


 CC-BY 4.0
CC-BY 4.0
1. Introduction
In the field of cancer therapy, photodynamic therapy (PDT) represents an interesting alternative approach besides chemotherapy, radiotherapy and surgery for some types of cancer, notably skin cancer or tumors accessible to light (directly or thanks to optical fiber) [1, 2, 3, 4]. It relies in using a photosensitizer (PS), which is activated by light and able to generate reactive oxygen species (ROS) and singlet oxygen (1O2). It implies that the PS in its singlet excited state (1PS) can reach its triplet state (3PS) through intersystem crossing (ISC, 1PS→3PS). Then, energy transfer from the 3PS with ground state molecular oxygen in its triplet state 3O2 leads to the generation of 1O2 (PDT of type 2). Alternatively, electron transfer processes involving the 3PS lead to the formation of ROS and cytotoxic free radicals (PDT of type 1). Ultimately, these strong oxidizing agents may lead to the death of cancer cells through necrotic and apoptotic mechanisms [5, 6, 7, 8, 9]. The important aspect of PDT is that only cells containing PS and irradiated with visible light are susceptible to be destroyed. To date, porphyrinoids (including porphyrins, chlorins, phthalocynanines) are among the best performing PS for PDT because of their inherent chemical stability and suitable photophysical properties [10, 11, 12, 13]. These tetrapyrrolic aromatic compounds strongly absorb light in the visible region and, more importantly, they are efficient PS to generate 1O2. For example, the 1O2 quantum yield of 5,10,15,20-meso-tetraphenylporphyrin (H2TPP) in toluene is 𝛷𝛥 = 68% [14]. Of course, there are some important drawbacks like their poor water solubility or their lack of selective accumulation in cancer cells, but these drawbacks may be circumvented by appropriate functionalization of the porphyrin core [15, 16, 17]. Interestingly, some porphyrins were functionalized with peripheral ligands allowing the coordination of metal ions at their periphery [18]. Some of the obtained complexes found relevant applications in the field of PDT [19]. For example, porphyrins functionalized with peripheral platinum(II) or ruthenium(II) complexes were used for dual chemo- and phototherapy [20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32]. Platinum(II) complexes have anticancer activity and also, significantly improve the photodynamic effect by promoting the generation of 1O2 through a heavy atom effect [20, 21, 22, 23, 24, 25, 26, 27, 28]. Ruthenium(II) complexes were also reported for dual chemo- and phototherapy [29, 30, 31, 32]. Moreover, some ruthenium(II) complexes proved to be efficient PS for two-photon excitation [33, 34, 35]. Although gold(I) complexes are promising candidates to develop new metallodrugs for cancer therapy, examples of porphyrinoids bearing peripheral gold(I) complexes for combined chemo- and phototherapy are relatively scarce. Gold(I) complexes of porphyrins functionalized with peripheral phosphine ligands were synthesized and biological studies showed that these compounds present rather low cytotoxicity [36]. However, water-soluble complexes of this type proved to be efficient PS for PDT. N-heterocyclic carbenes (NHC) are phosphine analogs and are attractive ligands to design new metallodrugs. During the last decade, we reported the synthesis of several porphyrin derivatives equipped with peripheral NHC-metal complexes [37, 38, 39, 40, 41], including gold(I) complexes [42, 43, 44, 45]. The photodynamic properties of imidazolium salt A and gold(I) complexes B–D depicted in Scheme 1 were investigated and we observed the important role played by the ligand trans to the NHC [45]. Indeed, the excellent photodynamic properties observed for the PS D functionalized with mannose are due to active targeting of cancer cells overexpressing mannose receptors at their surface. We also observed that imidazolium salt A is an efficient PS for PDT, while gold(I) complex B and C are inefficient for chemo- and phototherapy. The aim of this study is to draw more detailed conclusions about the potential of porphyrins equipped with peripheral NHC-gold(I) complexes for dual chemo- and phototherapy. For this purpose, we report here the synthesis and the biological properties of porphyrins bearing imidazolium salts and NHC-gold(I) complexes at their periphery.
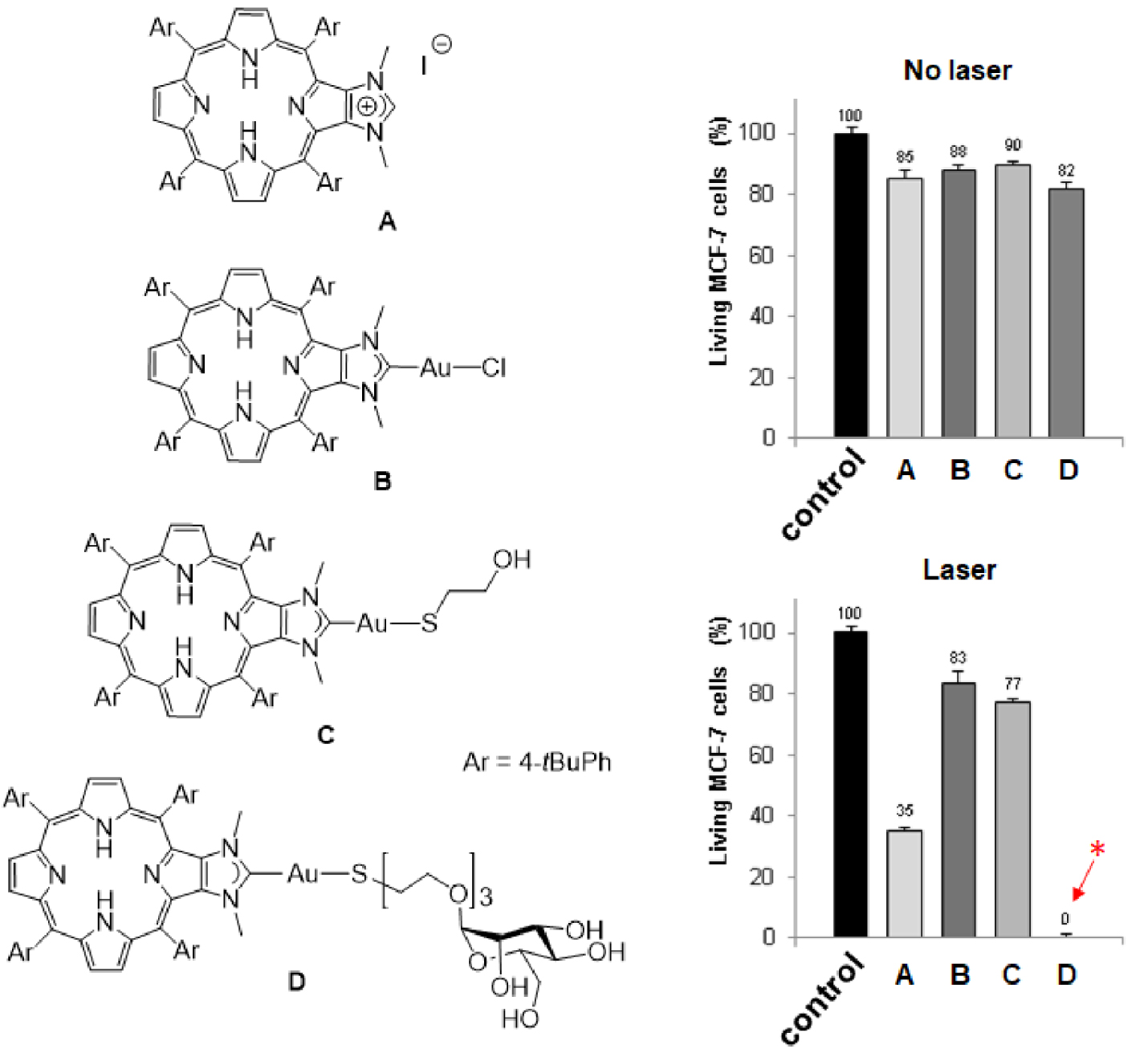
Left: Structures of compounds A–D. Right: Cytotoxic (no laser) and photodynamic effect (laser) of imidazolium salt A and gold(I) complexes B–D. The cells were incubated or not (control experiment in black) with 10 μm of PS for 4 h and then, submitted or not to laser irradiation (𝜆 = 405 nm, 18.75 J⋅cm−2, 10 min). Cells were allowed to grow for two days and cell viability was quantified with MTT assay. Data are mean values standard deviation from three independent experiments. *No living cells detected [45]. Masquer
Left: Structures of compounds A–D. Right: Cytotoxic (no laser) and photodynamic effect (laser) of imidazolium salt A and gold(I) complexes B–D. The cells were incubated or not (control experiment in black) with 10 μm of PS for 4 h and then, submitted ... Lire la suite
2. Results and discussion
2.1. Synthesis of porphyrins meso-functionalized with imidazolium salts
Imidazolium salts are routinely used as NHC precursors. NHC could be generated upon deprotonation of an imidazolium salt with a base and trapped with a metal cation such as Ag+ or Au+, for example. We first investigated compounds with imidazolium salts directly N-linked to one meso position of a porphyrin core. The first imidazolium salt, namely free-base porphyrin 3, was obtained in a three-step procedure starting from the porphyrin 1 previously described in [46]. The reaction of porphyrin 1 with imidazole and NaH in DMF (140 °C) afforded porphyrin 2 in 72% yield after column chromatography [46]. Then, demetalation of porphyrin 2 in acidic conditions followed by methylation of the peripheral imidazole group with CH3I afforded porphyrin 3 in an overall yield of 77% after purification (Scheme 2). The 1H NMR spectrum of porphyrin 3 showed the expected signal of the imidazolium proton H2 at 𝛿 = 10.44 ppm and the signal of the two inner NH protons at 𝛿 = −2.95 ppm. High-resolution ESI-TOF mass spectrometry (positive mode) featured the expected molecular mass peak at m∕z = 647.2926 Da with an isotopically resolved profile in agreement with the calculated distribution for the monocationic species [M–I]+ (calcd m∕z = 647.2918 Da). The iodide counter-ion was observed by ESI-TOF mass spectrometry (negative mode) at m∕z = 126.91 Da. Porphyrin 3 was then metalated with Zn(OAc)2 ⋅ 2H2O to obtain the corresponding zinc(II) porphyrin 4-I in 80% yield. Metal insertion within the porphyrin core was confirmed by 1H NMR, UV–visible absorption spectroscopy and mass spectrometry (see ESI). To avoid the presence of iodide in the coordination sphere of gold(I) complexes, iodide was substituted by tetrafluoroborate, which is a non-coordinating anion. For this purpose, reaction of porphyrin 4-I with AgBF4 in acetone in the dark gave porphyrin 4-BF4 in 85% yield after column chromatography. Tetrafluoroborate anion was observed by 19F NMR spectroscopy with the two expected signals at 𝛿 = −149.20 and −149.26 ppm (caused by the two boron isotopes 10B and 11B, respectively) and by mass spectrometry (negative mode).
