 ± 1.3 (800) | — | 49.5 ± 1.3 (800) |
| 13 | — | 21.8 ± 4.5 (250) | — | 10.4 ± 6.0 (250) |
| 14 | — | 39.8 ± 2.7 (500) | 385 | 66.1 ± 2.9 (800) |
| 15 | — | 36.3 ± 1.2 (800) | 760 | 51.5 ± 0.1 (800) |
| 16 | — | 50.3 ± 3.2 (1000) | — | 28.7 ± 4.2 (1000) |
| 17 | — | 22.5 ± 3.4 (500) | — | 24.7 ± 2.6 (500) |
∗Numbers given in parentheses represent maximal concentrations tested in μM.
Among the 17 tested 1,3-thiazoles, all the compounds showed some inhibitory activity on both enzymes. IC50 values were calculated for compounds 3, 7, 8, and 9 for the AChE (Figure 3) and 5, 6, 14, 15 for the BChE (Figure 4). Compound 7 showed the most potent inhibition of AChE with an IC50 value of 91 μM and three times more selective toward AChE over BChE (Table 1). The lead inhibitor for BChE is compound 6 with an IC50 value of 195 μM.
Dose-inhibition curves for inhibition of AChE by compounds 3, 7, 8 and 9. Data are mean ±stdev (n = 3).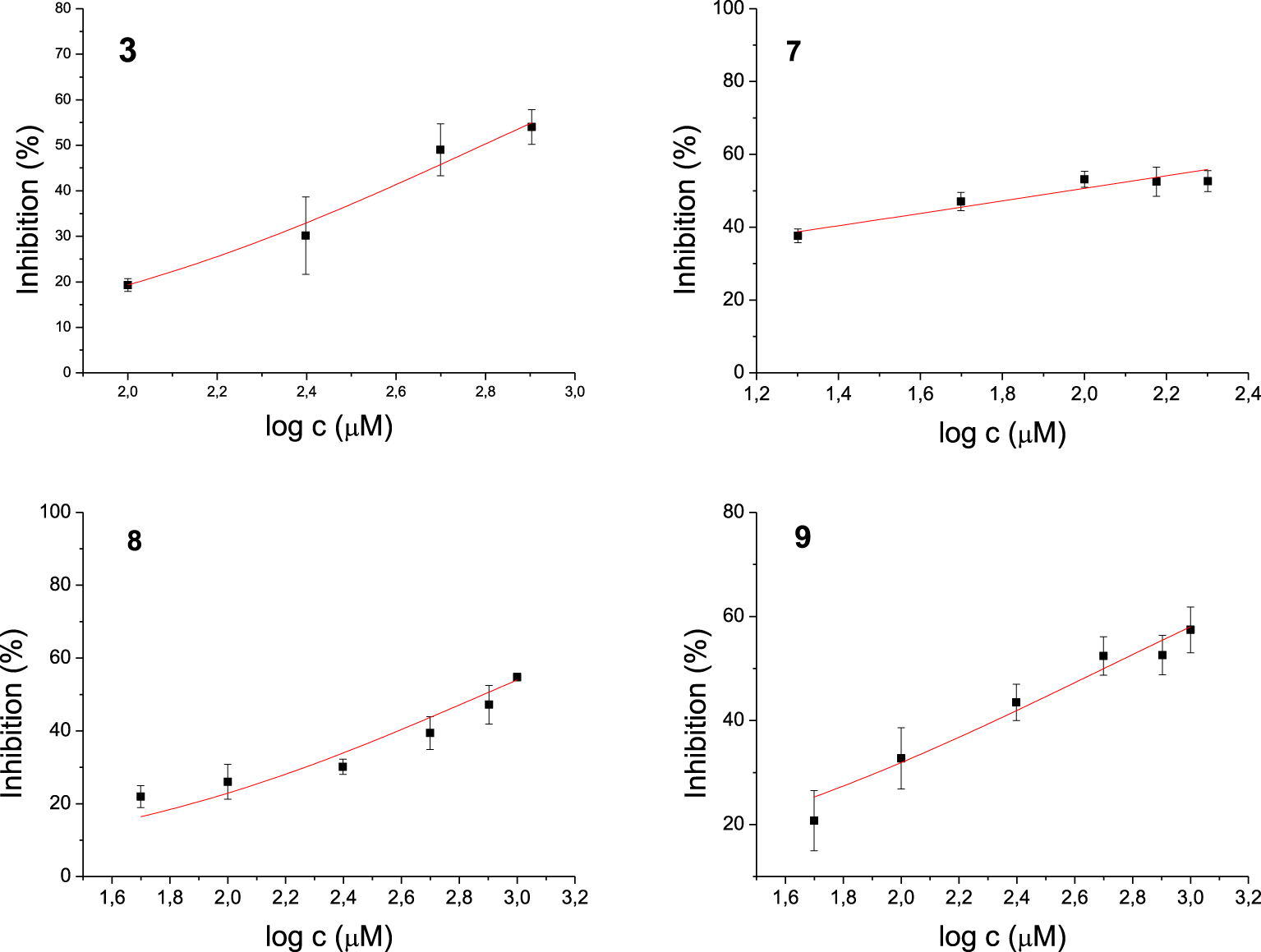
Dose-inhibition curves for inhibition of BChE by compounds 5, 6, 14, and 15. Data are mean ± stdev (n = 3).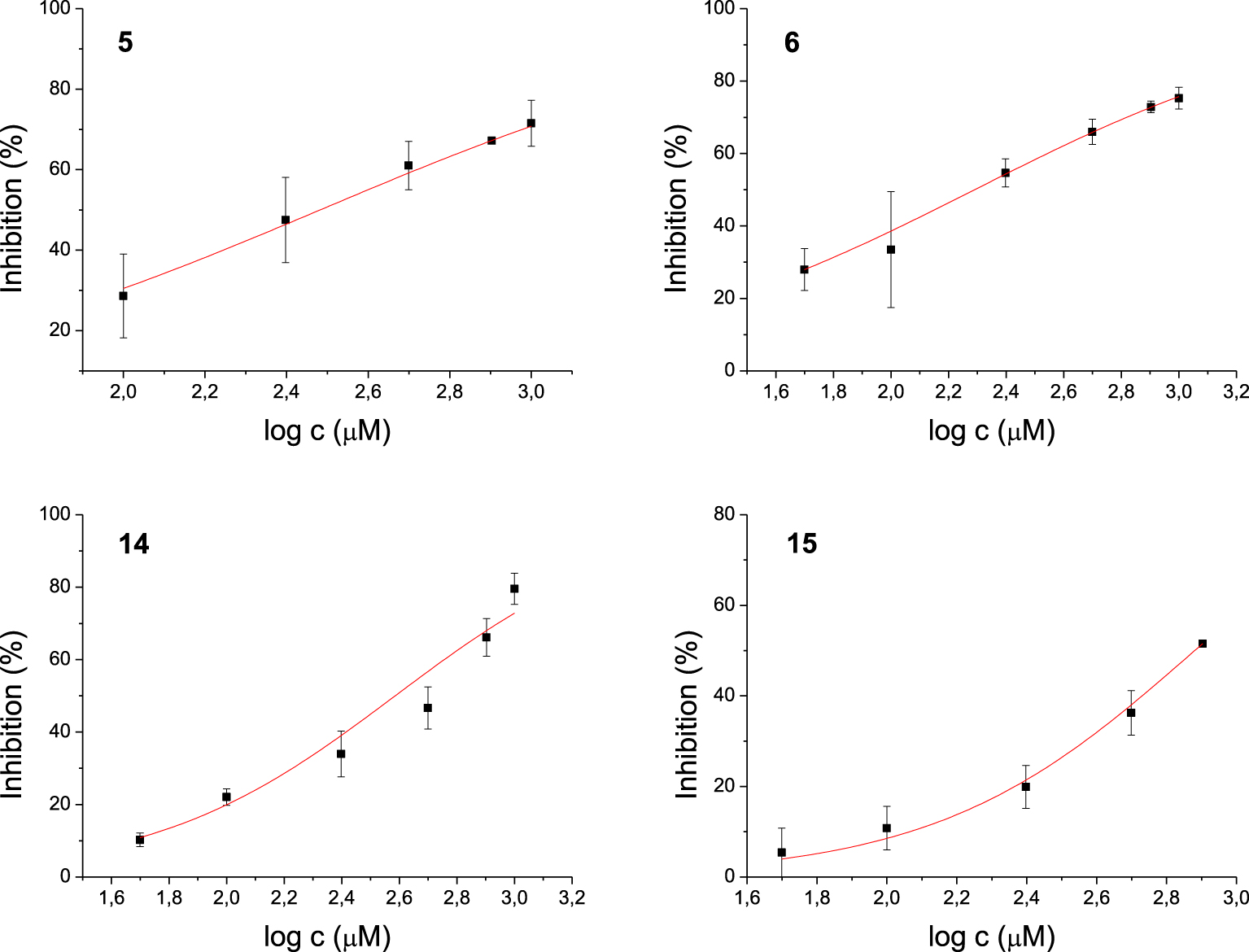
Each of amides 1–10 has a different substituent; among them, four derivatives achieved IC50 for enzyme AChE. Of these four, derivatives 7, 8, and 9 possess N-heterocyclic substituent at acyl part, while compound 3 bears electron-withdrawing group o-trifluoromethylbenzene. The most potent inhibitor of AChE is compound 7, containing a quinoxaline system. The strongest inhibitors of BChE among amides were 5 with 4,4-difluorocyclohexane and 6 with phenyl substituent. Although these compounds differ only in the substituent on the acyl moiety, this structural difference cannot be easily related to IC50 values. Furthermore, all active amides are selective inhibitors with the inhibition preference only for one enzyme. Among the four tested amines, only 14 with o-trifluoromethylbenzene as a substituent showed selective inhibition against BChE.
Comparison of analogously substituted amides and amines leads to the conclusion that there are no significant differences except for phenyl substituted compound 6 and benzyl substituted 11, with amide 6 showing a more positive effect on BChE inhibition.
Four 1,3-thiazole derivatives contain 4-metoxybenzene substituents: amide 2, amine 13, and their N-methylated counterparts amide 15 and amine 16. In this series, the only compound that showed inhibitory activity is amide 15 (only against BChE). It seems that the possibility of hydrogen bonding due to the presence of –NH group (as in amide 2) does not play a role in the inhibitory activity toward BChE. However, in the case of amines, N-methylation had the opposite effect: compound 14 was active against BChE, while in its methylated counterpart 17, the activity was drastically reduced [8].
