



 CC-BY 4.0
CC-BY 4.0
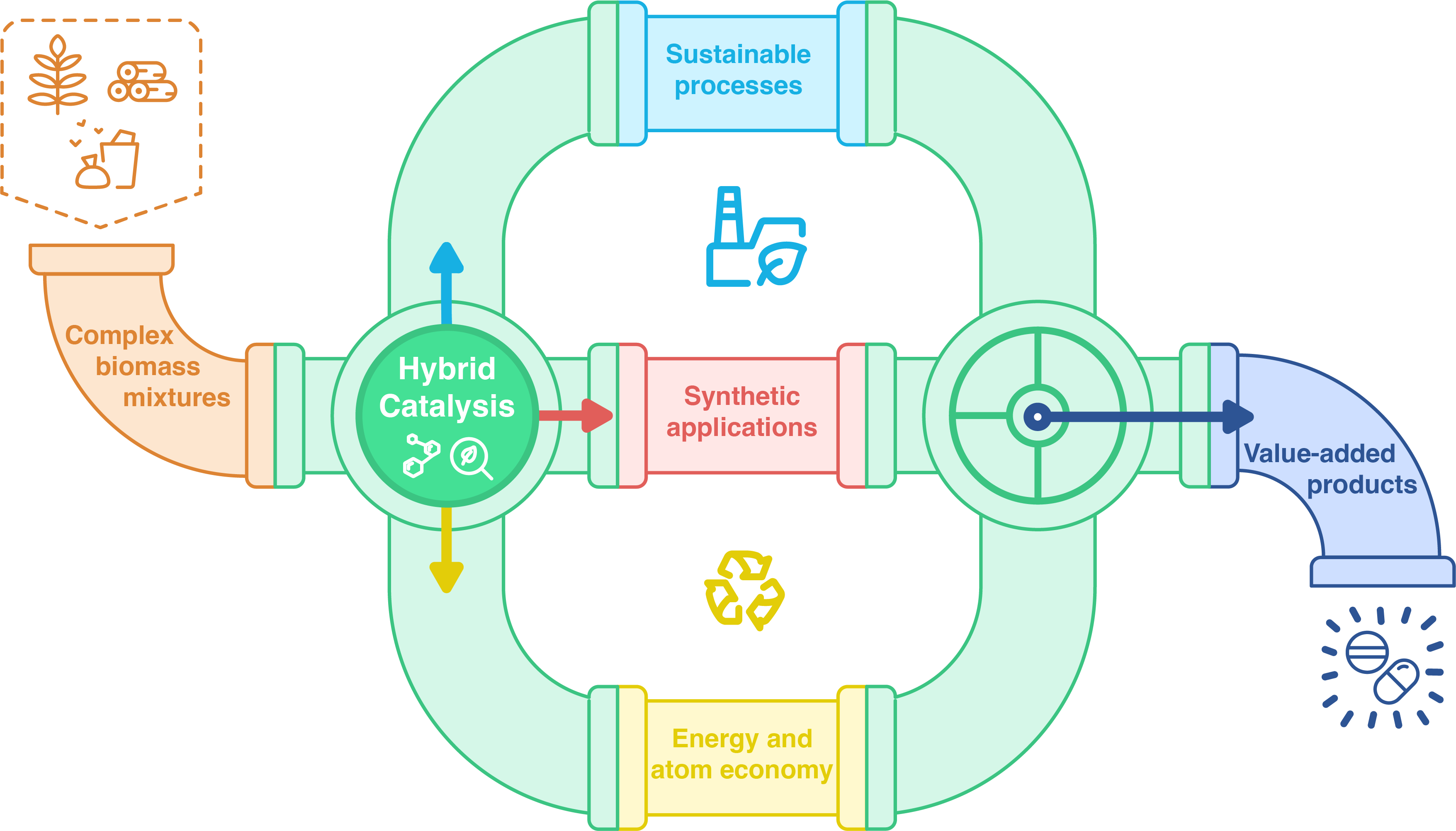
1. Hybrid catalysis for biomass valorization
The exploitation of limited natural resources such as oil reserves has damaged nature and biodiversity for the last decades [1], leading to an accelerated exhaustion of these resources. It is estimated that natural oil reserves will be consumed by the second half of the century if the current average exploitation is maintained [2]. With this in mind, it is necessary to adopt sustainable consumption and production models [3]. In order to achieve this objective, chemistry is shifting toward more sustainable practices, notably following the Green Chemistry principles proposed by Anastas and Warner at the end of the twentieth century, which include the use of alternative and renewable prime material sources rather than limited fossil fuels [4, 5]. Hence why, biomass, largely available in unexploited waste, stands out as a highly potential alternative for a new carbon source, especially as it is renewable [6]. Due to its complex composition, biomass presents significant challenges as a substrate for chemical transformations. Catalysis, therefore, has a pivotal role in addressing these challenges. However, conventional catalysts, initially designed for transforming purified products are ill-suited for handling complex mixtures [7]. Biological catalysts such as enzymes, on the contrary, are far more effective with these types of substrates. Indeed, many organisms already use enzymes to break down, depolymerize, and/or transform biomass molecules [8]. For example, cell-wall degrading enzymes (CWDE) are used by bacteria and fungi to feed on lignocellulose, or enzymes of the hydrolase family that are specific for degrading certain triglycerides and fatty acids in the case of vegetable or animal oils. Nevertheless, the use of biocatalysts remains a challenge at present, again due to the complexity of biomass sources which may contain a large number of inhibitors, including molecules produced by plants themselves to defend against pathogen enzymes. Thus, to incorporate biomass as a starting material, innovative catalysts capable of targeting only specific molecules in complex mixtures and that could alleviate the issue of the presence of inhibitors, need to be developed. Rather than working on the development of new, more robust, and more specific catalysts, an alternative solution could be to combine existing catalysts with complementary skills. A smart design leveraging the synergistic potential of catalysts of different natures—biological and non-biological—could significantly enhance chemical transformations. This approach holds particular promise for optimizing existing chemical processes, which often face limitations preventing their maturity and scalability at industrial levels. For illustration, in the case of high energy consumption for heating, where combination of catalysts logically allows to reduce the overall energetic cost by heating just one reactor [9]. Furthermore, such combined orthogonal processes could pave the way for entirely new families of molecules with valuable properties, by enabling the coupling of reactions that are otherwise difficult to conciliate. More specifically, the integration of chemical and biological catalysts could harness their complementarity and/or orthogonality for selectivity. This synergy may result in multi-step catalytic systems with high yields, driven by reaction equilibrium shifts and, in certain cases, the removal of inhibitors either inherent to the biomass or generated during the reaction. This innovative catalytic approach, which has gained significant attention over the past decade [10], is commonly referred to as “hybrid catalysis” (Figure 1).

Main fields of application of hybrid catalysis (enhancement of sustainable development by reducing atom economy, energetic and economic cost; transformation of complex mixtures through the complementary selectivities of the combined catalysts).
Usually, the term “hybrid catalysis” refers to the combination of chemical and biological catalysis. This combination illustrates the main criteria followed when designing a hybrid catalytic system: joining catalysis whose properties, but also limitations, are very distinct. Although hybrid catalysis can technically be composed of as many types and numbers of catalytic stages as desired, to our knowledge, examples with more than two of them are rarely found currently. The first example of a hybrid catalytic system for biomass valorization was reported in 1980 by Makkee et al., where d-glucose was transformed into d-mannitol through the combination of a biocatalytic isomerization and a chemocatalytic hydrogenation [11]. It is important to note that although the starting molecule, d-glucose, was biosourced, there was no question of biomass valorization when this study was carried out. Moreover, although the value of combining these two types of catalysts was demonstrated, particularly in a one-pot reaction, there was no real interest, at that stage, for the treatment of complex mixtures. Regardless of its ingenuity, this first approach only allowed obtaining 46% of d-mannitol, whereas 92% of d-glucose was converted into d-fructose. To improve the efficiency of the system, the process was later optimized by the same authors by combining the same biocatalytic isomerization of d-glucose to d-fructose as seen before with heterogeneous copper-based catalyst supported on silica (Figure 2). The key characteristic of this metallic catalyst is its substrate specificity, as it prefers interacting with d-fructose, and therefore here offers a solution to selectively act on one single substrate in a mixture of isomers, reflected in the increased d-mannitol yield (66%) [12].