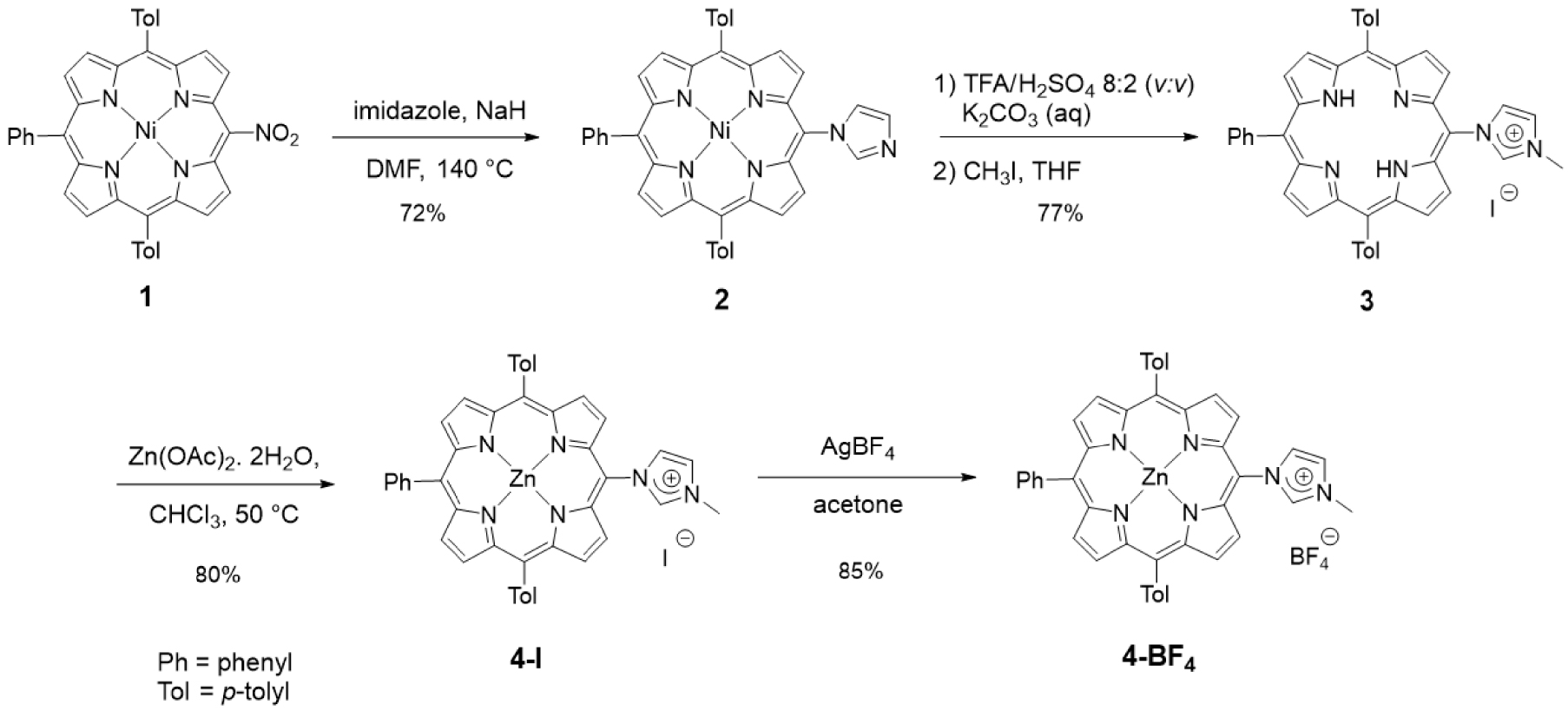
Synthesis of porphyrins 3, 4-I and 4-BF4.
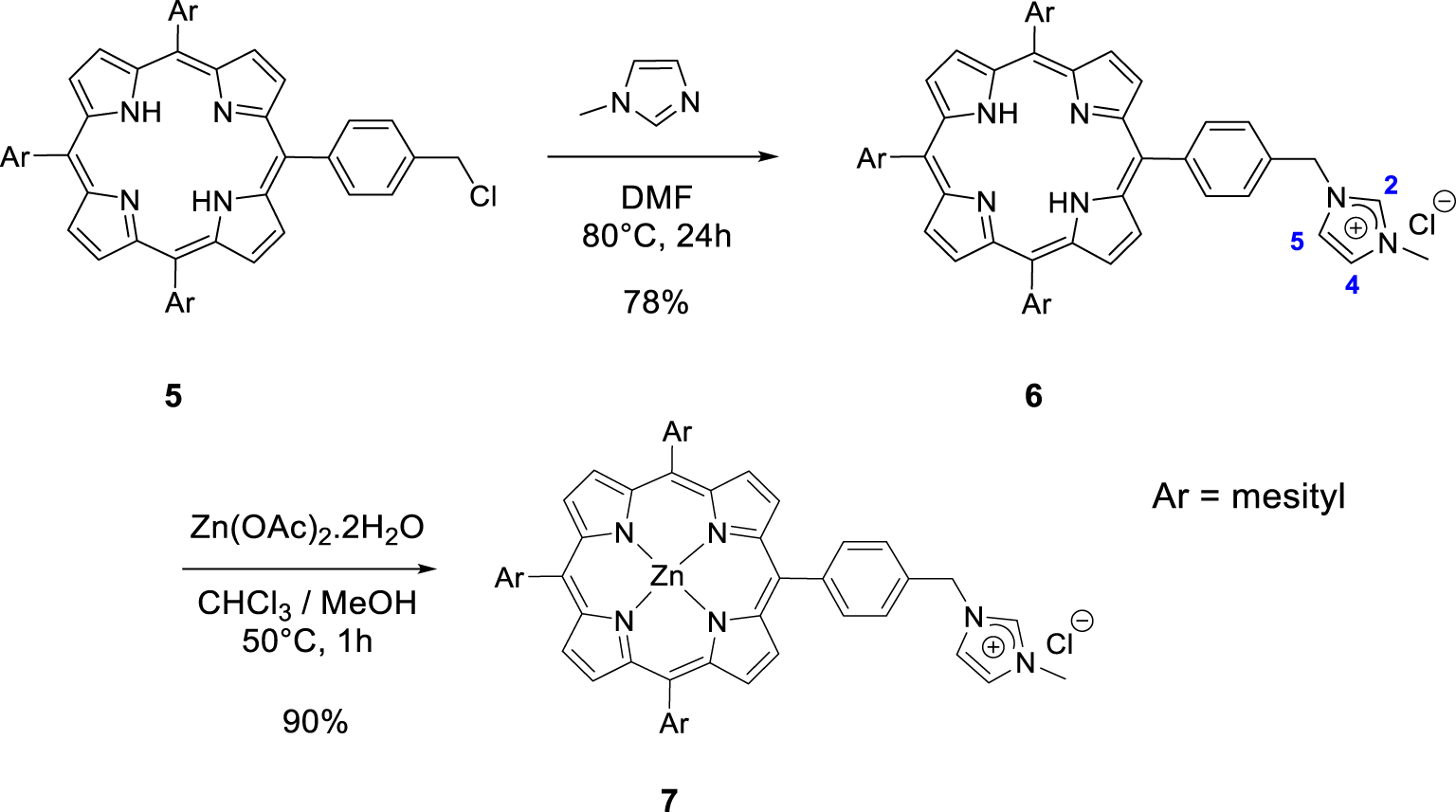
Synthesis of the porphyrins 6 and 7.
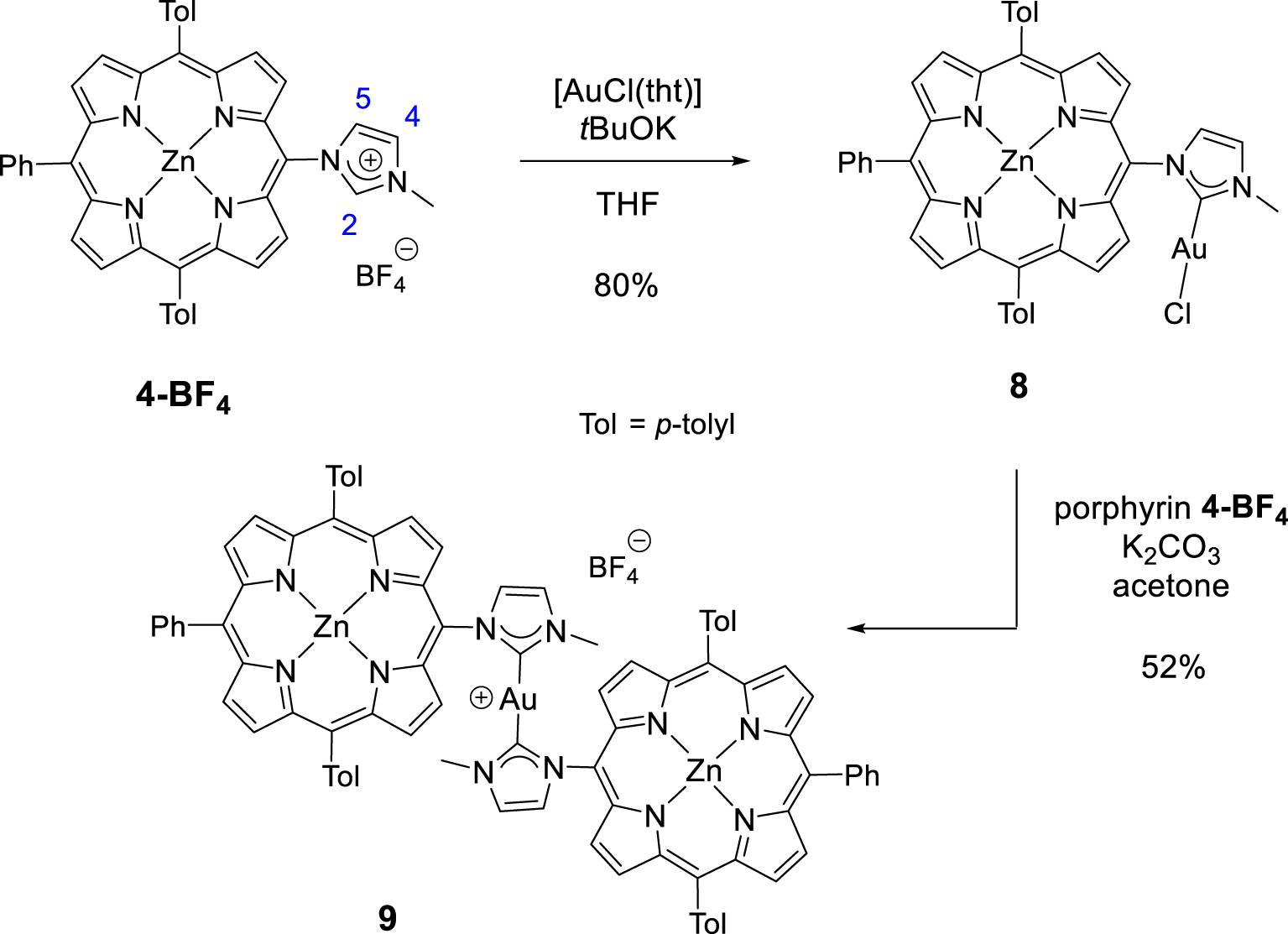
Synthesis of gold(I) complexes 8 and 9.