2.3. Molecular docking studies
To obtain insight into the structures of the complex between the active site of cholinesterases and selected thiazole derivatives, we used molecular docking. According to experimental results shown in Table 1, among all tested thiazoles, molecule 7 (containing quinoxaline) stands out as the best potential inhibitor of acetylcholinesterase, with the lowest IC50 value. Weaker but still promising inhibitory potential toward AChE is observed for compounds 9, 3, and 8. Therefore, we performed docking of molecules 3, 7, 8, and 9 into the empty active site of AChE (coordinates taken from PDB structure 2ACE [14]) using the Autodock program suite [15]. The number of requested genetic algorithm dockings was 10, resulting in ten different conformations of the thiazole within the active site. According to the roughly estimated energies obtained by docking, molecule 7 again shows better inhibitory potential than other tested thiazoles, with the lowest value of estimated free binding energy and inhibition constant (Table 2). The other three thiazoles have mutually comparable values of estimated free energies of binding and Ki, as already found by the experiment.
Results of molecular docking of selected thiazoles into the active site of AChE
| Compound | Estimated free energies of binding (lowest/highest) | Estimated inhibition constants, Ki (lowest/highest) | Number of distinctive conformational clusters | Distribution of conformations within clusters | ||
|---|---|---|---|---|---|---|
| 3 | −4.93 | −4.79 | 242 | 308 | 5 | 5, 2, 1, 1, 1 |
| 7 | −5.52 | −5.47 | 90 | 97 | 1 | 10 |
| 8 | −4.42 | −4.32 | 572 | 680 | 3 | 5, 3, 2 |
| 9 | −4.24 | −4.05 | 782 | 1040 | 2 | 8, 2 |
Free energies of binding given in kcal⋅mol−1, inhibition constants in μM.
More importantly, compound 7 takes ten mutually similar poses; all conformations belong to the same conformational cluster with quinoxaline “anchored” to the acyl pocket, where it is stabilized by residues Phe288 and Phe290. The remaining investigated molecules show more variety in taking poses within the active site. Again, the range of estimated values of binding free energy and Ki is the smallest for compound 7. To scrutinize the results suggested by molecular docking, we performed additional geometry optimizations of the active site docked with selected thiazoles, utilizing a quantum mechanical cluster-continuum approach [16]. We used the procedure we applied earlier to test potential AChE and BChE inhibitors [17]. For each investigated system, the structure with the lowest estimated free energy and Ki was chosen and prepared for QM optimization. During the optimization, the α-carbon atoms of residues in the active site were held fixed, enabling the active site’s architecture in the enzyme to be preserved [16] (details in Supporting Information). The Gaussian 16 program package was used [18], and structures were optimized at the CPCM/B3LYP/6-31G(d) level of theory, with the dielectric constant 𝜀 = 4. The estimate of the relative thermodynamical stability of investigated systems was assessed by calculating the final energy at CPCM/B3LYP+D3/6-311+G(2df,p)//CPCM/B3LYP/6-31G(d) model, where D3 stands for empirical dispersion correction (the Grimme’s D3 atom pairwise dispersion correction [19]). The optimized structure of the complex [7-AS] formed between molecule 7 and the active site of AChE is presented in Figure 5. It is visible that the quinoxaline moiety strongly interacts with aromatic residues in the active site: the π–π stacking is observed with Phe288, Phe290, Tyr121, Phe330, and His440. Each of these five residues is placed in proximity of quinoxaline, within a distance from 3.9 to 4.8 Å. Additional stabilizing interaction is the hydrogen bond between the H-donating –NH group of the 7 and the oxygen of Tyr121. The length of this hydrogen bond in the optimized structure of [7-AS] is 2.19 Å, as shown in Figure 5.
Optimized structure of the complex between the active site of AChE and molecule 7, [7-AS] Hydrogen atoms of residues are not shown for clarity.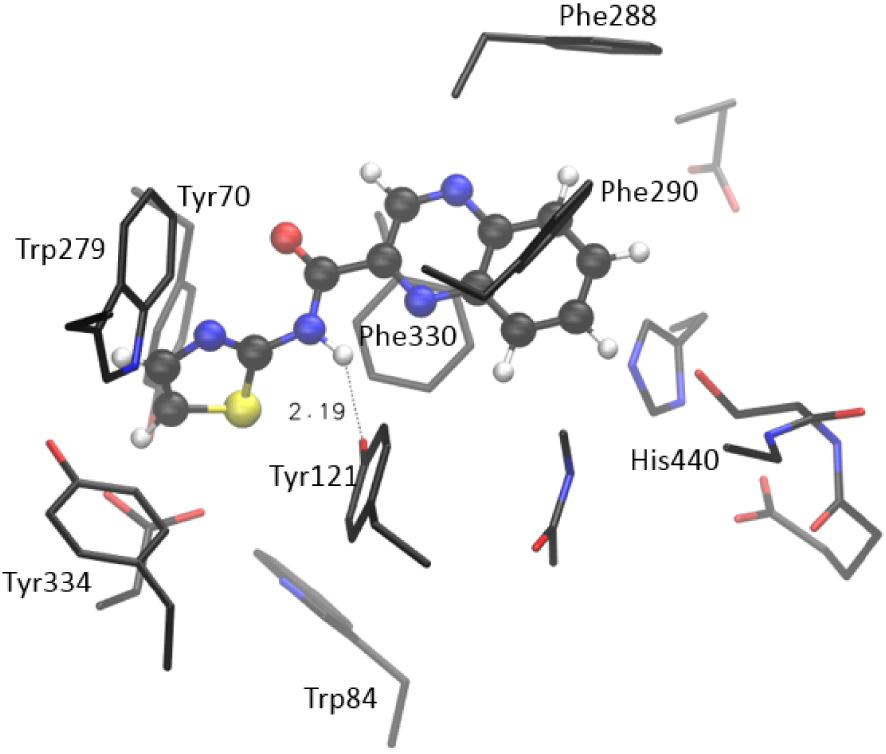
The steric placement of compound 7 enables additional favorable stacking between the thiazole part of the molecule and remaining aromatic residues: Tyr334, Tyr70, Trp84, and Trp 279. All these amino acids are placed within the ∼4 Å from the thiazole moiety of 7. These interactions are responsible for the relative thermodynamic stability of the complex: the calculated enthalpy for the formation of the complex is −23.0 kcal⋅mol−1. (This value represents the enthalpy of the complex formation reaction: Molecule 7 + Active site → [7-AS], see Supporting Information for details.)