One-pot/one-step hybrid process designed by Makkee et al. where a biocatalytic isomerization accesses d-fructose from d-glucose, preferred substrate by a copper catalyst for its subsequent hydrogenation into d-mannitol. Redrawn from Heuson et al. [13].
These pioneer works were a milestone in the design of new catalytic systems and led to the exploration of new combinations of chemo- and biocatalysts to enhance new reaction pathways with uncommon, and sometimes unexpensive, starting materials. Biomass has emerged as an excellent candidate to employ hybrid catalysis for several reasons. One reason is that biomass is largely composed of low energy chemical bonds (e.g., esters, amides, sometimes ethers with higher energy bonds) [14], it makes biomass suitable for reactions with enzymes under mild conditions, unlike fossil fuel-based chemicals, almost exclusively composed of carbon and hydrogen [15]. The second reason is that, biomass provides a wide variety of molecules that are hardly discriminated against by chemical catalysts due to their often low selectivity, leading to the formation of complex product mixtures. In this instance, while offering access to a large number of starting structures, the use of biomass as a starting material also calls for selectivity, potentially offered by enzymes, in order to limit downstream processing steps. Additionally, this complex nature also means that pretreatment systems are needed to avoid potentially toxic or undesirable species, or simply to make the desired starting substrates available, especially in cases where a single product is targeted. For instance, in lignocellulosic biomass, it is mostly cellulose that is converted [16]. The possibility of exploding the entire lignocellulose matrix in a single combined process (with the catalytic valorization steps included directly afterwards), would therefore be advantageous both in terms of atom economy and access to a wider range of new biosourced molecules. Hybrid catalysis could be very helpful in coupling the single-molecule valorization step with a pretreatment one, using a more robust and less specific catalyst, for example.
Indeed, to better understand all the potential of hybrid catalysis, the main attributes and resulting complementarities should be considered when combining catalysts (Figure 3). Biocatalysis, is characterized by its high chemo-, regio- and stereoselectivity [17]. These properties are provided by the tridimensional structure of enzymes, which can selectively host the substrates in their active sites, usually depending on how well they can sterically and electronically interact with each other [13]. This high selectivity is of great interest in the pharmaceutical industry since it can lead to enantiopure compounds [18]. Besides, biocatalytic systems can be conducted in a reaction cascade system, which provides an interesting set of chemical tools and reactions in the same pot, leading to especially high energy- and atom-saving processes [17].

Complimentary catalytic properties of chemo- and biocatalysis, leading to the synergistic properties of hybrid catalysis.
As an example, Merck recently developed a new biocatalytic cascade system for the synthesis of molnupiravir by combining five different enzymes, allowing not only the production of the target antiviral, but also the regeneration of the phosphate needed in the reaction on an industrial scale [19]. This system shortens the previous reported chemical synthesis from ten to only three steps and increases the yield from approximately 10% to 70%. Despite the interesting possibilities of such elegant biocatalytic cascades, the limited stability and high price of enzymes become a bottleneck in their incorporation on an industrial scale [20]. Meanwhile, heterogeneous chemocatalysts can enhance the recovery of the catalysts from the reaction mixture, as well as providing good stability under harsh working conditions, making them more suitable for industrial applications [21]. Regarding their properties, they are mostly related to their electronic structure [13], while leading to lower selectivity due to a very limited steric control of the catalytic center, makes them very versatile and consequently very well suited for complex mixtures. In addition, chemocatalysts tolerate better the exposition to toxic molecules and can then be interesting candidates for “depolluting” reaction media from inhibitors, to which biocatalysts are particularly sensitive and therefore vulnerable [22]. This potential use is perfectly demonstrated by Smeets et al. [23], who were able to design a zeolite-based multifunctional material, for the conversion of allylic alcohol into glycidol, employing the toxic and in situ produced hydrogen peroxide. Here, the TS-1 zeolite was able to both host the concerned enzyme, e.g., glucose oxidase, and simultaneously act as an inorganic catalyst. This hybrid material therefore allowed the production and removal, in the same process, of a substrate that can also be a potential inhibitor. Since the substrate here employed is glucose, this approach could be extended to biomass-containing mixtures, usually rich in polysaccharides. However, these approaches are not yet widespread. Another example of product protection and/or inhibitor removal is the recent development of nonionic surfactants by Novartis [24], capable of hosting water nonsoluble compounds, which could be either the product of the reaction, a chemocatalyst or any other potentially harmful species for the biocatalyst, allowing, at last, the biocatalyst to be protected from them. In addition, as it will be illustrated later in this article, it is also possible to design new types of reactors offering access to a more spatially isolated compartmentalization system while still offering the possibility for molecules to circulate from one compartment to another, using a liquid or solid permeable membrane. In essence, compartmentalization techniques that could behave as decontamination strategies enhance the cohabitat of chemo- and biocatalysts, or avoid inhibition phenomena, and are here again very interesting for biomass valorization. Therefore, chemo- and biocatalysis should no longer be considered as incompatible but rather complimentary. Their unique properties can overcome their respective limitations, making them capable of addressing the afore-mentioned challenging fields of biomass valorization and new compound synthesis.
However, the number of hybrid processes designed to directly run on biomass is still limited. In the case of lignocellulose, the few systems available are mainly aimed at the synthesis of furfural and its derivatives, which can be obtained by the dehydration of the sugars present in it. Mainly derived from hemicellulose, furfural is indeed considered as one of the top 30 platform chemicals [25], as it provides access to many derivatives with very different applications in polymer, pharmaceutical or food industries. Nonetheless, direct and efficient furfural synthesis from untreated biomass remains, again, mostly undeveloped, especially when it comes to generating its functionalized derivatives such as 5-hydroxymethylfurfural (HMF), furandicarboxylic acid (FDCA), or even furfurylamines like 5-aminomethyl-2-furancarboxylic acid (AMFC). Indeed, the processes to reach these compounds typically need tedious separation steps and control of the reactivity of the aldehyde moiety of furfural, which has been found to be particularly unstable since it can undergo multiple reactions (acetalization, acylation, aldolization, amination, halogenation or redox reactions, among others) [25, 26]. Therefore, such high reactivity not only does it make furfural a good platform molecule, but also a particularly excellent precursor for the above cited molecules. However, it also complexifies its related processes and storage, requiring fully integrated systems where furfural is present, ideally, only present in transient form before being converted into a more stable molecule. In this context, advances in hybrid catalysis have offered several solutions for the direct conversion of lignocellulosic biomass into furfural and its derivatives that otherwise would be difficultly accessible by conventional catalysis. A good example illustrating this upgrading of lignocellulosic biomass is the work of He et al. [27, 28, 29, 30, 31]. These authors first developed a one-pot/three-step process involving the initial acidic depolymerization of various biomass sources (chestnut shell, corncob, sorghum stems, rice straw or even bamboo shoot shells), into sugars, typically d-xylose. In a second step, a tin-based catalyst was used to convert the sugar into furfural, which was finally selectively employed in a third step by different biocatalysts (e.g., reductases, alcohol dehydrogenases, transaminases) to branch a whole range of chemical groups leading to higher value-added products (e.g., alcohols, carboxylic acids, amines). More recently, the authors have improved their process by combining the first two steps, where cellulose and hemicellulose are directly chemocatalytically converted into furfural, which can then be transformed, in a second step, by the selected biocatalyst (Figure 4). In the case of furfurylamine production, a transaminase was employed for example, either as the isolated enzyme or as a whole cell system [32, 33]. Different variants of the process have been designed, employing a similar variety of biomass sources, adjusting it to the desired products. In most of them, a biobased carrier (rice stalk, nutshell, etc.) was employed for the preparation of the concerned heterogeneous tin catalyst. The similar composition of the carrier and the lignocellulosic substrates improves their interactions, mainly due to their similar hydrophobicity and therefore favored adsorption on the surface. In addition, the catalyst supported on rice stalk showed 592-, 17- and more than 2-fold larger specific surface area, pore volume and pore size, respectively, compared to the previously employed material, which might increase the substrate load and therefore provide higher catalytic performance. The reaction media appears as biphasic systems, like in the case of the preparation of furfurylamine, which was performed in a toluene/water solution, working at 170 °C and 35 °C for the chemocatalytic and biocatalytic step, respectively. The authors highlight the utility of this arrangement as it allows the avoidance of the possible inhibition of the biocatalyst by furfural, as it proved to be toxic at high concentrations [32]. The use of toluene might be a drawback when scaling up, and therefore greener alternatives should be explored. This one-pot/two-steps system has been further explored by adding oxalic acid as the cocatalyst in the first step, followed by the biocatalytic step by using whole cells in deep eutectic solvent (ethylamine/hydrochloride/glycine/water) [33]. The motivation behind this approach was again to tackle several problems related to furfural (e.g., low solubility in water, degradation or cross-polymerization) by enhancing its extraction while keeping the above-mentioned energetic advantages of working in one single reactor. Here, adding an organic acid provides a partial pretreatment of the mixture, adding valuable properties to the hybrid system beyond the catalysis itself and demonstrating flexibility and polyvalency.