Then, we synthesized imidazolium-based porphyrin compounds with a spacer between the meso position of the porphyrin core and the imidazolium salt. For this purpose, free-base porphyrin 5 was used as starting material. The synthesis of this compound is described in [47]. Nucleophilic substitution on the chlorine atom of porphyrin 5 with 1-methylimidazole in DMF (80 °C) afforded porphyrin 6 in 78% yield (Scheme 3). The 1H NMR spectrum of the porphyrin 6 clearly showed the expected signal of the imidazolium proton H2 at 𝛿 = 11.17 ppm. The signals of imidazolium protons H4 and H5 were also observed as apparent triplets at 𝛿 = 7.45 and 7.31 ppm. Porphyrin 7 was obtained in 90% yield by reacting free-base porphyrin 6 with Zn(OAc)2 ⋅ 2H2O.
2.2. Synthesis of gold(I) complexes
NHC-gold(I) complexes can be synthesized following different procedures. For example, complex 8 was synthesized in one step by reacting porphyrin 4-BF4 with [AuCl(tht)] (tht = tetrahydrothiophene) in the presence of tBuOK in dry THF. The mono(NHC)-gold(I) complex 8 was obtained in 80% yield after column chromatography and recrystallization (Scheme 4). The absence of signal corresponding to the imidazolium proton H2 in the 1H NMR spectrum of complex 8 confirmed the formation of the CNHC–Au(I) bond. Imidazolium protons H4 and H5 are shifted upfield by 𝛥𝛿 ∼ 0.40 ppm as a consequence of the formation of the CNHC–Au(I) bond. The signal of the CNHC bound to AuI was observed at 𝛿 = 175.1 ppm in the 13C{1H} NMR spectrum of complex 8. This chemical shift is in good agreement with those reported in literature for [(NHC)AuCl] complexes [48, 49]. Complex 9 was prepared in one step according to the reaction conditions reported by Richeter [42, 43, 44, 45]. The deprotonation of the porphyrin 4-BF4 with K2CO3 in acetone in the presence of one equivalent of the complex 8 afforded the homoleptic complex 9 in 52% yield after column chromatography (Scheme 4). High-resolution ESI-TOF mass spectrometry (positive mode) featured the expected molecular mass peak at m∕z = 1617.3612 Da with an isotopically resolved profile in good agreement with the calculated distribution of the monocationic species [ZnP–Au–ZnP]+ (calcd m∕z = 1617.3610 Da). NMR spectroscopy also confirmed the formation of the complex 9. Notably, in the 13C{1H} NMR spectrum, the signal of the CNHC bound to AuI was observed at 𝛿 = 188.8 ppm, a chemical shift in good agreement with those reported in literature for homoleptic monocationic complexes [(NHC)AuI(NHC)]+ [48, 49]. The 1H NMR spectra of complexes 8 and 9 are displayed in Figure 1. Interestingly, in both cases, four separated doublets were observed for the ortho and meta protons of the tolyl groups. This can be explained by the restricted rotation of the Cmeso–NNHC bond due to the presence of the sterically demanding NHC-gold(I) complexes. As a consequence, different chemical environments are experienced by the meso aryl protons. The N–Me protons signal of complex 9 at 𝛿 = 2.04 ppm is significantly shielded (𝛿 = 4.23 ppm for complex 8) indicating that the N–Me group of one porphyrin sits on top of the second porphyrin ring and is exposed to its ring current. Finally, the two gold(I) complexes were characterized by diffusion-ordered spectroscopy (DOSY) 1H NMR. As expected, the diffusion coefficient of 9 (1.15 × 10−10 m2⋅s−1) is lower compared to 8 (1.54 × 10−10 m2⋅s−1) because it contains two porphyrins.
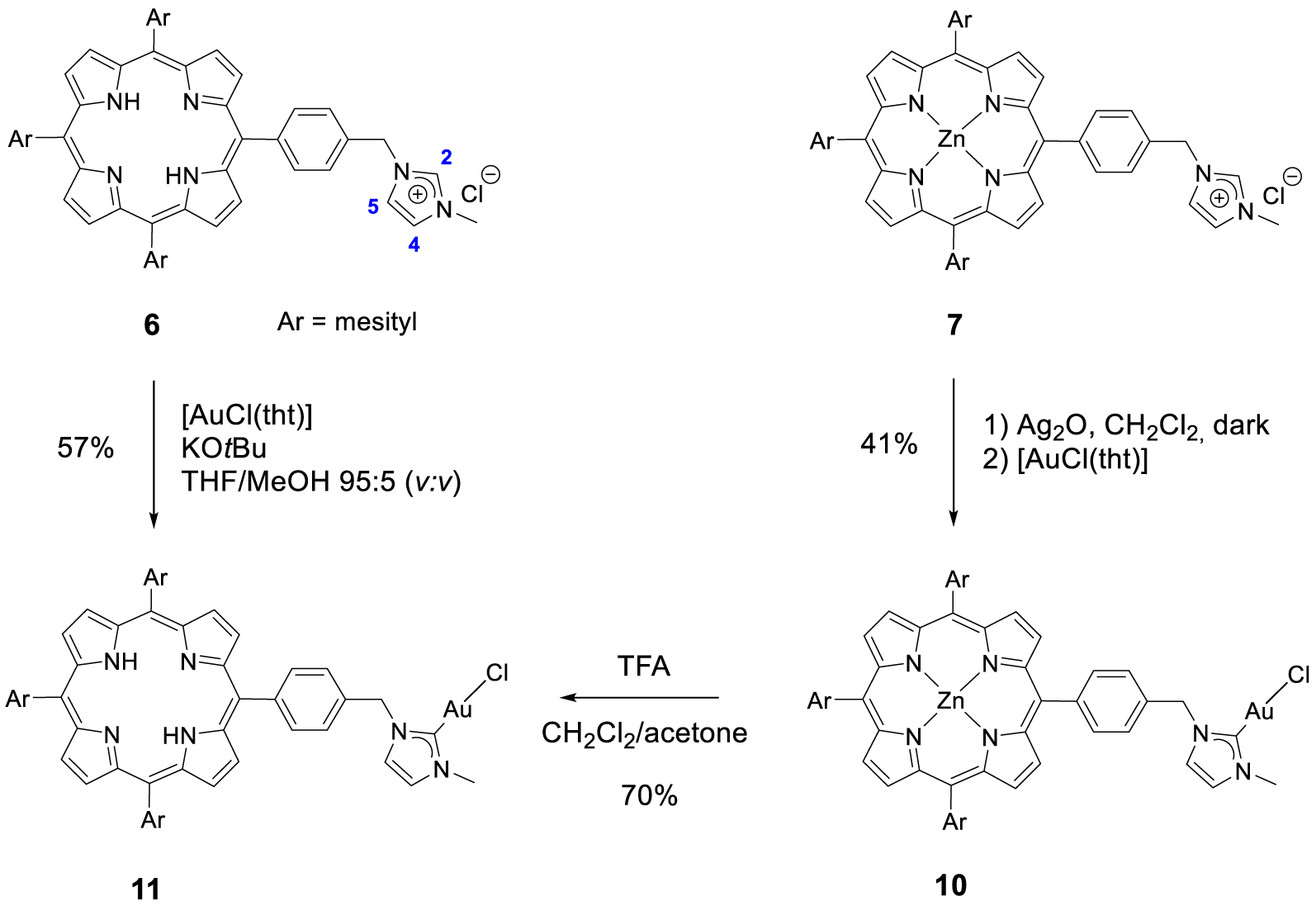
Synthesis of gold(I) complexes 10 and 11.
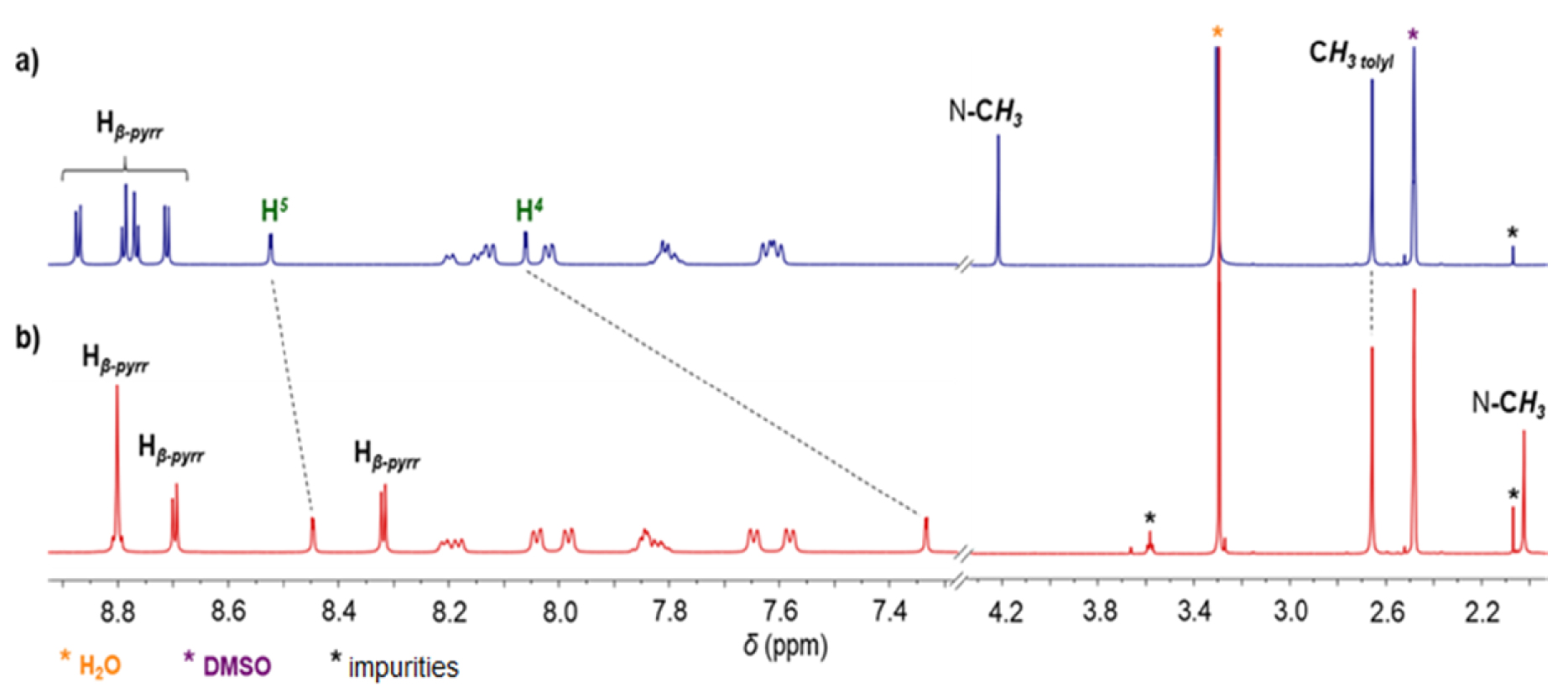
1H NMR spectra (400 MHz, DMSO-d6, 298 K) of gold(I) complexes 8 (a) and 9 (b).