It must be mentioned that the experimental measurements were performed using the AChE isolated from an electric eel (EeAChE), while the crystal structure of AChE isolated from Torpedo californica (TcAChE) was used for modeling. To verify findings made by molecular docking into TcAChE, we subsequently performed docking of compounds of interest into the active site of AChE from electric eel, using coordinates taken from PDB structure 1EEA [20]. The complex obtained by docking compound 7 into the active site of EeAChE was again more stable than the one docked with 3, with estimated free energies of binding for the most stable conformation of −5.24 and −4.86 kcal⋅mol−1 for 7 and 3, respectively (with the TcAChE, the values were −5.52 and −4.93 kcal⋅mol−1, as shown in Table 2). The structures of active sites of TcAChE and EeAChE docked with 7 are shown in Figure 6, and active sites docked with 3 are presented in Figure 7.
The active site of AChE isolated from Torpedo californica (coordinates taken from 2ACE.pdb) docked with molecule 7 (left), and the active site of AChE isolated from electric eel (coordinates from 1EEA.pdb) docked with the same molecule (right). The starting structure for DFT geometry optimization was taken from the complex obtained with TcAChE.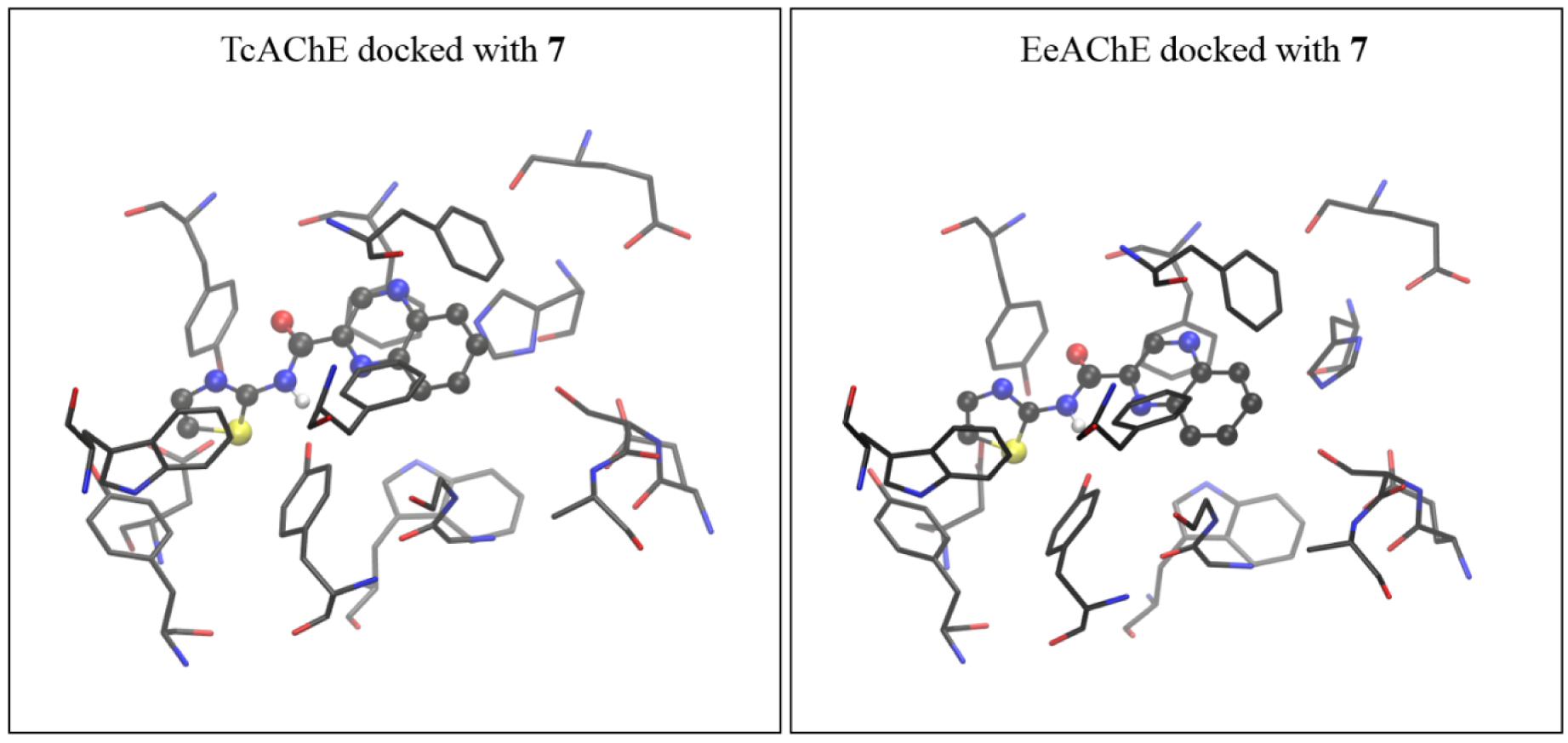