General one-pot/two-steps hybrid approach for different biomass conversion (e.g., corncob, rice stalk) into value-added furfural derivatives by combining a tin-based heterogeneous catalyst supported on a biobased carrier and a biocatalyst.
Overall, these examples demonstrate particularly well the versatility of the combination of two catalysts of different nature, once the common coupling conditions are defined. Typically, the authors were able to combine a broad number of different biocatalysts with the same chemical catalyst under similar conditions, giving them access to a complete range of molecules, while simplifying the development of the associated processes. The potential of combining a relatively universal chemical catalyst acting first on the biomass, with variable polymer compositions from different sources, was highlighted. The same intermediate, in this case furfural, can then be transformed by different enzymes.
Such versatility enabled by hybrid catalysis was also reflected in the work of Zhang et al. [34], who managed to functionalize asymmetric C–H bonds on a wide alkane substrate scope through a photobiocatalytic hybrid one-pot/two-steps reaction system. In the first stage, sulfonate anthraquinone was employed for the photo-oxidation of different alkanes (cyclohexane and toluene derivatives, mainly) to ketones, which were subsequently converted, in a second biocatalytic stage, to different functional groups (carboxylic acids, amines, alcohol cyanides and esters) by an enzyme (e.g., transaminase, reductase, lyase) (Figure 5). Although the incompatibility of both catalytic steps imposed a two-step manner way of working, a single aqueous phase at 30 °C could be employed for the whole reaction, notably thanks to the sulfonate group present in the photo-organocatalyst, which favors the latter soluble in water. As a result, the green character of the process is favored as it does not require undesired and often toxic organic solvents. Another remarkable aspect relies on the fact that this coupled catalytic system not only accepts a broad substrate scope, comprising in this case of 14 different substrates, but also leads to the formation of a large and often chiral range of products. Hybrid catalysis provides, again, one single process applicable to numerous substrates that leads to many different products of higher value, notably enantiomerically enriched. This last aspect illustrates, indeed, the additional complementarity of the catalysts, with the association of a nonstereoselective step with a highly stereoselective one, allowing to reach a wider product range in a relatively simple approach.

Photobiocatalytic one-pot/two-steps hybrid approach for C–H functionalization of different alkanes, alkenes and toluene derivatives. The system combines sodium anthraquinone sulfonate (SAS) as a photo-organocatalyst with various enzyme families, in aqueous medium at 30 °C. Redrawn from Zhang et al. [34].
Additionally, the catalytic singularity of hybrid technologies can also give rise to unexplored reaction pathways in organic chemistry, allowing new and often structurally rich products to be reached. The synthetic potential of hybrid catalysis relies on the conjunction of the numerous reactions enhanced by chemocatalysts (e.g., metal-catalyzed cross-couplings, organocatalytic asymmetric synthesis, oxidoreductions) and the high selectivity of biocatalysts, resulting, for instance, in the delicate creation of new stereogenic centers. Among the most targeted chemical structures by synthetic chemists, special attention has been drawn to nitrogen-containing structures, as they are often found in building blocks used to access pharmaceutical compounds [17]. As an example, a N-acetyl-d-glucosamine-derived chiral alcohol was prepared by Hao et al. employing a chemobiocatalytic approach in a stereo- and especially chemoselective manner [35]. The process combined tyrosine hydrochloride for the dehydration of N-acetyl-d-glucosamine at 160 °C, and a carbonyl reductase for the asymmetric reduction of a ketone at 30 °C (Figure 6).

One-pot/two-steps process for the asymmetric synthesis of acetamido-5-(hydroxyethyl)furan that combines a chemocatalytic dehydration with tyrosine chloride (TyrCl) as catalyst and biocatalytic reduction of N-acetyl-d-glucosamine. Redrawn from Hao et al. [35].
Compared to the previous processes, where the intermediate is purified, the one-pot/two-steps hybrid catalysis led to an increase in the enantiomeric excess of the furane derivative from 91% to over 99%. In addition, the 99% of the E factor (6025) is related to solvent waste from product purification, which again highlights the sustainable character of this one-pot transformation. Nine different substrates were obtained in good to excellent yields, showing the versatility of the system to reach multiple high value-added building blocks from inexpensive chitin. Overall, this work reinforces the potential of valorizing inexpensive biomass through hybrid catalysis to access new compounds with high enantiopurity and to reduce the environmental impact. Another interesting example in the framework of stereocontrolled synthesis is the recent work of Gastaldi et al. [36], who prepared, from achiral alkynes, chiral polyhydroxylated a rare analog of monosaccharides through a one-pot/two-steps process at 60 °C. A gold N-heterocyclic carbene, responsible for the hydration of the alkyne to the corresponding ketone, was combined with an aldolase, which enhanced the formation of a C–C bond in the presence of an aldehyde and led to two new stereogenic centers. A family of five different chiral monosaccharides were formed in excellent yields, all of them being high value-added products with potential applications in food and pharmaceutical industries. As in the previous example, the reaction was performed as a one-pot/two-steps, due to the different temperatures and pH working ranges of the catalysts, 60 °C and pH 3 for the chemocatalytic step, and room temperature and pH 7 for biocatalytic one, respectively. Striving to reach a one-step process, an attempt to increase the pH of the chemocatalytic step was done, which unfortunately derived in a quick decrease of the ketone production. In addition to the synthesis of new products, the hybrid process again shows a major advantage in terms of atom economy, as it presents a fourfold lower value of E factor (0.5) with respect to the previous conventional synthesis, and also helps avoiding the isolation of the freshly formed hydroxyacetone, which is particularly unstable given its tendency to polymerize and/or undergo aldol condensation [37]. With the same hydration step and further alkyne transformations, Li et al. have very recently reported the hybrid regioselective conversion of alkynes to chiral alcohols also using a gold catalyst [38]. Similarly to the previous example, the reaction occurs in a one-pot/two-steps manner and combines the chemocatalytic hydration step with the biocatalytic reduction of the intermediate ketone to a chiral alcohol by using a carbonic anhydrase. Again, a step differentiation was required so the pH could be increased from acidic to alkaline (pH 8) and so the temperature could be decreased from 65 °C to room temperature, which contributed to the energy and atom efficiency by not requiring any separation or exogeneous energy-demanding process for the second step. This process afforded a wide scope of differently substituted chiral alcohols with excellent enantioselectivities (>99%) and good to excellent yields (most of products obtained in between 60% and 90% yield). Most remarkably, within this work, the same authors have developed two other parallel hybrid systems for the synthesis of chiral alcohols that allow the enlargement of the substrate scope to alkanes and alkenes [38]. When alkanes were employed as starting materials, a photocatalytic oxidation followed by a biocatalytic reduction was achieved, while in the case of alkenes, a metal-catalyzed oxidation was performed, followed by a biocatalytic reduction. Therefore, Li et al. was able to design three hybrid reactions to produce chiral alcohols from nonfunctionalized alkanes, alkenes, and alkynes, which widely enlarged the substrate scope and led to a notable increase in the final value of the obtained products (Figure 7). In all cases, the one-pot/two-steps setup was maintained to avoid incompatibility issues, mainly related to pH, temperature, or light-exposure working conditions.

Three parallel one-pot/two-steps hybrid approaches for synthesis of chiral alcohols depending on starting from alkanes, alkenes and alkynes, respectively. Redrawn from Li et al. [38].
Among these approaches, the photobiocatalytic one is particularly interesting from a sustainable perspective, as it allows the use of visible light as an energy source. This case echoes the above-mentioned work of Zhang et al. [34], which already showed the potential of photocatalysts to oxidize C–H bonds to carbonyls for their potential transformation by biocatalysts. Expanding this reactivity presents therefore huge potential as different chemical functions can be obtained from alkanes, which could also be extrapolated to biomass-derived substrates, such as fatty acids or lignin derivatives.