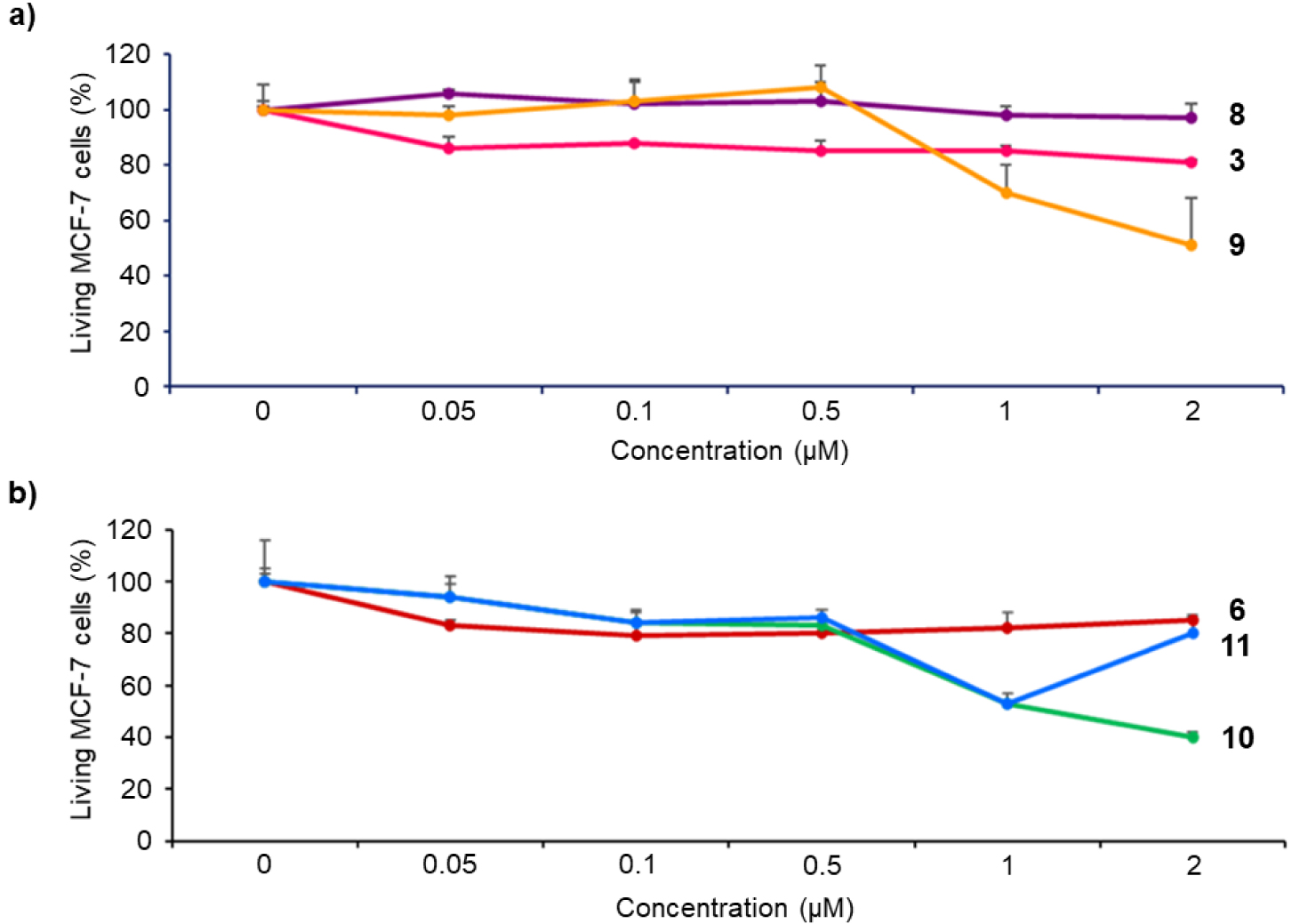
Cytotoxicity of imidazolium salts 3 and 6, and their corresponding gold(I) complexes 8–11 on MCF-7 cancer cells. The cells were incubated without (control) or with 0.05, 0.1, 0.5, 1 or 2 μM of the different compounds for 72 h. Cell viability was quantified with MTT assay. Data are mean values standard deviation from three independent experiments.
Gold(I) complex 10 was prepared following a strategy that is widely used for the synthesis of NHC gold(I) complexes: the reaction of the porphyrin 7 with 1 equation of Ag2O in dichloromethane afforded the corresponding silver(I) complex which was not isolated and used straightforward for the transmetallation reaction with [AuCl(tht)]. The corresponding gold(I) complex 10 was obtained in 41% yield after column chromatography and recrystallization (Scheme 5). The absence of signal for proton H2 in the 1H NMR spectrum of complex 10 and the signal of the CNHC bound to AuI observed by 13C{1H} NMR spectroscopy at 𝛿 = 172.3 ppm confirmed the formation of the expected gold(I) complex. Gold(I) complex 11 containing a free-base porphyrin could not be synthesized following a similar strategy because silver(I) may be complexed by the porphyrin core. However, gold(I) complex 11 could be obtained by two different pathways. First, it is possible to remove the zinc(II) by treating the complex 10 with a TFA/CH2Cl2 mixture. These acidic conditions do not degrade the peripheral gold(I) complex as it was previously shown by us with analogous complexes [45]. Complex 11 was obtained in 70% yield after neutralization with NaHCO3 and column chromatography. It is also possible to react free-base porphyrin 6 with one equivalent of [AuCl(tht)] with tBuOK, in a THF/MeOH mixture in the dark to obtain the corresponding complex gold(I) complex 11 in 57% yield after column chromatography (Scheme 5). The formation of complex 11 was first confirmed by 1H NMR spectroscopy and the disappearance of the signal at 𝛿 = 11.17 ppm corresponding to the imidazolium proton H2. In its 13C{1H} NMR spectrum, the carbene signal observed at 𝛿 = 172.5 ppm demonstrated the formation of the CNHC–Au(I) bond.
2.3. Cytotoxicity and phototoxicity studies
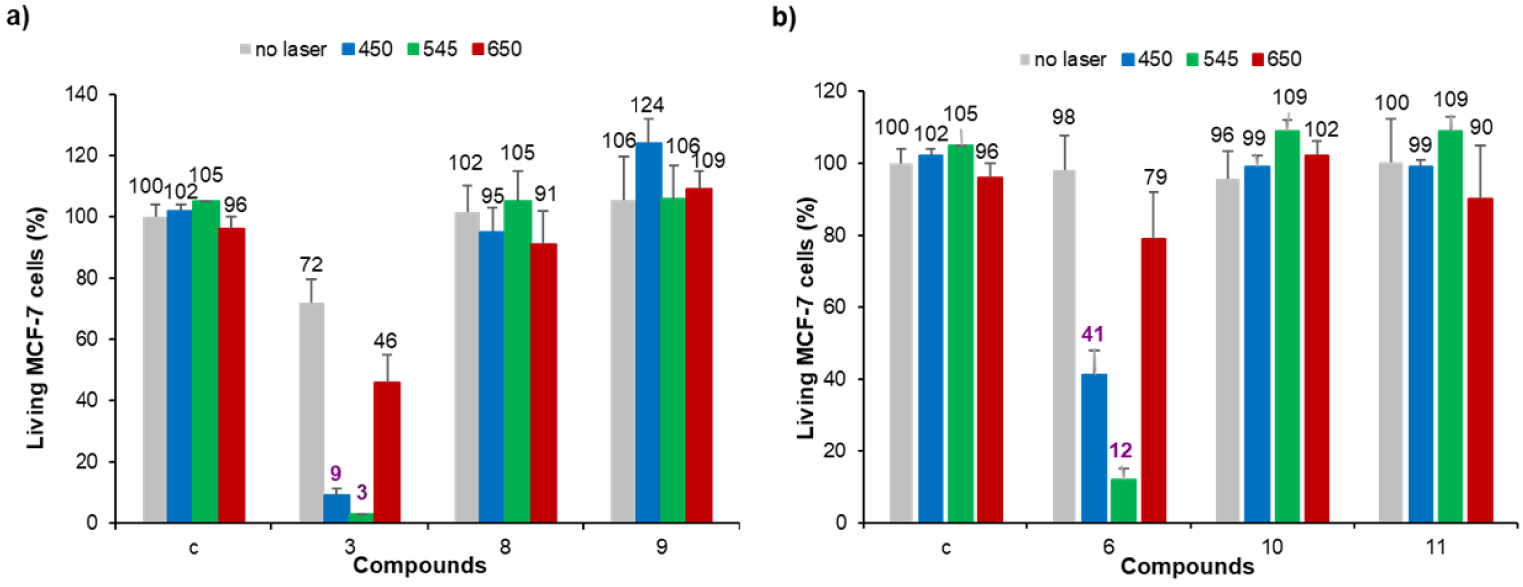
Photodynamic effect of imidazolium salts 3 and 6, and their corresponding gold(I) complexes 8-11 on MCF-7 cancer cells. The cells were incubated with 0.5 μM of each compound for 24 h and then submitted to laser irradiation at 𝜆 = 450, 545 or 650 nm for 10 min. Cells are allowed to grow for 48 h and cell viability was quantified with MTT assay.