The active site of AChE isolated from Torpedo californica (coordinates taken from 2ACE.pdb) docked with molecule 3 (left), and the active site of AChE isolated from electric eel (coordinates from 1EEA.pdb) docked with the same molecule (right). The starting structure for DFT geometry optimization was taken from the complex obtained with TcAChE.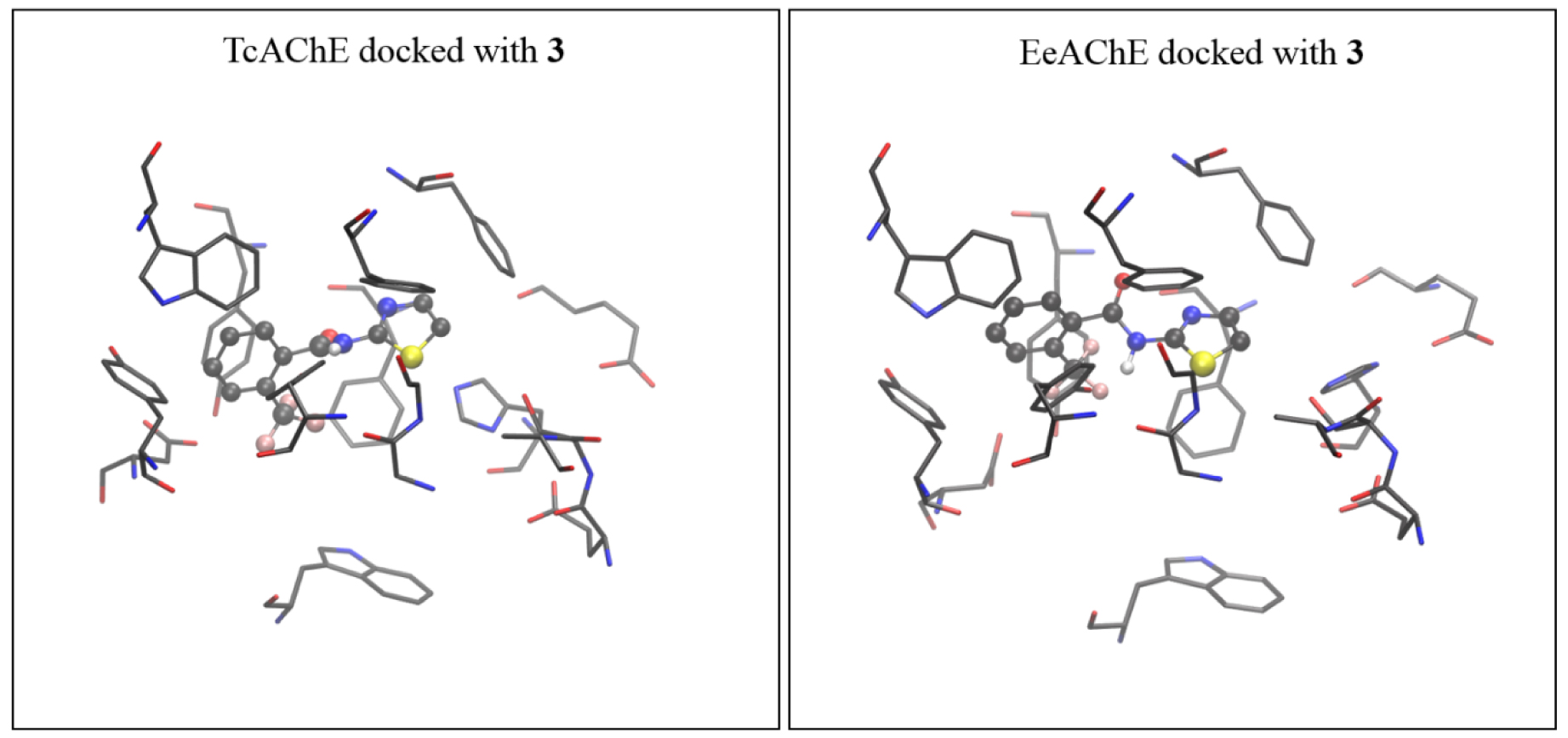
It turns out that the geometries of TcAChE and eeAChE docked with molecules 7 and 3, respectively, are very similar. Therefore, we feel that using the active site assessed from TcAChE docked with 7 and 3 as the starting geometries for more computationally demanding DFT geometry refinements did not affect the final results.
The optimized structure of the complex [3-AS], formed between molecule 3 and the active site of AChE, shown in Figure 8, is obtained starting from the most stable pose given by the docking. Unlike the positioning of the 7, the thiazole moiety of 3 is oriented toward His440.