All these studies can be seen as great proof of concept regarding the advantages that can be provided by hybrid catalysis in organic chemistry. Firstly, it affords the possibility to combine catalysts in one single reactor instead of sequential systems, and therefore reducing significantly the energetic and economic cost of the process. Secondly, as it is frequent for one-pot systems, the isolation of intermediates is not necessary. This leads to more efficient atom use, as evidenced in the afore-mentioned E factor values, it results in safer processes because reactive species are rapidly transformed, minimizing uncontrolled side-reactions and undesired byproducts. Over these technical aspects, the synthetic potential should be highlighted. In all the reported works, innovative reactional approaches were highly stereoselective. Particularly interesting for the pharmaceutical industry, hybrid catalysis can therefore provide high value-added structures in a relatively convenient and economic way. Other hybrid processes, which will be detailed in the next part of this work, alternatively focus more on introducing highly reactive and functionalizable chemical groups on building blocks to access new platform molecules. Whatever the final objective, hybrid catalysis could be the key to unlocking numerous simplified, and more importantly, efficient synthetic strategies, especially for future biorefineries.
Beyond all the above-mentioned synthetic and industrial interests, technical adaptability of hybrid catalysis with the reaction should also be mentioned. The main aspects to consider are the catalysts involved, their allocation in the reactors and the sequence of the reaction steps. The choice of the catalysts depends on the nature of the reaction, the requirements of the process and the energy source. The most relevant hybrid approaches for biomass conversion were recently classified by Heuson and Dibenedetto based on the nature of the biocatalysts involved, and the way they were combined [13]. But, as a final illustration of both the complexity and advantages of hybrid catalysis, we finally propose to detail the constraints that can govern the combination of catalysts of different natures and operating conditions through a series of examples we recently developed around the conversion of glucose into new platform molecules, which notably involve the development of a completely new type of reactor specifically designed to overcome the inhibition of the catalysts involved in the transformation.
2. Application: from glucose to furfurylamides through hybrid processes
In line with the advantages that we have detailed previously for hybrid catalysis, we have developed several approaches to exploit two main advantages: the design of more economical processes and the discovery of completely new synthetic routes to access new families of molecules. We will illustrate these two aspects through the conversion of glucose into HMF, a process that has already been implemented but which we have tried to improve in a hybrid way. Then through the production of furfurylamines, and then of furfurylamides again through a hybrid route, families of molecules with interesting potential yet to be identified.
2.1. From d-glucose to 5-hydroxymethylfurfural through a two-pots/one-step system
As we have already seen from several examples cited above, one d-glucose-derived platform molecule that is at the heart of all concerns is 5-hydroxymethylfurfural (HMF) [39]. Indeed, HMF can be further valorized by different reactions, including hydrogenation, which leads to 2,5-dimethylfuran (2,5-DMF) [40], a promising fuel additive, or to a precursor to terephthalic acid by a dehydrative Diels–Alder reaction with ethylene to form p-xylene, followed by an oxidation [41, 42]. Alternatively, HMF oxidation can also yield 2,5-furandicarboxylic acid (FDCA), a biosourced alternative to terephthalic acid with significant potential for polyester plastic production [43, 44]. However, d-glucose, despite its abundance, is not the preferred substrate for efficient HMF production compared to the more reactive and costly d-fructose. This preference is due to the fact that fructose, with its fructofuranose tautomer, dehydrates to a furanic ring with lower energy barriers than d-glucose [45]. Therefore, isomerizing d-glucose into d-fructose is a crucial step that cannot be bypassed. One solution for this additional step could be an enzymatic d-glucose isomerization, which is already widely used in industrial production of high-fructose corn syrup (HFCS) [46]. This enzymatic reaction remains the method of choice due to its high efficiency and moderate cost. Although this process requires, high-purity d-glucose, specialized buffers and multiple ion-exchange resins to remove metallic impurities in food-grade HFCS, it is still favored over chemical isomerization, despite renewed interest in the latter [47, 48, 49, 50]. The enzymatic process, however, is limited by its thermodynamic equilibrium (Keq ≈ 1), restricting d-glucose conversion [51, 52]. While this equilibrium is not an issue for HFCS production (which tolerates d-glucose/d-fructose mixtures), it is less than ideal in the case of HMF production as the remaining d-glucose cannot be converted into the final product. One solution is then to shift the isomerization equilibrium by directly coupling the first reaction with d-fructose dehydration, as the latter is irreversible. Nevertheless, this requirement introduces challenges, as catalyst compatibility between the isomerization system and the dehydration system remains problematic. However, Huang et al. proposed a very interesting one-pot/two-steps hybrid process combining a thermophilic glucose isomerase immobilized on aminopropyl-functionalized mesoporous silica (FMS) with a heterogeneous Brønsted acid catalyst (propylsulfonic-FMS-SO3H) in a THF/H2O (4/1 v/v) solvent mixture [53]. But, although isomerization with the enzyme at 363 K achieved a promising 61% d-fructose yield, subsequent dehydration required a higher temperature (403 K), which only led to a HMF 30% yield, and more importantly, caused complete denaturation of the glucose isomerase. Overall, these results highlight the difficulty of integrating these processes together.
As an alternative to what has been proposed, we thought it would be interesting to turn to an alternative two-pot/one-step process for this particular situation, as the separation of the reactions could allow different reaction conditions (pH, temperature, etc.) when run in parallel, while still benefitting from the possibility of continuously consuming d-fructose. Indeed, strategies have already been adopted to combine enzymatic isomerization of d-glucose to d-fructose and chemical dehydration. These approaches involve the separation of bio- and chemocatalysis and d-fructose transportation between the aqueous phase for isomerization and an organic phase for subsequent reactivity. Huang et al. first introduced this concept by adding sodium tetraborate to the aqueous isomerization medium, forming a fructoboronate complex through interaction with d-fructose [54]. This complex was then transported to a separate organic phase with the aid of a cationic quaternary ammonium salt, enhancing d-glucose conversion by shifting the isomerization equilibrium toward d-fructose. The dehydration of d-fructose in the organic phase led to an increase in HMF yield to 63%, compared to 28% without borate addition, and by improving the conversion of glucose from 53 to 88%. However, the selectivity of the complexation between d-glucose and d-fructose was suboptimal, which was later addressed by Delidovich et al., who optimized the chemical nature of the boronate species [55]. A global process exploiting this concept for d-glucose to HMF production has concomitantly been proposed by Alipour [56]. Therein, d-fructose is complexed with phenylboronic acid and transferred to an organic phase, which is then separated and brought into contact with an acidic ionic liquid phase to release free d-fructose. The d-fructose-rich ionic liquid is subsequently used for dehydration to HMF in a biphasic medium, with HMF being back-extracted into a final low boiling point organic phase. This method relies on four distinct media and intermediate phase separations which consequently limits its industrial potential.
Inspired by these sequential steps, we therefore sought to propose a more streamlined and integrated approach that reduces the number of steps and avoids the use of costly and difficult-to-recover ionic liquids. It would be composed of the following steps: (1) isomerization of d-glucose in a first aqueous phase at neutral or slightly basic pH, compatible with the enzyme; (2) complexation of the d-fructose produced with an arylboronic acid, allowing its solubilization from the first aqueous phase into a second, organic “transport” phase; (3) hydrolysis of the complex at the interface of a second, acidic pH aqueous “receiving” phase; in which (4) the d-fructose is dehydrated to HMF using an acid catalyst. This concept is illustrated in Figure 8.

General principle of the hybrid “two-pot/one-step” simultaneous process applied to the transformation of d-glucose to HMF (d-Glc = d-glucose, d-Fru = d-fructose, HMF = 5-hydroxymethylfurfural).