Biological properties of NHC-based gold(I) complexes have been extensively reported in literature [50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62]. The cytotoxicity of imidazolium salts and gold(I) complexes on MCF-7 breast cancer cells was first investigated. For this purpose, MCF-7 cancer cells were incubated for 72 h in the dark with each compound at different concentrations varying from 0.05 to 2 μM. After 72 h, MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was performed to establish the cell viability and the results obtained are summarized in Figure 2. Free-base porphyrins 3 and 6 containing imidazolium salts are rather non-toxic up to 2 μM with less than 15% cell death irrespective of the presence or not of a linker between the porphyrin core and the NHC. In contrast, gold(I) complexes’ cytotoxicity strongly depends on the presence or not of a linker between the porphyrin core and the NHC. Indeed, complex 8 without linker between the porphyrin core and the NHC is not cytotoxic at 2 μM. On the contrary, complex 10 with the meso-benzylic linker between the porphyrin core and the NHC shows a significant cytotoxicity with 60% cell death at 2 μM. Therefore, the closer are the porphyrin core and the NHC gold(I) complex, the lower is the observed cytotoxicity. This is in agreement with the low cytotoxicity observed for complex B (Scheme 1) where NHC ligand is fused to the porphyrin core. Compound 9 is a cationic bis(NHC) gold(I) complex and it seems that the positive charge tends to increase the observed cytotoxicity since ∼50% cell death was observed at 2 μM. Compared to complex 10, cytotoxicity of the corresponding gold(I) complex 11 including a free-base porphyrin is similar to a concentration up to 1 μM. The lower cytotoxicity observed for complex 11 at a concentration of 2 μM may be attributed to its low solubility in the cell culture medium.
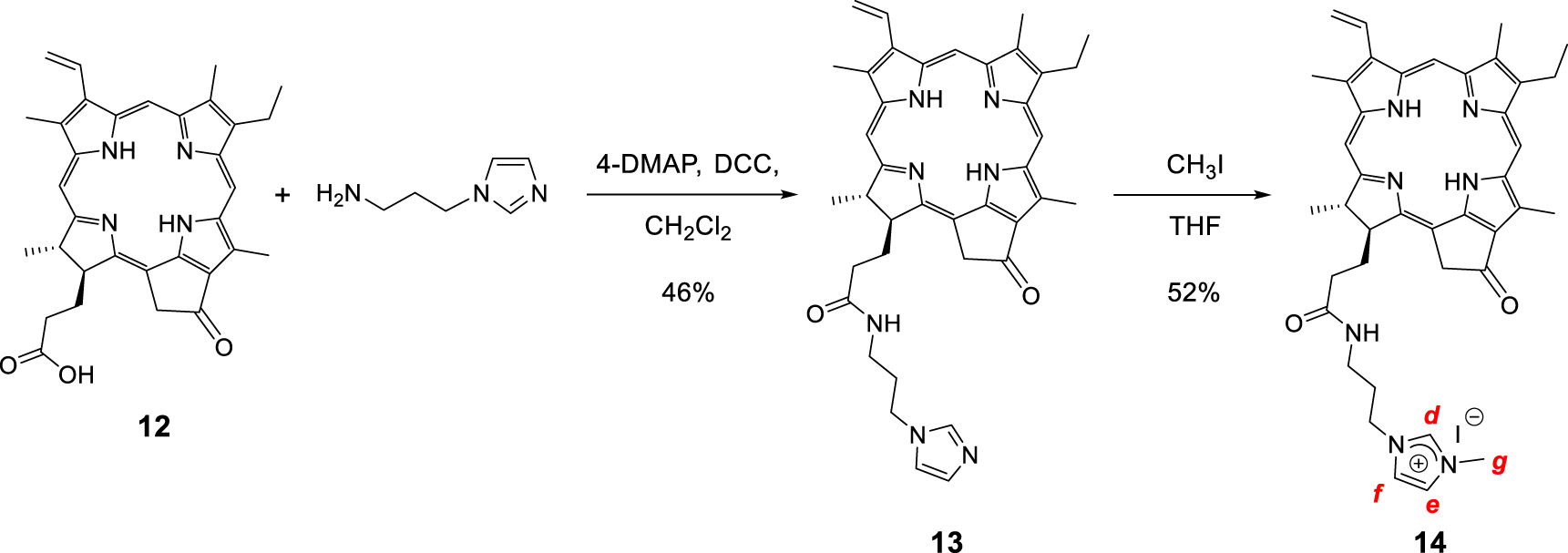
Synthesis of the monocationic chlorin 14.
Photodynamic properties of the different compounds were then investigated at 0.5 μM since no or moderate cytotoxicity was observed at this concentration. MCF-7 cancer cells were incubated with the different PS for 24 h and submitted to laser irradiation at 𝜆 = 450, 545 or 650 nm for 10 min. The results obtained after MTT assay to estimate cell viability are gathered in Figure 3. As can be seen in this figure, none of the gold(I) complexes showed significant photodynamic effect whatever the irradiation wavelength. This is in agreement with our previous finding that gold(I) complexes without any specific targeting agent are not suitable PS for PDT, although their 1O2 quantum yields may be improved through heavy atom effect [45]. On the contrary, both imidazolium salts 3 and 6 induced important cell death under laser irradiation at 𝜆 = 450 and 545 nm. The best photodynamic effect was observed for imidazolium salt 6 at 0.5 μM which induced ∼86% cell death after 10 min of irradiation at 𝜆 = 545 nm.1 The good solubility of imidazolium salts in aqueous media and the fact that these cationic species can strongly interact with the negatively charged cancer cell membranes can explain the observed enhanced photodynamic activity compared to the corresponding gold(I) complex. Nevertheless, photodynamic activity of both imidazolium salts 3 and 6 is weaker under irradiation at 𝜆 = 650 nm since less than ∼19–26% cell death was observed (see footnote 1). This prompted us to investigate the functionalization of pyropheophorbide a with imidazole and imidazolium groups and their use for PDT applications.
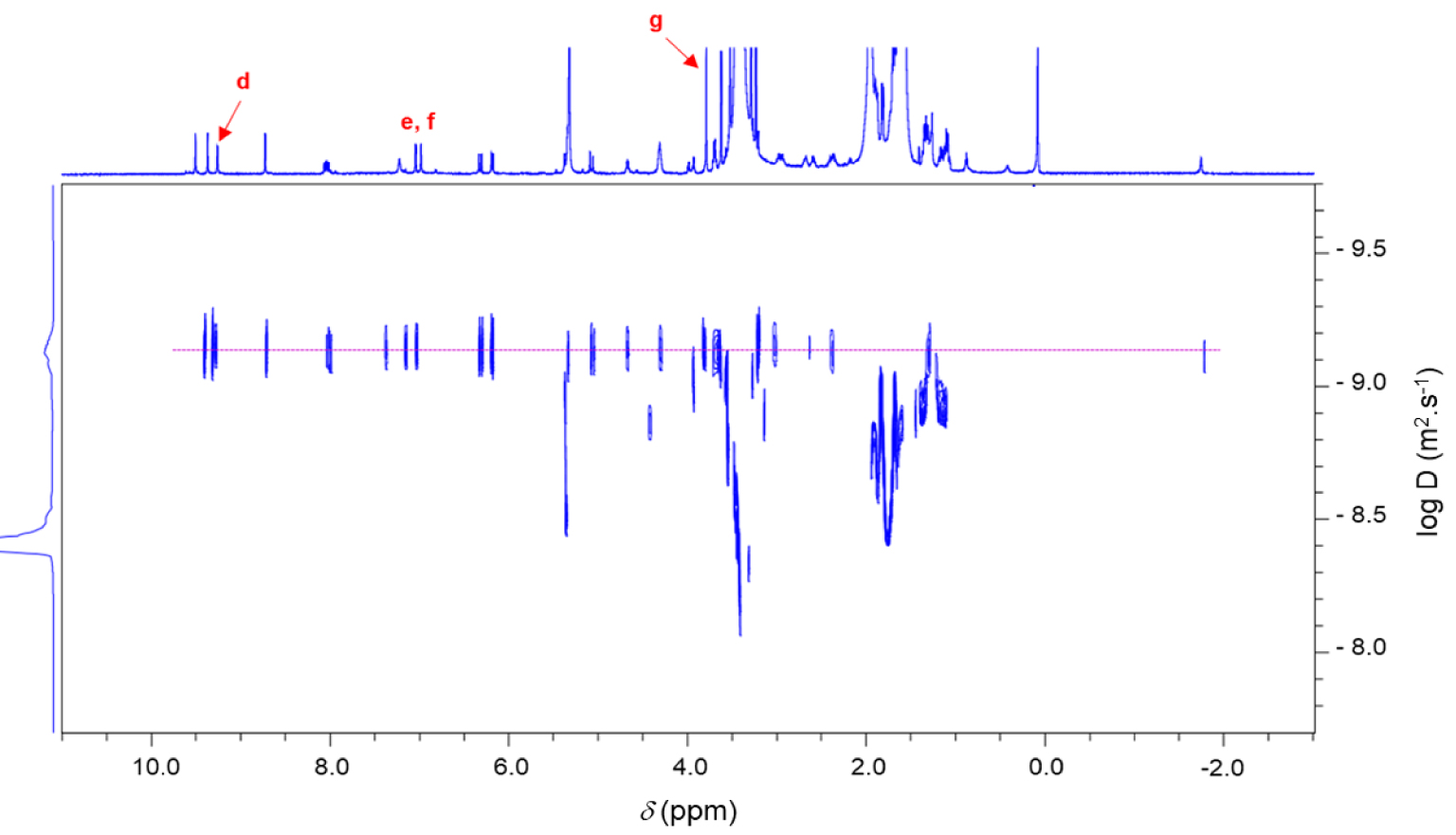
1H 2D DOSY NMR spectrum (600 MHz, CD2Cl2, 298 K) of the chlorin 14.
2.4. Synthesis and photodynamic properties of pyropheophorbide a functionalized with imidazole/imidazolium groups
Monocationic chlorin 14 was prepared in a two-step procedure starting from pyropheophorbide a12 as described in literature (Scheme 6) [63, 64, 65, 66]. First, the reaction of pyropheophorbide a12 with 1-(3-aminopropyl)imidazole in the presence of 4-dimethylaminopyridine (DMAP) and dicyclohexylcarbodiimide (DCC) in dry dichloromethane afforded the chlorin 13 in 46% yield [67]. Its 1H NMR spectrum clearly showed three triplets at 𝛿 = 7.18, 6.81, and 6.62 ppm corresponding to the protons of the imidazole ring. The signals of the propyl chain were also observed at 𝛿 = 3.54 ppm and between 2.90 and 1.90 ppm. Then, methylation of the chlorin 13 with excess of CH3I in dry THF afforded the monocationic chlorin 14 in 52% yield. Its 1H NMR spectrum clearly showed the deshielded signal of the imidazolium proton at 𝛿 = 9.26 ppm. The singlet observed at 𝛿 = 3.79 ppm corresponds to the protons of the N–CH3 group (Scheme 6). In the 1H 2D DOSY NMR spectrum of monocationic chlorin 14, alignment of the signals of the propyl bearing the imidazolium ring with the other signals of the chlorin core clearly confirmed the formation of the expected compound (Figure 4).