Optimized structure of the complex between the active site of AChE and molecule 3, [3-AS] Hydrogen atoms of residues are omitted for clarity.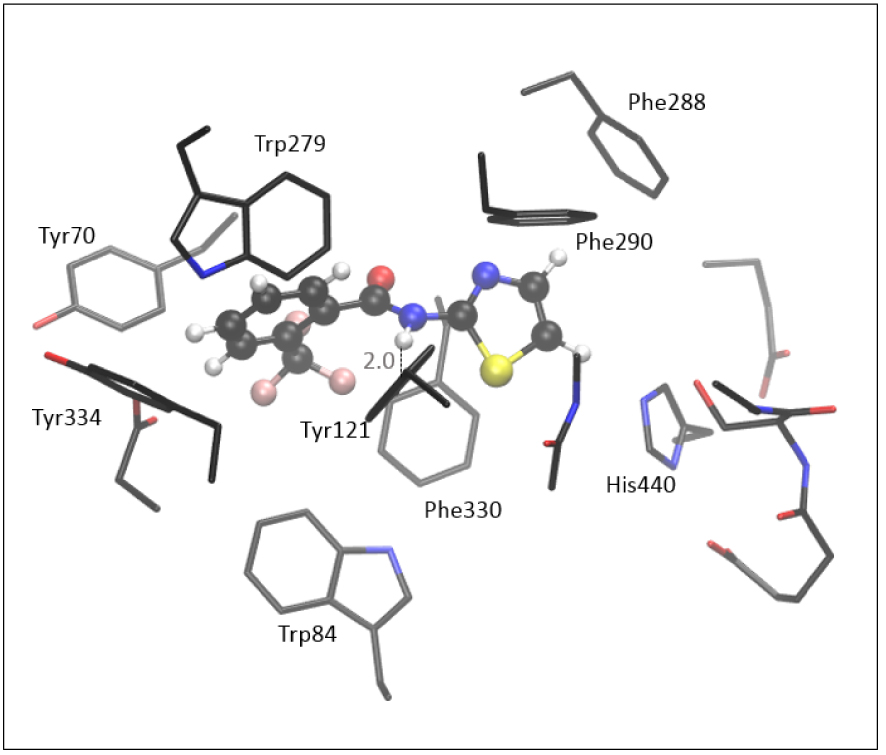
According to the experimental data (Table 1) and results assessed by molecular docking (Table 2), compound 3 has weaker inhibitory potential toward the AChE than the 7. The computationally assessed enthalpy of complex formation for molecule 3 is −10.8 kcal⋅mol−1, indicating that the [3-AS] is less stable than the complex with quinoxaline compound 7. It should be emphasized that these relative thermodynamic stabilities obtained by QM refinement are not an exact quantitative indicator of inhibitory potential; their primary purpose is to examine the reliability of results assessed by molecular docking. The stabilizing interactions in [3-AS] include stacking between the thiazole part of the molecule with residues Phe288, Phe290, His440, Phe330, and Tyr121, all of them being situated within 5 Å from the thiazole. The trifluoromethylphenyl interacts with Trp279, Tyr70, and Tyr334. As in other structures of the active site of AChE docked with amides, the H-bond between the hydrogen of –NH of amide 3 and the oxygen in Tyr121 is present in [3-AS] (Figure 8).
The experimental testing of the ability of thiazole derivatives to inhibit butyrylcholinesterase showed that the best candidates were compounds 5, 6, 14, and 15. The results of molecular docking of these molecules to the active site of BChE are presented in Table 3.
Results of molecular docking of selected thiazoles into the active site of BChE
| Compound | Estimated free energies of binding (lowest/highest) | Estimated inhibition constants, Ki (lowest/highest) | Number of distinctive conformational clusters | Distribution of conformations within clusters | ||
|---|---|---|---|---|---|---|
| 5 | −4.83 | −4.44 | 286 | 555 | 2 | 9, 1 |
| 6 | −4.51 | −4.42 | 495 | 580 | 2 | 7, 3 |
| 14 | −4.17 | −3.71 | 882 | 1520 | 4 | 3, 4, 2, 1 |
| 15 | −4.31 | −4.17 | 688 | 916 | 2 | 9, 1 |
Free energies of binding given in kcal⋅mol−1, inhibition constants in μM.
The active site of AChE contains nine aromatic residues, whereas there are six in the active site of BChE. Here, we did not perform additional geometry optimization and refinement of the active site docked with the tested thiazoles. Figure 9 shows the active site of BChE docked with molecule 5; four aromatic amino acids (Trp82, Tyr128, Tyr114, and His438) surround the thiazole subunit, and the difluorocyclohexyl fragment of 5 is positioned within the acyl pocket of the active site, at a distance of 4.3 and 4.4 Å from Leu286 and Val288. The H-bond is observed between the hydrogen of the –NH group and His438.
The structure of the active site of BChE docked with molecule 5. Hydrogen atoms are omitted for clarity.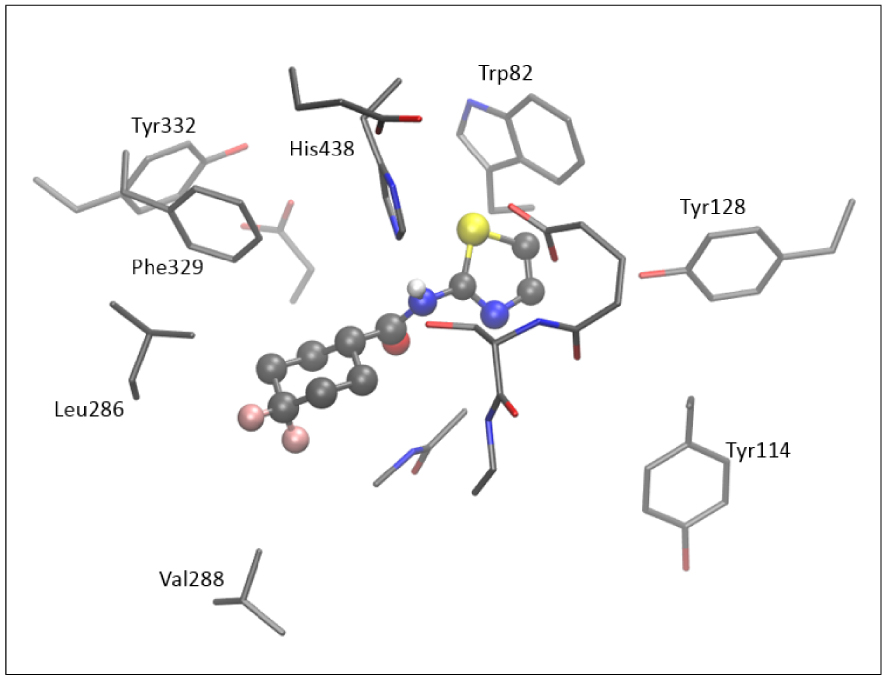
2.4. Structure–activity relationship
Obtained results of IC50 values of AChE and BChE inhibition were summarized in structure–activity relationship overview (Figure 10) together with our previous study concerning the newly synthesized 1,3-thiazoles 1–17 (Figure 2) and their anti-inflammatory activity on lipopolysaccharide (LPS)-stimulated tumor necrosis factor-alpha (TNFα) production and causing increased ACh production in human peripheral blood mononuclear cells (PBMCs).