Through a first study [57], we optimized the various parameters related to each of the steps independently, starting with the isomerization step with glucose isomerase. For this, we studied a temperature range from 323 K to 363 K and tried to find the optimum pH between 4.5 and 9. These parameters were applied to a commercially available immobilized glucose isomerase (Sweetzyme® IT Extra, d-xylose ketol-isomerase), to make the process as scalable as possible. As a result, we observed that the enzyme exhibited maximum activity around pH 7.5, at a temperature of 343 K. As the extraction of d-fructose by formation of a complex [d-fructose–boronic acid]− is favored if the pH of the aqueous solution containing d-fructose is larger than the pKa of boronic acid and the pKa of the main boronic acids are lower than 8.5, we chose pH 8. This condition allows us to overcome the pKa-related lock of the selected boronic acid to transport d-fructose to the organic phase while maintaining a relative activity of more than 80% of the enzyme. Under these conditions, a final yield of 55% in d-fructose isomerization could be obtained. We then turned our attention to optimizing the d-fructose extraction from the aqueous “giving” phase to the organic “transport” phase. For the latter, methylisobutylketone (MIBK) was selected over other solvents like methyl-tert-amyl ether and dimethyl carbonate based on solubility, toxicity, and boiling point. Despite similar d-fructose extraction yields, MIBK’s higher boiling point (117 vs. 86 °C for methyl-tert-amyl ether) made it more suitable for chemical catalysis at 80–90 °C. Then, inspired by a previous study [58], we chose the couple lipophilic arylboronic acid (carrier) along with a quaternary ammonium salt (Aliquat336®) as a phase transfer agent for d-fructose extraction into the MIBK. At the chosen pH conditions (pH 8), aryl boronic acid ArB(OH)2 is actually present under its hydroxylated anionic form, as a tetrahedral aryltrihydroxyborate ArB(OH)3− [59]. At the interface between the aqueous and organic phases, d-fructose further reacts with the arylborate to form a tetrahedral fructoboronate ester. The fructoboronate complex then forms an intimate ion pair with Aliquat336®, which enables its transportation to the organic phase [60]. We then studied the influence of the boronic acid structure to optimize the kinetics and maximize the selectivity of d-fructose complexation/transportation. Seven arylboronic acids, which differ through the electronic properties of their substituents and thus by the pKa, were screened in the complexation with d-fructose: 3,4-dichlorophenylboronic acid (3,4-DCPBA), 3,5-dichlorophenylboronic acid (3,5-DCPBA), 2,4-dichlorophenylboronic acid (2,4-DCPBA), 2,3-dichlorophenylboronic acid (2,3-DCPBA), 4-tert-butylphenylboronic acid (4-TBPBA), and 4-(trifluoromethyl)phenylboronic acid (4-TFMPBA). With an excellent extraction yield and rate (46.5% and 1.48 μmol/min, respectively) under our conditions, 3,4-DCPBA was chosen as the aryl boronic acid for the continuation of our studies. The 3,4-DCPBA/Aliquat336® molar ratio was then studied, again with a view to maximizing extraction speed and yield. The best yield was obtained with a 1/2 ratio, as the increase in Aliquat336® concentration meant that no more d-fructose could be extracted above it. Keeping a molar ratio of aryl boronic acid/Aliquat336® equal to 1/2 and at fixed initial d-fructose amount, both 3,4-DCPBA and Aliquat336® concentrations were then varied. From this screening, we found that the d-fructose/3,4-DCPBA/Aliquat336® ratio of 1/1/2 (with d-fructose used at 100 mM) was the most effective, even though the extraction yield could not be further improved compared to previously obtained results. The release of d-fructose into the aqueous “receiving” phase was also studied as a function of its pH. By changing the pH from 8 to 1, the release yield was increased from 20% to 100%, and over 91% at pH 5. We concluded that, as expected, the pH of the recipient phase should be as low as possible. Finally, the optimal conditions for d-fructose dehydration to HMF were studied at different temperatures, as for the isomerization, along with different catalyst-to-d-fructose mass ratios. For this study we chose, as an alternative to a strong homogeneous acid that cannot be recycled in processes, a commercially available acidic resin containing strong sulfonic groups. This resin, indeed, facilitates a potential reusability and recyclability of the catalyst. In addition, this reduces the risk of using and heating strong acids in liquid form. A temperature of 80 °C was found to be the most effective with this catalyst. At this temperature, no humins were detected, avoiding significant resin browning observed at 90 °C.
In the formerly optimized conditions, a sequential process was tested. The first step involved simultaneous d-glucose isomerization and d-fructose transport by using a fructoboronate complex. This was followed by the hydrolysis of the complex, releasing d-fructose into the aqueous receiving phase. Finally, the third step dehydrated the released d-fructose to HMF using the sulfonic resin. All these steps were performed one after the other, the organic phase being first isolated, before being put in contact with the aqueous “receiving” phase, without the chemocatalyst, the latter being added in a third time. As a result, only 57% of d-fructose (based on 100% of potential d-glucose to be converted) was extracted from the “giving” phase, followed by a release of 63% of the extracted d-fructose into the “receiving” phase, and 20% of this extracted d-fructose could be dehydrated into the final product, leading to a very small overall HMF yield of 5%. However, as each phase is decoupled from the previous/subsequent one, this sequential approach benefits little from the shift in the isomerization equilibrium. The latter is only pulled by the d-fructose extraction to the organic phase, which also has its own equilibrium. Not so surprisingly, low yield was obtained. Logically, we then attempted to couple the three phases and the two catalytic stages, into a single, very simple system, as described in Figure 9.

Illustration of the combined process for the conversion of d-glucose to HMF using a triphasic system, combining an immobilized glucose isomerase (IGI) and an acidic catalyst. Redrawn from Gimbernat et al. [57].
As a result, the isomerization of d-glucose was achieved with a 70% yield. Half of the produced d-fructose was successfully transported to the “receiving” phase, leading to 35% of d-glucose transformed to d-fructose available for further dehydration in the acidic phase, as for the sequential process. Therefore, it is with no surprise that we again obtained a very low HMF final yield of 4%. Despite showcasing the potential of such a chemoenzymatic catalysis cascade for direct HMF production from d-glucose, our process was clearly limited by additional factors, the most preeminent one being probably insufficient phase agitation.
This consequently led us to rethink completely the architecture of the reactor, toward a more complex, but also more efficient system to promote the continuous extraction of d-fructose toward the receiving phase. In order to propose radically different reaction conditions between the two aqueous phases (donor and recipient), while maximizing diffusion of the d-fructose formed from one to the other, we switched to a rather unusual “H”-type reactor topology (Figure 10) [61]. This reactor incorporates all the stages previously tested, but this time with two separate compartments that can be independently thermostated, linked by a bridge for circulation of the organic transfer phase. With this new reactor at our disposal, we repeated the tests with the different combined phases we had previously carried out, but unfortunately did not observe the increase in d-fructose transfer we hoped. Conversely, we observed a strong accumulation of the fructoboronic complex in the organic phase of the first pot. Given the small diameter of the solvent bridge, we concluded that the main limitation was the lack of circulation of the organic phase between the two pots. Therefore, we added a fin with baffles in the opposite direction in both compartments, allowing the flow of the organic phase created by the rotation of the two stirring systems to be redirected, so that the organic phase could circulate freely from one compartment to the other. This improvement enabled us to increase the rate of d-fructose release into the receiving aqueous phase by a factor of three. However, in order to further improve the process, we optimized a number of additional parameters. Firstly, we observed that, as previously mentioned, a pH difference between the two aqueous phases was important to enable efficient continuous extraction of d-fructose from the donor to the recipient phase. This is because, at basic pH, the fructoboronic complex can form and dissolve in the organic phase, which can then be hydrolyzed in the receiving phase at a more acidic pH, and the greater the pH difference, the more the equilibrium of the two dissolutions is exceeded in the receiving phase. However, following the introduction of the fin with baffles, we observed that the pH of the two phases tended to equilibrate, with the organic phase partially transferring protons from one phase to the other, helped by the new MIBK circulation, albeit rather slowly. We therefore investigated the possibility of buffering the two aqueous solutions in an attempt to slow down or even eliminate this phenomenon. To this end, we used a buffer at three different concentrations in each phase, to see whether the buffer strength also had a predominant role in this limitation, while maintaining a pH difference of 5.5 between the phases (pH 8.5 and 3 for the donor and recipient phases, respectively). As a result, with a sufficient buffer concentration (500 mM), we were able to limit the fall in pH difference to a value of 3.5 over 6 h of reactions, which showed promise for the efficiency of the hybrid reaction. Obviously, this efficiency decreases with decreasing buffer concentration. It should be noted that this type of strategy is obviously not transferable to a larger scale, and it would then be preferable to set up active pH control in each of the bases by adding base and acid respectively, or even to envisage a continuous system directly for the aqueous phases, with the organic bridge remaining static, to dilute the pH variation, while still maximizing the effects of equilibrium displacement.

H-type reactor concept for HMF production from d-glucose in a two-pot/one-step hybrid process combining a glucose isomerase and a sulfonic resin in three different phases. Redrawn from Heuson et al. [13].