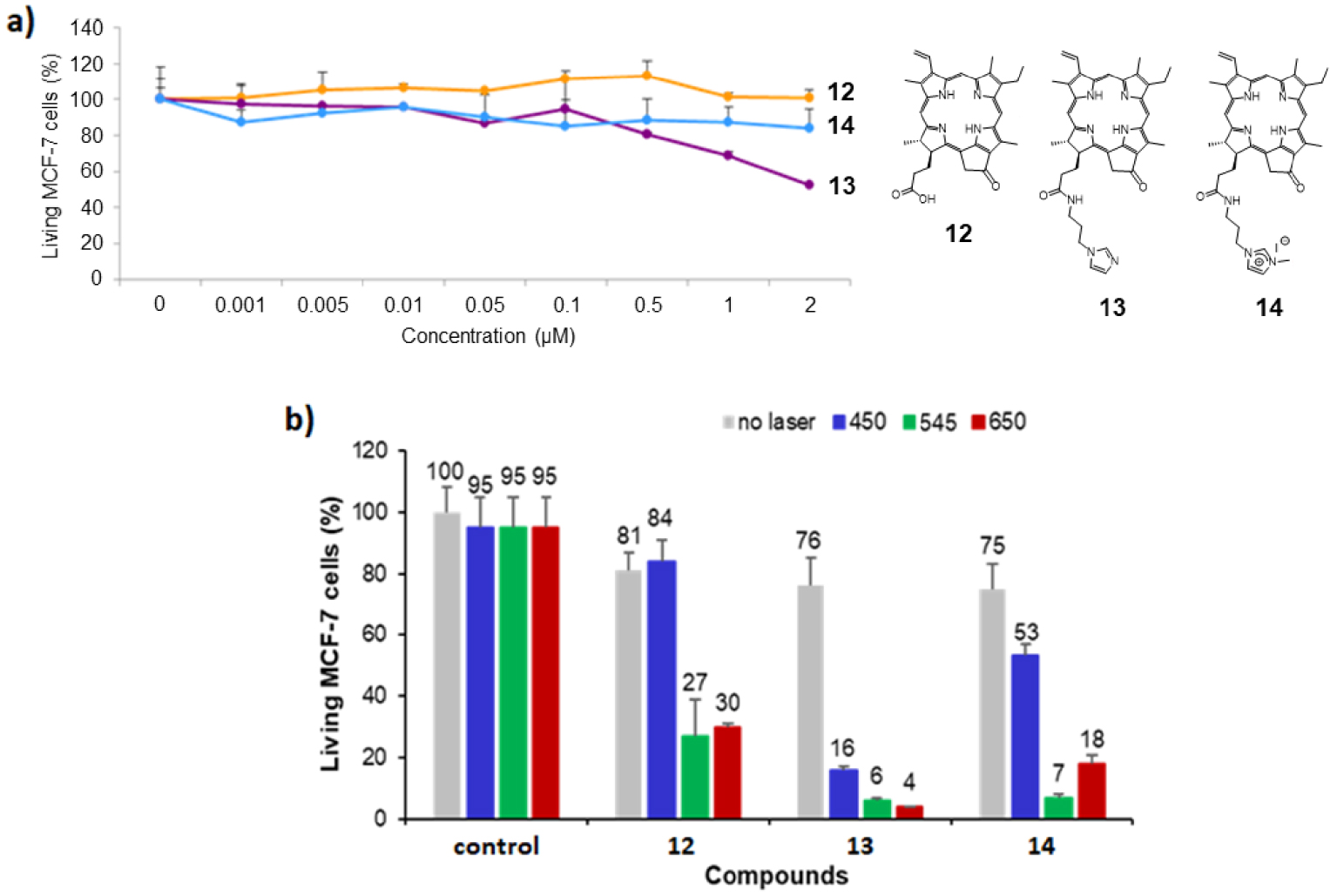
(a) Cytotoxic effect of chlorin derivatives 12–14 on MCF-7 cancer cells. The cells were incubated without (control) or with different concentrations of compounds 12–14 for 72 h. Cell viability was quantified with MTT assay. Data are mean values standard deviation from three independent experiments. (b) Photodynamic effect of the chlorin derivatives 12–14 under irradiation at 𝜆 = 450 nm (left), 545 nm (middle) and 650 nm (right). MCF-7 cancer cells were incubated with 0.5 μM of each chlorin for 24 h and then submitted to laser irradiation for 10 min. Cells were allowed to grow for 48 h and the living MCF-7 cells were quantified with MTT assay. Masquer
(a) Cytotoxic effect of chlorin derivatives 12–14 on MCF-7 cancer cells. The cells were incubated without (control) or with different concentrations of compounds 12–14 for 72 h. Cell viability was quantified with MTT assay. Data are mean values standard deviation from ... Lire la suite
The cytotoxicity studies of chlorin derivatives 12–14 on MCF-7 breast cancer cells are summarized in Figure 5a and revealed that pyropheophorbide a12 and imidazolium salt 14 were not cytotoxic on MCF-7 cells because they induce less than 10% cell death at a concentration of 2 μM. On the contrary, imidazole 13 exhibits significant cytotoxicity at 1 μM, with more than 30% cell death (∼45% cell death at 2 μM). According to these results, photodynamic activity of chlorin derivatives 12–14 was evaluated with PS concentration of 0.5 μM under irradiation at 𝜆 = 450,545 and 650 nm. The obtained data are summarized in Figure 5b. Upon irradiation at 𝜆 = 450 nm, only imidazole 13 exhibited a strong photodynamic effect by inducing 60% cell death. Imidazolium salt 14 revealed a slightly phototoxic activity with 22% cell death, whereas pyropheophorbide a12 was not phototoxic at all (Figure 5b, left). Upon irradiation at 𝜆 = 545 and 650 nm, all chlorin derivatives 12–14 turned out to be phototoxic (Figure 5b, middle and right). The significant phototoxicity of the three chlorins observed upon irradiation at 𝜆 = 650 nm constitutes the noteworthy result in this study, since synthetic imidazolium salts 3 and 6 induced less than ∼19–26% cell death upon irradiation at 𝜆 = 650 nm (Figure 3). Chlorin derivatives 12, 13 and 14, induced 51%, 72% and 57% cell death, respectively, and turned out to be better photosensitizers upon irradiation with red light (see footnote 1).
3. Conclusion
Porphyrins bearing imidazolium salts were used as NHC precursors for the synthesis of gold(I) complexes with the aim to combine their cytotoxicity with PDT. This study shows that porphyrins conjugated with NHC-gold(I) complexes are metallodrugs possessing anticancer properties, as long as the NHC ligand is not directly bonded to the porphyrin core: a spacer is probably needed to keep away the NHC-gold(I) complex from the bulky porphyrin core. Although heavy atom effect may be beneficial to improve 1O2 production, these porphyrins conjugated with gold(I) complexes are not suitable PS for PDT. Additional functionalization with targeting agents is necessary to ensure active targeting of the cancer cells and simultaneously better solubility in aqueous media [68, 69, 70]. By contrast, the corresponding porphyrins equipped with imidazolium salt moieties are suitable PS for PDT and this effect is attributed to the positive charges of the imidazolium cations, which are known to ensure strong interactions with negatively charged cancer cell membranes. These cationic porphyrins can induce significant cell death upon laser irradiation at 450 and 545 nm, but not at 650 nm. To improve light absorption and photodynamic effect at 650 nm, pyropheophorbide a derivatives equipped with pendant imidazole/imidazolium groups were synthesized. These hemisynthetic compounds proved to be efficient PS for PDT at 650 nm.
4. Experimental section
4.1. Materials and instruments
Reactions needing inert atmosphere were performed under argon using oven-dried glassware and Schlenk techniques. Dry THF was obtained by a PureSolve MD5 solvent purification system from Innovative Technology. Dry DMF was purchased from Sigma-Aldrich. Dry CH2Cl2 and THF were obtained by a PureSolve MD5 solvent purification system from Innovative Technology. Gold(I) complex [AuCl(tht)] (tht = tetrahydrothiophene) was prepared according to the procedure described in literature [71]. Pyrrole ( >99%) was purchased from TCI and distilled under reduced pressure before use. Imidazole (99.5%), iodomethane (99%) and zinc(II) acetate dihydrate ( >98%) were purchased from Sigma-Aldrich. Silver tetrafluoroborate (99%) and hydrogen tetrachloroaurate(III) trihydrate ACS 99.99%, Au 49.0% min was used as starting material and purchased from Alfa Aesar. TLC were carried out on Merck DC Kieselgel 60 F-254 aluminum sheets and spots were visualized with UV-lamp (𝜆 = 254∕365 nm) if necessary. Preparative purifications were performed by silica gel flash column chromatography (Merck 40–60 μM). NMR spectroscopy and MS spectrometry were performed at the Laboratoire de Mesures Physiques (LMP) of the University of Montpellier (UM). NMR spectra were recorded on Bruker 400 MHz Avance III HD or 600 MHz Avance III spectrometers at 298K. DMSO-d6, CD2Cl2 and CD3OD were used as received (purchased from Eurisotop, France). 1H and 13C{1H} NMR spectra were calibrated to TMS on the basis of the relative chemical shift of the residual non-deuterated solvent as an internal standard. Chemical shifts (𝛿) are expressed in ppm from the residual non-deuterated solvent signal and coupling constants values (n J) are expressed in Hz. Abbreviations used for NMR spectra are as follows: s, singlet; d, doublet; t, triplet; m, multiplet. Mass spectra (HRMS) were recorded on ESI-TOF Q instruments in positive/negative modes. UV–visible absorption spectra were recorded at 25 °C on a JASCO V-650 spectrophotometer in 10 mm quartz cells (Hellma). Molar extinction coefficients 𝜀 (L⋅mol−1⋅cm−1) are expressed as log 𝜀.
4.2. Synthesis and characterization of the different compounds
4.2.1. Porphyrin 1
The synthesis and characterization data of the porphyrin 1 were reported in [46].
4.2.2. Porphyrin 2
Porphyrin 1 (150 mg, 0.225 mmol, 1.0 eq) and imidazole (150 mg, 2.25 mmol, 10 eq) were dissolved in dry DMF (11 mL). The solution was purged under Argon atmosphere for 10 min. NaH (60% in suspension in oil, 85 mg, 2.22 mmol, 10 eq) was added and the reaction mixture was stirred at room temperature for 30 min. Then, the reaction mixture was stirred at 140 °C for 1 h. After cooling at room temperature, CH2Cl2 (150 mL) was added and the organic phase was washed (distilled H2O), dried (MgSO4) and concentrated. The residue was purified by column chromatography (SiO2, eluent from CH2Cl2 to CH2Cl2/MeOH (98:2)) to give the porphyrin 2 in 72% yield (112 mg). 1H NMR (400 MHz, CD2Cl2, 298 K): 𝛿 8.86 (d, 3 JH–H = 5.0 Hz, 2H, Hpyrrole), 8.84–8.76 (m 4H, Hpyrrole), 8.66 (d, 3 JH–H = 5.0 Hz, 2H, Hpyrrole), 8.37 (s, 1H, H2), 8.05 (s, 1H, H5), 8.02 (dd, 3 JH–H = 7.8 Hz, 4 JH–H = 1.6 Hz, Ho), 7.90 (d, 3 JH–H = 8.0 Hz, 4H, Ha), 7.79–7.64 (m, 3H, Hm), 7.52 (m, 5H, Hb and H4), 2.65 (s, 6H, CH3 tolyl). 13C {1H} NMR (150.9 MHz, CD2Cl2, 298, K): 𝛿 144.4, 144.2, 143.7, 142.4, 141.1, 138.5, 137.9, 134.2, 134.2, 134.1 133.2, 133.2, 129.1, 128.7, 128.5, 128.2, 127.5, 121.2, 120.9, 112.8, 21.7 (CH3 tolyl) ppm. UV–visible (DMSO): 𝜆max(log𝜀): 414 (5.39), 526 (4.26) nm. MALDI-TOF+MS: calcd for C43H30N6Ni: 689.2, found 689.2.