Structure–activity relationship of most active compounds.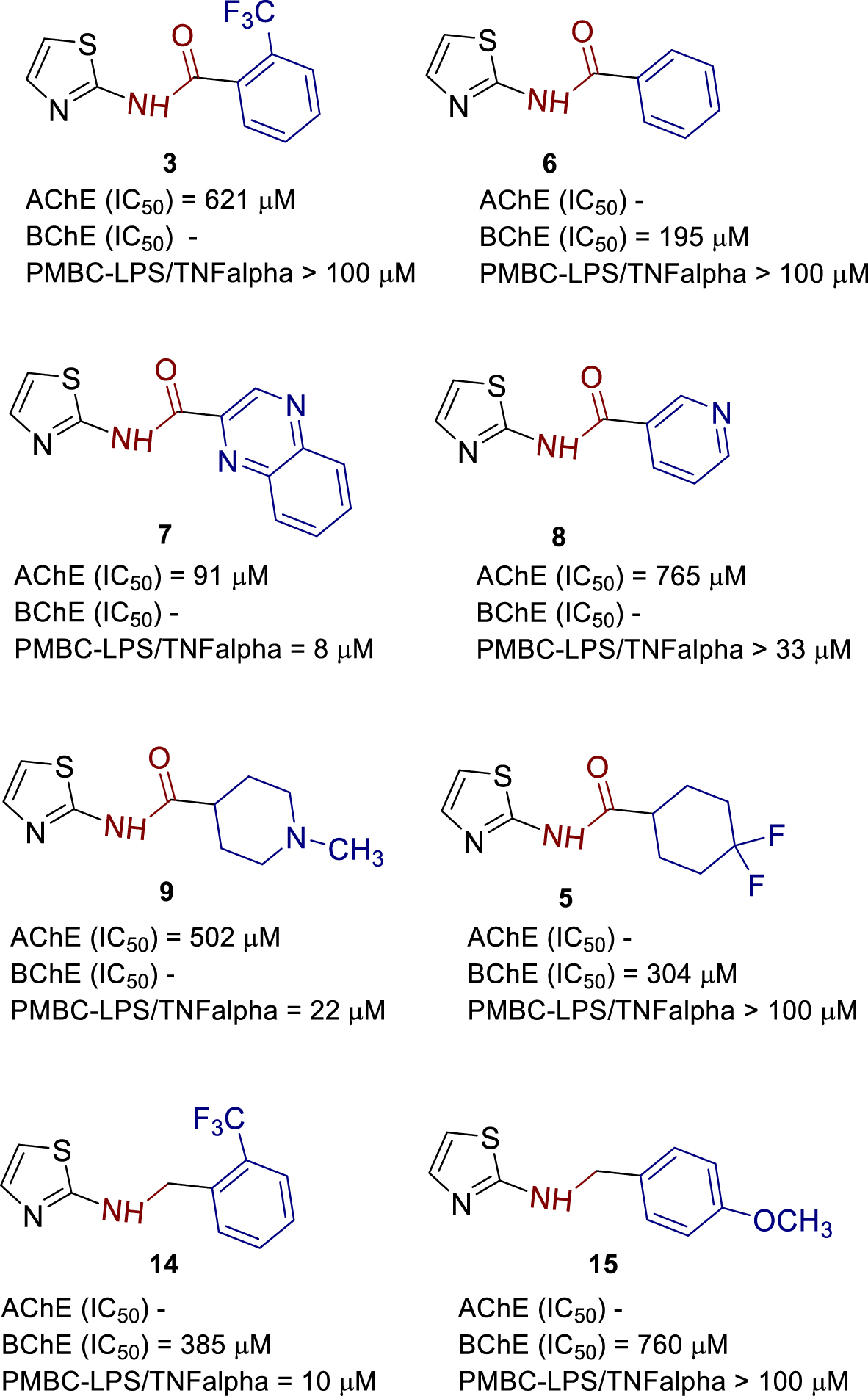
It is known that the cholinergic anti-inflammatory system, a part of the vagus nerve system, is a powerful tool by which the organism regulates the inflammation excess to ensure the elimination of the threat and prevent any potential tissue disruption or damage. The key player in this pathway is acetylcholine (ACh), and its actions are mainly bound to receptors α7nAChR and M1AChR [9]. The cholinergic pathway functions by releasing the ACh, which agonizes the α7nAChR receptor and causes Ca2+ influx into cells. Increased calcium levels in cells lead to activation of NFκB transcription factor and suppression of immune response. This mechanism can be interrupted by cholinesterases (ChE)—enzymes that degrade ACh to acetic acid and choline. Therefore, the inhibitors of AChE and BChE enzymes could also have anti-inflammatory effects [21, 22, 23, 24, 25, 26].