In parallel, we also restudied the concentration ratio of d-fructose/3,4-DCPBA, and determined that with this new system a new ratio of 1/0.25 was now more efficient (while maintaining the 3,4-DCPBA/Aliquat336® ratio at 1/2) for d-fructose extraction, while allowing a considerable reduction in the amount of 3,4-DCPBA and Aliquat336® used, which represents another advantage of the new reactor. With these latest optimizations in hand, we tested the new complete hybrid process for HMF production on a scale of 100 mM initial d-glucose. For this experiment, a temperature of 70 °C was set in the first pot (enzymatic isomerization step) while 80 °C was set in the second pot (chemical dehydration step), and isomerization, extraction and dehydration efficiencies were then followed over 32 h. At the end of the reaction, the isomerization, d-fructose extraction and HMF production yields reached 79%, 97%, and 31%, respectively. The extraction yield obtained thus demonstrates the efficiency of our new process and its associated optimized parameters for d-fructose transportation between the two aqueous phases, while the isomerization yield clearly highlighted an equilibrium shift as expected. The carbon balance in both aqueous phases was found to be equivalent to 90% in d-glucose, d-fructose and HMF, proving the efficiency of the release process. The only step that was really limiting here was therefore the dehydration, with a final yield of only 30.9% in the desired product. Although this is on a par with the best attempts previously described, such as that of Huang et al. [53], it seems crucial to now turn our attention to optimizing the dehydration step, probably by choosing a better catalyst than the sulfonic resin, or perhaps by considering a better process control so as to be able to further lower the pH and increase, if possible, the temperature of the second pot, while keeping humins formation as low as possible.
Nonetheless, this first proof-of-concept clearly highlights the effectiveness of the hybrid catalysis concept in obtaining molecules of interest more efficiently, in this case the transformation of d-glucose, that can easily be obtained from biomass, into the highly valuable HMF, under mild conditions. More precisely, it shows how the combination of different catalytic steps within the same system can greatly help shift the equilibrium of reactions that are normally highly reversible. Interestingly enough, it would also be possible to apply this process with the H-shape reactor and organic membrane to an unrefined d-glucose source, typically a lignocellulosic biomass extract, insofar as the MIBK could prevent or, at least, limit the diffusion of polar species that could poison the chemical catalyst toward the phase where dehydration takes place. If we take the concept a step further, why not consider direct cellulose and hemicelluloses depolymerization using an enzymatic cascade in the donor phase, insofar as the first stage takes place under conditions that are entirely compatible with the action of cell wall degrading enzymes. But more than just a synthesis proof-of-concept, this approach, with all the process-level optimization that it required, illustrates how different combinations of catalysts, in one-pot/two-steps or two-pots/one-step systems, can enable radically different catalytic functionalities to be accessed, and offer specific advantages in each case. This makes the combination of these processes highly complementary and interesting.
2.2. One-pot conversion of 5-hydroxymethylfurfural into 5-aminomethyl-2-furancarboxylic acid
As described above, HMF is a very interesting platform molecule as it can be transformed into a variety of value-added building blocks [62, 63, 64], including monomers useful for the production of biobased polymers such as 2,5-dihydroxymethylfuran (DHMF), 2,5-dicarboxaldehydefuran (DCAF), 5-hydroxymethyl-2-furancarboxylic acid (HFCA) and 2,5-furandicarboxylic acid (FDCA), thanks to a wide range of innovative catalytic pathways [65, 66, 67]. The conversion of HMF to FDCA, for example, has been the subject of intensive research in recent decades, and numerous approaches have been reported [67, 68, 69]. Noble-metal-based chemical catalysts demonstrate high efficiency in HMF oxidation, with supported noble metal nanoparticles exhibiting exceptional catalytic performance in liquid-phase oxidation under both alkaline and acidic conditions [70, 71, 72, 73, 74, 75, 76]. Moreover, enzymes were also applied. One of the earliest studies on biological oxidation of HMF was conducted by Koopman et al., who identified a novel HMF oxidoreductase from Cupriavidus basilensis HMF14. By introducing the hmfH gene into Pseudomonas putida S12, a whole-cell biocatalyst was created. This system produced 30.1 g/L of FDCA from HMF in fed-batch experiments, using glycerol as the carbon source, with an impressive yield of 97% [77]. For their part, Dijkman et al. utilized an HMF oxidase, expressed in E. coli, for a four-step oxidation of HMF to FDCA. This FAD-dependent enzyme led over 95% conversion of HMF within 24 h at 25 °C, in an aqueous phosphate buffer solution (pH 7) [78]. Still with the same objective, Carro et al. combined an aryl-alcohol oxidase (AAO) with an unspecific peroxygenase (UPO), both from Agrocybe aegerita in a cascade reaction [79]. AAO reduced O2 to generate H2O2, while enabling stepwise oxidation of HMF to DFF (2,5-diformylfuran) and FFCA (5-formylfuran-2-carboxylic acid). However, as AAO could not oxidize the carbonyl groups in FFCA directly, the authors combined it with the UPO to perform the conversion of FFCA into FDCA using H2O2, generated during the first step, to produce FDCA with a 91% yield after 116 h [80]. Finally, and although examples are rare, a few hybrid attempts have also been made. Liu and coworkers applied an immobilized laccase on magnetic nanoparticles with TEMPO as a chemomediator, obtaining a FDCA yield of 90.2% in 96 h at 35 °C [81]. What all these examples have in common is that they carry out only one reaction type, i.e., oxidation (alcohol to carbonyl and then carbonyl to acid). Obviously, it is much more complex to get catalysts to cohabit when the types of functionalization reactions sought are different, or even in some cases opposed, as is the case for oxidations and reductions. Nonetheless, with the same mindset, furfurylamines have been identified as valuable precursors for biobased polymers such as polyamides, polyimides, polyaspartimides, polyureas, polyhydroxyurethanes, polyimines, and polyenamines. Their monomers can be conveniently synthesized from biosourced furfural derivatives, further supporting their potential in sustainable material development [82]. These compounds also have several applications including the preparation of benzoxazine derivatives for flame-retardant resins [83, 84] and, after conversion to difurfuryl diisocyanates, the replacement of petroleum-based diphenylmethane diisocyanate in polyurethane systems [85, 86]. Furfurylamines are typically synthesized through the reductive amination of the carbonyl group in the furfural structure under mild conditions with inexpensive reagents [87]. However, such processes often require the use of protecting groups, numerous chemical steps, and toxic reductive agents [87, 88]. It is worth mentioning that the reduction combined with the potential oxidation of HMF’s hydroxyl groups, to obtain the corresponding ω-amino aldehyde or acid, is obviously particularly challenging. To circumvent these issues, several chemocatalytic pathways have been recently developed [87, 88, 89, 90, 91, 92]. However, these are not ideal for the synthesis of furfurylamines derived from HMF, owing to the sensitivity of the furan ring to reductive conditions and the tendency of these compounds to form secondary and tertiary amines [65, 66, 67, 93]. An efficient alternative to perform amination of HMF derivatives is the use of transaminases, their primary catalytic function being the nonreductive amination (transfer of an amine) of carbonyl groups in water at low temperatures [34]. Recently, several ω-transaminases were employed for the synthesis of several furfurylamines from HMF derivatives [93, 94]. Dunbabin et al. reported yields up to 92% for the aminated products, starting from different HMF and furfural derivatives. Among them, they achieved the synthesis of 5-hydroxymethylfurfurylamine (HMFA), 5-aminomethyl-2-furancarboxaldehyde (AMFA), furan-2,5-diyldimethanamine (FDMA), and the 5-aminomethyl-2-furancarboxylic acid (AMFC). However, in the case of AMFC, they performed the synthesis directly on 5-aldehyde-2-furancarboxylic acid (AFCA) instead of HMF, as the only amine that can be directly produced from HMF, in a single step with one catalyst, is HMFA, the other furfurylamines requiring a coupled oxidation step, whether before or after the amination. It is precisely here that hybrid catalysis can be of great interest by proposing the combination of an oxidation catalyst with a transaminase, as has already been done for other applications we described earlier, to gain access to other more complex derivatives of HMF-derived furfurylamines. We have paid particular attention to the synthesis of AMFC, as it is a promising building block for polymer synthesis. Being an ω-amino acid, it can be used to produce unnatural peptides such as cyclopeptides [95, 96], but could more generally be integrated in aromatic polyamides. Thus, we attempted to develop the combination of an oxidative metal chemocatalyst and a transaminase for the direct synthesis of AMFC from HMF.