4.2.3. Porphyrin 3
Porphyrin 2 (125 mg, 0.18 mmol, 1.0 eq) was dissolved in a TFA/H2SO4 4:1 (v:v) mixture and the reaction was stirred for 45 min at room temperature. Then, the mixture was poured into ice and water and neutralized by K2CO3(s). The organic phase was extracted with CH2Cl2, dried (MgSO4), and concentrated. The residue was purified by column chromatography (SiO2, eluent CH2Cl2). Recrystallization from CH2Cl2/n-hexane afforded the free base corresponding porphyrin in 87% yield (100 mg). Then, the obtained compound (90 mg, 0.14 mmol, 1.0 eq) was dissolved in dry THF (25 mL) and CH3I (2.2 mL, excess) was added. The reaction was stirred at 40 °C for 18 h under argon. Recrystallization from CH2Cl2/n-hexane afforded the porphyrin 3 as a purple solid in 89% yield (98 mg).
1H NMR (400 MHz, CD2Cl2, 298 K): 𝛿 10.44 (t, 3 JH–H = 1.7 Hz, 1H, H2), 9.06–8.98 (m, 5H, Hpyrrole and H5), 8.90–8.86 (m, 4H, Hpyrrole), 8.40 (t, 3 JH–H and 4 JH–H = 1.7 Hz, 1H, H4), 8.24–8.19 (m, 2H, Ho), 8.11 (d, 3 JH–H = 7.3 Hz, 4H, Ha), 7.92–7.82 (m, 3H, Hm and Hp), 7.68 (d, 3 JH–H = 7.3 Hz, 4H, Hb), 4.31 (s, 3H, N–CH3), 2.69 (s, 6H, CH3 tolyl), −2.95 (s, 2H, NH) ppm. 13C{1H} NMR (150.9 MHz, DMSO-d6, 298 K): 𝛿 142.5, 140.9, 137.9, 137.5, 134.4, 139.3, 134.3, 134.2, 132.7, 131.9, 130.2, 128.5, 128.2, 128.0, 127.1, 123.3, 123.1, 121.9, 108.7, 36.8 (NCH3), 21.2 (CH3 tolyl) ppm. UV–visible (DMSO): 𝜆max(log𝜀) = 413 (5.32), 525 (4.19), 615 (3.37) nm. HR ESI-MS (positive mode): calcd for
4.2.4. Porphyrin 4-I
Porphyrin 3 (50 mg, 0.065 mmol, 1.0 eq) was dissolved in CHCl3(5.5 mL). A solution of Zn(OAc)2 ⋅ 2H2O (19.8 mg, 0.090 mmol, 1.4 eq) in MeOH (1.5 mL) was added and the reaction was stirred at 50 °C for 1 h. After evaporation of the solvent, recrystallization from CH2Cl2/n-hexane afforded the porphyrin 4-I in 80% yield (44 mg). 1H NMR (400 MHz, DMSO-d6, 298 K): 𝛿 10.40–10.36 (br s, 1H, H2), 8.95 (t, 3 JH–H and 4 JH–H = 1.8 Hz, 1H, H5), 8.92 (s, 4H, Hpyrrole), 8.86–8.78 (m, 4H, Hpyrrole), 8.38 (t, 3 JH–H and 4 JH–H = 1.8 Hz, 1H, H4), 8.22–8.14 (m, 2H, Ho), 8.10–8.02 (m, 4H, Ha), 7.88–7.76 (m, 3H, Hm and Hp), 7.64 (d, 3 JH–H = 7.4 Hz, 4H, Hb), 4.30 (s, 3H, N–CH3), 2.68 (s, 6H, CH3 tolyl) ppm. 13C{1H} NMR (150.9 MHz, DMSO-d6, 298 K): 𝛿 150.6, 150.1, 149.5, 146.9, 142.2, 142.2, 139.1 137.1, 134.3, 134.2, 134.1, 132.7, 133.7, 132.7, 132.3, 130.4, 128.0, 127.9, 127.6, 127.5, 126.8, 123.4, 123.0, 122.2, 108.9, 36.8 (N–CH3), 21.2 (CH3 tolyl) ppm. UV–visible (DMSO): 𝜆max(log𝜀) = 426 (5.37), 516 (3.42), 599 (3.60) nm. ESI-MS (positive mode): calcd for C44H33N6Zn+: 709.21, found: 709.28. ESI-MS (negative mode): calcd for I−: 126.91, found: 126.96.
4.2.5. Porphyrin 4-BF4
Porphyrin 4-I (160 mg, 0.19 mmol, 1.0 eq) was dissolved in acetone (50 mL). Then, a solution of AgBF4 (39.5 mg, 0.20 mmol, 1.1 eq) in acetone (2.5 mL) was added dropwise and the reaction mixture was stirred for 1 h under argon at room temperature in the dark. After evaporation, the crude product was purified by column chromatography (SiO2, eluent from CH2Cl2 to CH2Cl2/MeOH 95:5, (v:v)). Recrystallization from CH2Cl2/n-hexane afforded the porphyrin 4-BF4 in 85% yield (125 mg). 1H NMR (400 MHz, DMSO-d6, 298 K): 𝛿 10.39 (br dd, 1H, H2), 8.95 (t, 3 JH–H and 4 JH–H = 1.8 Hz, 1H, H5), 8.91 (s, 4H, Hpyrrole), 8.84–8.76 (m, 4H, Hpyrrole), 8.37 (t, 3 JH–H and 4 JH–H = 1.8 Hz, 1H, H4), 8.21–8.15 (m, 2H, Ho), 8.08–8.03 (m, 4H, Ha), 7.83–7.79 (m, 3H, Hm and Hp), 7.63 (d, 3 JH–H = 7.4 Hz, 4H, Hb), 4.30 (s, 3H, N–CH3), 2.68 (s, 6H, CH3 tolyl) ppm. 13C{1H} NMR (150.9 MHz, DMSO-d6, 298 K): 𝛿 150.6, 150.1, 149.5, 146.9, 142.2, 142.2, 139.1 137.1, 134.3, 134.2, 134.1, 132.7, 133.7, 132.7, 132.3, 130.4, 128.0, 127.9, 127.6, 127.5, 126.8, 123.4, 123.0, 122.2, 108.9, 36.8 (N–CH3), 21.2 (CH3 tolyl) ppm. 19F (400 MHz, DMSO-d6, 298 K): 𝛿 −149.26 (s), −149.20 (s) ppm. UV–visible (CH2Cl2): 𝜆max(log𝜀) = 420 (5.79), 518 (3.18), 548 (4.40) nm. ESI-MS (positive mode): calcd for C44H33N6Zn+: 709.21, found: 709.28. ESI-MS (negative mode): calcd for
4.2.6. Porphyrin 5
The synthesis and characterization data of the porphyrin 5 were reported in [47].
4.2.7. Porphyrin 6
Porphyrin 5 (160 mg, 0.2 mmol, 1.0 eq) was dissolved in dry DMF (15 mL) and 1-methylimidazole (1.1 mL, 100 eq) was added. The solution was stirred at 80 °C for 24 h under argon atmosphere. Alkylation completion was monitored by TLC. Once finished, the solvent was evaporated under reduced pressure. Recrystallization from CH2Cl2/n-hexane gave the porphyrin 6 in 78% yield (138 mg). 1H NMR (400 MHz, CD2Cl2, 298 K): 𝛿 11.17 (br s, 1H, H2), 8.76 (d, 3 JH–H = 4.8 Hz, 2H, Hpyrrole), 8.67 (d, 3 JH–H = 4.8 Hz, 2H, Hpyrrole), 8.63 (br s, 4H, Hpyrrole), 8.28 (d, 3 JH–H = 8.0 Hz, 2H, Ho), 7.86 (d, 3 JH–H = 8.0 Hz, 2H, Hm), 7.45 (t, 3 JH–H and 4 JH–H = 1.8 Hz, 1H, H5), 7.31 (dd, 3 JH–H and 4 JH–H = 1.8 Hz, 1H, H4), 7.29 (broad s, 6H, Hmes meta), 5.94 (s, 2H, CH2), 4.16 (s, 3H, N–CH3), 2.61 (s, 9H, CH3 mes para), 1.83–1.86 (2s, 18H, CH3 mes ortho), −2.62 (s, 2H, NH) ppm. 13C{1H} NMR (150.9 MHz, CD2Cl2, 298 K): 𝛿 143.9, 139.9, 139.8, 138.7, 138.6, 138.5, 135.8, 133.3, 128.3, 127.9, 123.9, 122.4, 121.0, 118.6, 118.6, 118.4, 22.0, 21.9, 21.7 ppm. UV–visible (CH2Cl2): 𝜆max(log𝜀) = 418 (5.41), 515 (4.05), 548 (3.55), 590 (3.52), 648 (3.31) nm. HR ESI-MS (positive mode): calcd for C58H55N
4.2.8. Porphyrin 7
Porphyrin 6 (110 mg, 0.126 mmol, 1.0 eq) was dissolved in CHCl3 (12 mL). A solution of Zn(OAc)2 ⋅ 2H2O (41.6 mg, 0.189 mmol, 1.5 eq) in MeOH (2.6 mL) was added and the solution was stirred at 50 °C for 1 h. Then, the solvent was evaporated under reduced pressure. Recrystallization from CH2Cl2/n-hexane afforded the porphyrin 7 in 90% yield (106 mg). 1H NMR (400 MHz, DMSO-d6, 298 K): 𝛿 9.46 (t, 4 JH–H = 1.7 Hz, 1H, H2), 8.64 (d, 3 JH–H = 4.6 Hz, 2H, Hpyrrole), 8.56 (d, 3 JH–H = 4.6 Hz, 2H, Hpyrrole), 8.54–8.50 (m, 4H, Hpyrrole), 8.22 (d, 3 JH–H = 8.1 Hz, 2H, Ho), 8.07 (t, 3 JH–H and 4 JH–H = 1.7 Hz, 1H, H5), 7.87 (t, 3 JH–H and 4 JH–H = 1.7 Hz, 1H, H4), 7.77 (d, 3 JH–H = 8.1 Hz, 2H, Hm), 7.30 (broad s, 6H, Hmes meta), 5.78 (s, 2H, CH2), 3.98 (s, 3H, N–CH3), 2.57 (s, 9H, CH3 mes para), 1.78, 1.77, 1.75 (3s, 18H, CH3 mes ortho) ppm. 13C{1H} NMR (150.9 MHz, DMSO-d6, 298 K): 𝛿 149.0, 149.0, 148.9, 148.8, 143.2, 139.1, 139.0, 138.4, 138.4, 137.1, 136.9, 136.8, 134.6, 133.9, 131.5, 130.5, 130.0, 127.6, 126.2, 124.2, 122.8, 118.4, 117.7, 51.9, (–CH2), 36.0 (N–CH3), 21.6, 21.5, 21.0 (CH3 ortho, CH3 para) ppm. UV–visible (CH2Cl2): 𝜆max(log𝜀) = 420 (5.63), 549 (4.22) nm. HRESI-MS (positive mode): calcd for C58H53N6Zn+: 897.3623, found: 897.3632.