It is noteworthy to point out that out of 17 tested thiazoles, compound 7 with fused quinoxaline system (Figure 10) showed the most potent inhibition of AChE with an IC50 value of 91 μM. Selectivity over BChE (Table 1) was three times. Compound 7 also showed inhibition of LPS-induced TNFα with an IC50 value of 8 μM which demonstrates that compound 7 can modulate inflammatory response through inhibition of AChE. Replacing quinoxaline with pyridine AChE inhibition was decreased eight times, selectivity over BChE retained, and TNFα inhibition decreased four times, which led to the conclusion of importance to AChE inhibition of π–π stacking as well as pKa modifying.
By combining computational studies with biological evaluations, compound 14 showed inhibition of BChE with IC50 value of 385 μM, selectivity over AChE, and inhibition of LPS-induced TNFα with IC50 value of 10 μM that is in the range of compound 7. SAR suggests that 1,3-thiazole-2-amino linker with flexible benzyl type of coupled substituent is preferred for BChE selectivity over AChE.
These promising data on the series of small fragments provided us with a good starting point to further design and synthesize 1,3-thiazoles derivatives with computational studies as potent and selective ChE inhibitors with anti-inflammatory activity and acceptable physicochemical properties drug-like compounds.
3. Materials and methods
3.1. General
The amidation test reactions and the reductive amination reactions [8] were carried out on a magnetic stirrer with a heating body, while part of the amidation reactions was carried out on a shaker with a heating body. Chromatographic separations were performed on a Biotage instrument using puriFlash silica gel-filled columns (4 g, 15 μm and 12 g, 15 μm). All products obtained were identified and characterized by ultra-high-efficiency liquid chromatography (UPLC) with mass and UV detectors and NMR. 1H NMR and 13C NMR spectra were recorded on Bruker Ultra Shield 300, Bruker Ultra Shield 400, Bruker Ultra Shield 500, and Bruker Ultra Shield 600, depending on the amount of compound and the desired spectra. Ultra-high performance liquid chromatography monitoring the range of the reaction and determining the purity of the final product was performed on a Waters Acquity Ultra Performance LC with an SQD mass spectrometer (UPLC-MS/UV).
3.2. Cholinesterase inhibitory activity
Acetylcholinesterase (eeAChE, E.C. from electric eel), butyrylcholinesterase (BuChE, E.C. from equine serum), Tris-HCl buffer, acetylthiocholine iodide (ATChI), S-butyrylthiocholine iodide (BTChI), and galantamine hydrobromide were purchased from Sigma-Aldrich (St. Louis, MO), 5,50-dithiobis-(2-nitrobenzoic acid) (Ellman’s reagent, DTNB) was purchased from Zwijndrecht (Belgium). The AChE/BChE inhibitory potentials of 1,3-thiazoles 1–17 were determined using modified Ellman’s method [27]. Briefly, 180 μL Tris HCl buffer (50 mM, pH 8.0), 10 μL of AChE/BChE (final concentration 0.03 U/mL, prepared in 20 mM Tris HCl buffer, pH 7.5), and 10 μL tested solution (final concentrations, depending on solubility, in a range 20–1000 μM) were mixed and pre-incubated for 5 min at 4 °C. The reaction was then initiated with the addition of 10 μL of DTNB (final concentration 0.3 mM prepared in Tris buffer) and 10 μL of ATChI/BTChI (final concentration of 0.5 mM prepared in Tris buffer). AChE/BChE activity was measured using a 96-well microplate reader (IRE 96, SFRI Medical Diagnostics) at 405 nm for 6 min at room temperature. Non-enzymatic hydrolysis was measured as blank for control measurement without inhibitors. The non-enzymatic hydrolysis reaction with added inhibitor was used as a blank for the samples. The enzyme was replaced by an equivalent buffer amount. The experiment was run in triplicate. Percentage enzyme inhibition was calculated according to the equation: Inhibition(%) = [(AC − AT)∕AC] × 100: where AC is the activity of enzyme without test sample and AT is the activity of the enzyme with a test sample, and represented as mean values ± standard deviation. Data were used to calculate IC50 value by a nonlinear fit of compound concentration (log) values vs. response. Samples were dissolved in ethanol. The inhibitory activity of ethanol was measured, and its contribution to inhibition was subtracted.
4. Conclusion
Thiazole derivatives 1–17 were investigated in order to screen their cholinesterase inhibition activity. Some 1,3-thiazole fragments were found to be potent with IC50 values in the μM range by inhibiting AChE or BChE selectively. The connection of their cholinesterase inhibition and inhibiting LPS-induced TNFα were also detected. Concerning their anti-inflammatory activity in our previous research, amide 7 with quinoxaline system showed the best activity and without cytotoxic effect. In addition, compound 14 showed selective BChE over AChE inhibition with anti-inflammatory activity in the range of compound 7. Encouraged by promising data obtained on small fragments, a new series of 1,3-thiazole derivatives is planned to be developed in drug-like molecules with both anti-inflammatory and cholinesterase inhibition activity aiming IC50 values in the nM range with satisfactory ADME-Tox properties.
Conflicts of interest
Authors have no conflict of interest to declare.
Acknowledgements
Presented synthetic work has been performed at Fidelta Ltd., Zagreb. We thank the University of Zagreb Computing Centre (SRCE) for granting computational time on the ISABELLA cluster.