To achieve this synthesis, we took full advantage of the high-throughput screening capabilities of the REALCAT platform in Lille, enabling us to select the best catalysts, both for the chemical catalyst and the enzyme, from a panel of catalysts of different natures, particularly regarding supported metal nanoparticles. Indeed, depending on the metal used and the properties of the support, the selectivity of the catalyst is particularly different, which can lead to the production of undesirable byproducts. In a parallel study, for example, we demonstrated that under certain conditions, several of our supported metal catalysts were capable of performing oxidative deamination of furfurylamines [97], which is obviously problematic in the context of AMFC synthesis, since the latter is then converted back into purely oxygenated derivatives. Another challenge was to find a chemical catalyst capable of operating at a relatively neutral pH, since this is generally the optimum pH for transaminases. Indeed, many supported noble metal nanoparticle catalysts are more active at more basic pH, which is typically the case with gold nanoparticles, for example [36]. After screening 15 catalysts containing nanoparticles of gold, palladium, platinum, ruthenium and even some bimetals, immobilized on different types of supports, metal oxides for the majority of them, we were able to demonstrate that one of the combinations, namely platinum immobilized on silica, was able to carry out the oxidation of HMF to AFCA (the ω-aldehyde acid derivative) directly using the oxygen present in the head-space gas phase of the reaction. This catalyst proved highly effective at pH 8 and at 60 °C, enabling a 100% substrate conversion in less than 24 h. Notably, this temperature could in theory also allow for the use of different transaminases, several of which have already been described as thermostable [98]. A negative aspect of this catalyst, however, was its lack of selectivity for the oxidation of the alcohol or aldehyde function of the substrate. Indeed, when oxidation step takes place before the amination step, apart from the desired intermediate (AFCA), a byproduct from the complete oxidation of the two functions to the acid (FDCA) is also found. Despite the previously mentioned interest of FDCA and the possibility of directly incorporating it into polymerization matrices, its formation naturally limits the obtainable yield of AMFC. Therefore, the use of this catalyst should ideally take place sequentially to the amination, which would avoid the adoption of a one-pot/one-step system. In our process, this represented the main limitation since at the time of the study, we still did not have any thermostable transaminases capable of carrying out the amination of AFCA, and therefore had to introduce the enzyme into the reaction medium in a second step, once the reaction had returned to room temperature. Nonetheless, we could employ one of the transaminases classically described in the literature for furfural amination, e.g., Chromobacterium violaceum. Notably, we had immobilized it beforehand on silica too, in order to enhance its removal from the medium by simple filtration like for the chemical catalyst, simplifying the posttreatment of the process. As a final result, by using two different amine donors, methylbenzylamine and isopropylamine, which are formed through transamination of the thermodynamically stable acetophenone and the easily removable acetone product, respectively shifting the reaction equilibrium. Following this approach, we reached a process leading to a 77% yield in AMFC in 52 h and with FDCA as the only byproduct (Figure 11).

One-pot/two-steps hybrid synthesis of 5-aminomethyl-2-furancarboxylic acid (AMFC) combining an oxidation step catalyzed by platinum nanoparticles immobilized on silica leading to the formation of an intermediate 5-aldehyde-2-furancarboxylic acid (AFCA), which is then aminated by the immobilized CvTA transaminase in a second step in the same reaction medium. Redrawn from Lancien et al. [99].
Interestingly, this process has paved the way for other attempts by various research groups. We should mention the one from Giri and coworkers [100], who recently reported a robust transaminase from Shimia marina (SMTA) that enables the scalable amination of HMF to biobased furfurylamines with high activity and broad substrate specificity. In their case, they succeeded in developing a similar cascade reaction, only enzymatically, coupling an aldehyde reductase from Synechocystis sp. PCC 6906 (SAHR), in a one-pot/two-steps system, nearly yielding a quantitative production of AMFC in 30 h. However, as the authors themselves acknowledged, obtaining the AFCA intermediate by a purely enzymatic route generally requires the use of several consecutive oxidases (two in their case) as well as the regeneration of expensive cofactors (which involve an additional catalase and horseradish peroxidase in their process), which drastically increases the complexity and the cost of the system, and consequently, limits the industrial application of this approach. This demonstrates, again, the potential of hybrid catalysis, particularly when chemical catalysts are used to replace the costliest steps of cofactor regeneration and inhibitor suppression (e.g., H2O2).
2.3. Taking the concept a step further: one-pot/two-steps hybrid production of furfurylamides from AMFC
Recently, willing to take the concept a step further, we sought to directly convert AMFC into a new family of molecules. More precisely, we wanted to take advantage of the polyfunctional character of AMFC, and even more precisely of the presence of two ionizable functions, one of them positively (amine) and the other one negatively (carboxylic acid), to try to produce amphiphilic molecules with different properties, by attaching a fatty chain to one of these two functions. Among the many types of new bonds that can be formed on these two functions to introduce a fatty chain, one of them seemed particularly promising, as it could potentially be applied to both functionalities of the molecule: the creation of an amide bond from a fatty alcohol or acid. This type of bond also seemed relevant because of its rigidity and resistance to hydrolysis, guaranteeing a certain stability of the produced compound. More generally speaking, the synthesis of amides is one of the most important transformations in agrochemical, pharmaceutical and polymer industries [101, 102]. In recent decades, there has been growing interest in amide-producing processes after the ACS Green Chemistry Institute pointed out the need for more sustainable approaches to amide production as a key research area for a more environmentally friendly chemistry [103]. The most common approach for producing amides is the nucleophilic addition of an amine to an activated carboxylic acid. However, the activating agents involved in these reactions often lead to the generation of waste and hazardous byproducts [104, 105]. It should be noted, however, that many enzymes are also capable of forming amide bonds, such as hydrolases (e.g., lipases, esterases, and acylases), nitriles hydratases, and transglutaminases that have already been used at an industrial scale under milder conditions compared to the traditional organic synthesis routes [106]. It is worth mentioning that other and much less exploited classes of enzymes are also available to form these bonds, notably acyl-coenzyme A ligases (ACLs) [107, 108]. ACLs catalyze ATP-dependent formation of acyl-CoA thioesters from carboxylic acids in a two-step reaction: (1) formation of adenylate derivative from carboxylic acid and ATP and (2) nucleophilic attack of coenzyme A (HSCoA) [109]. Diversion of the native reaction by adding an extra amine nucleophile in absence of HSCoA leads to the formation of amides [110, 111, 112]. One of the main advantages of this reaction, unlike the use of hydrolases for example, is that the amine used for nucleophilic substitution, responsible for the formation of the final amide bond, is not taken over by the enzyme at all, since the substitution takes place spontaneously thanks to the intrinsic reactivity of the adenylate intermediate. Thus, virtually any amine could be used, substituted by an infinite number of substituents, as long as it is sufficiently nucleophilic to attack the adenylate, implying that the products formed could therefore be highly chemomodulated. Using these enzymes, it would therefore be possible to either; (1) activate the AMFC acid and transform it into an amide by attacking a free amine in the medium; or (2) consider using AMFC as a nucleophilic amine, by activating another carboxylic acid, which might also be derived from biomass. In either case, if the other reagent contains a fatty chain, it is possible to obtain a relatively apolar AMFC derivative, but one which retains a polar head, ionizable in acidic or basic media depending on the strategy chosen. When the amide is formed from the carboxylic acid of AMFC, it should be noted that it would also be possible to form a quaternary ammonium at its free amino group, leading to a charged molecule. In any case, to our knowledge, none of these molecule families has ever been reported, which makes them a very promising subject for study. Having at our disposal a library of ACLs produced from previous work, we verified that AMFC was indeed a substrate for one of the ACLs. Unfortunately, none of the eight enzymes we tested, which had shown the most promise in previous assays (activity, stability, etc.), showed activity toward AMFC, as these enzymes tend to accept aliphatic substrates with different chain lengths. This stopped, for now, the acid functionalization pathway for AMFC, and we therefore turned our attention to the possibility of using the latter as a nucleophilic amine. Still seeking to branch out into a variety of fatty chains, and given the estimated substrate selectivity of our enzymes, we then screened our library on fatty mono- and diacids, ranging from four to ten carbon atoms. It should be noted that we did include in the panel some aromatic substrates whose ring was distant from the carboxylic acid. Different selectivity was observed for short and long chains depending on the enzyme (Figure 12). Considering the possibility of using these enzymes in hybrid processes, we carried out the assays at 60 °C, a temperature which had proved interesting in our previous study with the chemical catalysts tested. One particular TsACL displayed a broad substrate range, being very active toward all the tested monocarboxylic acids excepted the decanoic acid and revealed to be almost the only ACL accepting a diacid (e.g., succinic acid). Thus, we can consider that the production of AMFC dimers linked by a fatty chain enables to obtain two polar heads instead of just one on the same molecule. It should also be noted that these screenings were carried out directly with AMFC as the nucleophilic amine, which gives an indication of the yields, even though the assays were carried out on an analytical scale. Also, looking more specifically at TsACL, yields between 53% and 71% for aliphatic acids and 39% for succinic acid, respectively, can be obtained, which is already an excellent result in itself. However, determined to go one step further by proposing the use of a wider range of substrates, we were interested to see if we could couple these enzymes with oxidative catalysts, similar to those we had previously developed. This time, however, fatty alcohols would directly be used instead of fatty acids, given the higher stability of the alcohols for storage purposes rather than the acids. The idea was then to carry out a first oxidation step, once again using immobilized metal nanoparticles to transform a fatty alcohol into the corresponding acid, then to make the ACL act on the formed acid to enable it to be attacked by AMFC, in a one-pot process (Figure 12).

General scheme for the synthesis of AMFC-based amides through the implementation of a one-pot/two-steps process combining supported gold nanoparticles as oxidative chemocatalyst and a CoA ligase as biocatalyst. Redrawn from Bisel et al. [113].
To bring this concept to fruition, a screening of chemical catalysts, assisted by the REALCAT platform’s robots, was carried out to confirm their ability to oxidize alcohols into their corresponding acid, under conditions compatible with those of the enzymes. To this end, the reaction was performed at 60 °C and a family of gold nanoparticles immobilized on a variety of supports with acidic, basic or neutral properties tested. As expected, nanoparticles immobilized on the basic support, and more particularly on calcium oxide (CaO), proved the most active, with a total conversion in less than 24 h, and perfect selectivity for the desired acid. These assays were first carried out on butanol, and then extended to the evaluation of the activity measurement for the best catalyst (Au/CaO) to the alcohol panel corresponding to the acids accepted by TsACL. Here, the catalyst demonstrated an equivalent activity on all substrates, with total conversion to the desired aliphatic acids, as well as to 1,4-butanediol leading to succinic acid. This highlights the highly versatile nature of this new catalyst, making it an ideal candidate for our hybrid system. On the other hand, recycling tests of this catalyst led to leach out observation, with the basic sites gradually being eliminated, leading to its gradual deactivation. This effect was first observed, albeit more slowly, in water, and then much more rapidly when using a buffer to prepare the combination of the two catalysts. We screened different buffers potentially compatible with ACLs (MOPS, TRIS and CAPS) at different pH values ranging from 8 to 11, and it was observed that the catalyst activity was greater at more basic pHs, as well as the recycling activity. Surprisingly, it was also observed that TRIS and CAPS were strongly degraded by the catalyst, although the degradation products have not yet been identified. In the end, it was the 100 mM MOPS buffer that provided the best catalyst activity, with around 80% conversion in 24 h for pH 8 and 9. We also took advantage of this assay to test the activity of the catalyst in the presence of MgCl2 and MnCl2, two cofactors of the enzyme, knowing that the enzyme can use both quite indifferently. It was found that the activity of the chemical catalyst was surprisingly greatly diminished in the presence of Mn2+, whereas the Mg2+ ion did not allow us to see any significant difference. Having demonstrated the activity of the catalyst in a buffered medium, the coupling of our two catalysts could be achieved. A first assay was carried out on butanol as substrate, which was first converted to butanoic acid, then acidified to give 5-(butyramidomethyl)furan-2-carboxylic acid by introducing TsACL, with AMFC and ATP in the medium in a second step. Under the previously optimized conditions, a 65% yield of the desired product was obtained in 48 h, with alcohol-to-acid conversion always quantitative, the limiting step being the enzymatic amide formation, although the obtained yield is in line with the one from the enzyme screening. We then extended our one-pot/two-steps process to other alcohols and obtained the corresponding amides in similar yields to butanol for pentanol and hexanol, and lower yields (26%) for octanol, perhaps due to alcohol solubility issues, the latter having been slightly less oxidized (88% in acid formation) than the shorter-chain alcohols by the chemical catalyst. Interestingly, we also managed to perform the hybrid cascade on 1,4-butanediol, with a modest 24% yield. Nevertheless, the presence of two products was confirmed by NMR as mono- and disubstituted succinic acid, with a preference for the second one (16% yield vs. 8% for the monosubstituted). This creates opportunities for additional valorization pathways and applications for AMFC. To sum up, this tandem heterogeneous enzymatic catalysis, performed under mild aqueous conditions, marks a significant advancement through the integration of gold nanoparticles and enzymes within a one-pot reaction. Concretely, it represents the first association of an ACL with a chemocatalyst, showing how such strategies can offer access to valuable amides. A limitation of the present process though, is the use of large excess of ATP, but this could be easily circumvented by setting up an ATP regeneration system previously reported in the use of ACL enzymes [114]. Another avenue for improvement is the implementation of a fully integrated process in a single step, ideally using catalyst compartmentalization techniques to limit their direct interaction. This aspect might be crucial as, after trying to set up the one-pot/one-step hybrid system, the chemical catalyst seemed to be poisoned by enzyme—its oxidation capacity falling to zero as soon as the enzyme was introduced into the medium. Indeed, it is not a concept detailed in the present article but the production of “multicatalytic hybrid materials” combining the active centers of the catalysts in a finely compartmentalized manner, represents one of the major promises of hybrid process. As already mentioned in several previous reviews, these new materials could represent an excellent solution for avoiding cross-poisoning, while maximizing the synergistic effects of the catalysts, and simplifying processes [115]. In the meantime, a two-pot/one-step system could also be considered, especially as the molecules generated here have probably a different partition coefficient with respect to an organic transfer phase, depending on their oxidation state and level of functionalization.
3. Conclusion
In conclusion, through these three examples, as well as with the important work accomplished by the community in hybrid catalysis over the last decade, we have tried to show that the combination of catalysts of different natures, and more particularly chemical and biological ones, could be highly relevant for the valorization of biosourced molecules. Indeed, the aforementioned recent developments in hybrid catalysis have shown the possibility of directly transforming untreated biomass materials into value-added products. In this field, the production of furfural is one of the most targeted molecules found in the literature; as many of its derivatives can have potential applications in different industries (pharmaceuticals, polymers, food, for instance). It is worth mentioning that the developed systems are not only simple in terms of substrate and product treatment, notably without needing any additional pretreatment or intermediate isolation and purification steps, but also more efficient in terms of atom and energy economy, thanks to a one-pot set-up. In parallel, research in hybrid catalysis has also enhanced the exploration of new synthetic pathways. As a result, structurally rich molecules, often chiral and therefore interesting to access pharmaceutical molecules, can now be accessed through such approaches. The wide substrate scopes and the subsequent obtainment of a range of products show the versatility of hybrid approaches, which from a future industrial application perspective, could be very interesting. All these aspects are reflected on the presented application, showing the whole workflow to develop a series of hybrid approaches where biosourced d-glucose is converted into HMF, then to AMFC, a furfurylamine, and then to amphiphilic furfurylamides that were never produced to the best of our knowledge. Still, despite this high potential, catalyst combination remains very limited due to their cross-poisoning or incompatibility between reaction conditions. Smart processes, based on innovative chemical engineering, can nonetheless help overcome these limitations with new equipment (two-pot/one-step process), or in the ideal situation, tailor-made compartmentalized hybrid materials. Among all, hybrid catalysis stands as a highly interdisciplinary and emerging subject, which will increasingly require close collaboration between different scientific fields, notably chemistry and biology, while also keeping an engineering perspective to enhance further efficient applications.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Acknowledgments
This work was supported by an ANRT grant with Sanofi (CIFRE 2024/0663, PhD grant for Sara Arteche Echeverría). The authors thank the REALCAT platform funded by the French government grant managed by the French National Research Agency (ANR) as part of the “Investments for the Future” program (ANR-11EQPX-0037). A CC-BY public copyright license has been applied by the authors to the present document and will be applied to all subsequent versions up to the Author Accepted Manuscript arising from this submission, in accordance with the grant’s open access conditions.