4.2.9. Gold(I) complex 8
Porphyrin 4-BF4 (60 mg, 0.075 mmol, 1.0 eq) and [AuCl(tht)] (26.5 mg, 0.083 mmol, 1.1 eq) were dissolved in dry THF (6.3 mL). A solution of tBuOK (9.3 mg, 0.083 mmol, 1.1 eq) in THF/MeOH (2.3 mL/10 drops) was added dropwise and the reaction mixture was stirred at room temperature under argon for 18 h. After evaporation, the crude was purified by column chromatography (SiO2, eluent CH2Cl2). Recrystallization from CH2Cl2/n-hexane gave the gold(I) complex 8 as a dark pink solid in 80% yield (72 mg).
1H NMR (600 MHz, DMSO-d6, 298 K): 𝛿 8.87 (d, 3 JH–H = 4.6 Hz, 2H, Hpyrrole), 8.80–8.76 (m, 4H, Hpyrrole), 8.71 (d, 3 JH–H = 4.6 Hz, 2H, Hpyrrole), 8.52 (d, 3 JH–H = 1.9 Hz, 1H, H5), 8.21–8.11 (m, 4H, Ho), 8.06 (d, 3 JH–H = 1.9 Hz, 1H, H4), 8.02 (dd, 3 JH–H = 7.3 Hz and 4 JH–H = 2.1 Hz, 2H, Ha) 7.82–7.78 (m, 3H, Hm and Hp), 7.64–7.58 (m, 4H, Hb), 4.23 (s, 3H, N–CH3), 2.67 (s, 6H, CH3 tolyl) ppm. 13C{1H} NMR (150.9 MHz, DMSO-d6, 298 K): 𝛿 175.1 (CNHC), 150.2, 150.0, 149.3, 148.1, 142.4, 139.3,136.9, 134.15, 134.1, 134.0,133.0, 132.1, 131.9, 130.2, 128.4, 127.3, 126.6, 122.4, 121.5, 121.2, 113.7, 38.1 (CH3 tolyl), 21.1 (N–CH3) ppm. 1H DOSY NMR (600 MHz, CD2Cl2, 298 K): 1.54 × 10−10 m2⋅s−1. UV–visible (CH2Cl2): 𝜆max(log𝜀) = 420 (5.75), 548 (4.38) nm. HR ESI-MS (positive mode): calcd for C44H32AuClN6Zn: 946.1911, found: 946.1923.
4.2.10. Gold(I) complex 9
Complex 8 (45 mg, 0.048 mmol, 1.0 eq) and porphyrin 4-BF4 (34.6 mg, 0.044 mmol, 0.90 eq) were dissolved in acetone/MeOH (18 mL/0.5 mL). Then, K2CO3 (6.7 mg, 0.048 mmol, 1.0 eq) was added and the reaction mixture was stirred at room temperature for 16 h under argon in the dark. After evaporation, the crude was purified by column chromatography (SiO2, eluent from CH2Cl2 to CH2Cl2/MeOH 95:5 (v:v)). Recrystallization from CH2Cl2/n-hexane afforded the gold(I) complex 9 in 52% yield (40 mg).
1H NMR (600 MHz, DMSO-d6, 298 K): 𝛿 8.80 (s, 8H, Hpyrrole), 8.70 (d, 4H, 3 JH–H = 4.5 Hz, Hpyrrole), 8.45 (d, 3 JH–H = 1.8 Hz, 2H, H5), 8.32 (d, 3 JH–H = 4.5 Hz, 4H, Hpyrrole), 8.22–8.17 (m, 4H, Ho), 8.04 and 7.98 (2dd AB system, 3 JH–H = 7.7 and 2.0 Hz, each 4H, Ha), 7.89–7.79 (m, 6H, Hm and Hp), 7.65 and 7.58 (2dd AB system, 3 JH–H = 7.7 and 2.0 Hz, each 4H, Hb), 7.34 (d, 3 JH–H = 1.8 Hz, 2H, H4), 2.68 (s, 12H, CH3 tolyl), 2.04 (s, 6H, N–CH3) ppm. 13C{1H} NMR (150.9 MHz, DMSO-d6, 298 K): 𝛿 188.8 (CNHC), 150.2, 150.0, 149.8, 149.5, 147.6,142.3, 139.2,136.9, 134.1, 134.0, 133.9,132.7, 132.3, 132.0, 129.7, 128.3, 127.7, 127.4, 126.7, 122.4, 121.5, 113.6, 35.1 (CH3 tolyl), 21.0 (N–CH3) ppm. 1H DOSY NMR (600 MHz, CD2Cl2, 298 K): 1.15 × 10−10m2⋅s−1. UV–visible (CH2Cl2): 𝜆max(log𝜀) = 420 (5.75), 548 (4.38) nm. High-resolution ESI-MS (positive mode): calcd for
4.2.11. Gold(I) complex 10
Porphyrin 7 (40 mg, 0.043 mmol, 1.0 eq) was dissolved in dry CH2Cl2 (10 mL). Then, Ag2O (9.9 mg, 0.043 mmol, 1.0 eq) was added and the reaction mixture was vigorously stirred at room temperature under argon protected from light. After 16 h, [AuCl(tht)] (13.7 mg, 0.043 mmol, 1.0 eq) was added and the mixture was stirred for 18 h under the same conditions. The solvent was evaporated under reduced pressure and the crude compound was purified by column chromatography (SiO2, eluent CH2Cl2). Recrystallization from CH2Cl2/n-hexane gave the gold(I) complex 10 as a red-purple solid in 41% yield (20 mg). 1H NMR (400 MHz, CD2Cl2, 298 K): 𝛿 8.84 (d, 3 JH–H = 4.6 Hz, 2H, Hpyrrole), 8.73 (d, 3 JH–H = 4.6 Hz, 2H, Hpyrrole), 8.69 (d, 3 JH–H = 1.2 Hz, 4H, Hpyrrole), 8.23 (d, 3 JH–H = 8.0 Hz, 2H, Ho), 7.69 (d,3 JH–H = 8.0 Hz, 2H, Hm), 7.29 (broad s, 6H, Hmes meta), 7.26 (d, 3 JH–H = 2.0 Hz, 1H, H5), 7.13 (d, 3 JH–H = 2.0 Hz, 1H, H4), 5.74 (s, 2H, CH2), 3.96 (s, 3H, N–CH3), 2.62 (s, 9H, CH3 mes para), 1.84–1.82 (2s, 18H, CH3 mes ortho) ppm. 13C {1H} NMR (150.9 MHz, CD2Cl2, 298 K): 𝛿 172.3 (CNHC), 150.4, 150.4, 150.3, 150.2, 143.8, 139.7, 139.6, 139.5, 139.4, 138.1, 135.4, 135.3, 138.4, 135.6, 135.5, 132.4, 131.6, 131.5, 131.1, 128.1, 126.5, 123.2, 121.4, 119.7, 119.4, 119.3, 55.5 (CH2), 38.9 (N–CH3), 22.0, 21.9, 21.7 (CH3 mes ortho, CH3 mes para) ppm. UV–visible (CH2Cl2): 𝜆max(log𝜀) = 420 (5.75), 549 (4.36) nm. MALDI-TOF+MS: calcd for C64H55Au4N12Cl4: 1128.29, found 1128.30.
4.2.12. Gold(I) complex 11
Procedure A. Porphyrin 6 (40 mg, 0.043 mmol, 1.0 eq) was dissolved in dry THF (10.5 mL). Then, [AuCl(tht)] (15.4 mg, mmol, 1.1 eq) was added and the mixture was degassed with argon for 10 min. tBuOK (6 mg, mmol, 1.1 eq) was dissolved in MeOH (0.5 mL) and added dropwise to the reaction mixture, which was stirred at room temperature for 18 h under argon in the dark. Then, the solvent was evaporated under reduced pressure and the residue was purified by column chromatography (SiO2, eluent CH2Cl2). Recrystallization from CH2Cl2/n-hexane afforded the gold(I) complex 11 as a purple solid in 57% yield (28 mg). Procedure B. Porphyrin 10 (30 mg, 0.027 mmol) was dissolved in acetone (12 mL) under argon. Then, a CH2Cl2/TFA 4:1 mixture (6 mL) was slowly added and the reaction mixture was stirred for 30 mn. After neutralization with NaHCO3, the solvent was evaporated and the residue purified by column chromatography (SiO2, eluent CH2Cl2). Recrystallization from CH2Cl2/n-hexane afforded the gold(I) complex 11 as a purple solid in 70% yield (20 mg). 1H NMR (400 MHz, CD2Cl2, 298 K): 𝛿 8.77 (d, 3 JH–H = 4.8 Hz, 2H, Hpyrrole), 8.67 (d, 3 JH–H = 4.8 Hz, 2H, Hpyrrole), 8.63 (s, 4H, Hpyrrole), 8.22 (d, 3 JH–H = 8.0 Hz, 2H, Ho), 7.69 (d, 3 JH–H = 8.0 Hz, 2H, Hm), 7.29 (s, 6H, Hmes meta), 7.24 (d, 3 JH–H = 1.9 Hz, 1H, H5), 7.12 (d, 1H, 3 JH–H = 1.9 Hz, H4), 5.73 (s, 2H, CH2), 3.95 (s, 3H, N–CH3), 2.61 (s, 9H, CH3 mes para), 1.86–1.84 (2s, 18H, CH3 mes ortho), −2.62 (s, 2H, NH) ppm. 13C{1H} NMR (150.9 MHz, CD2Cl2, 298 K): 𝛿 172.5 (CNHC), 143.0, 139.9, 138.9, 138.8, 138.6, 138.4, 135.6, 135.5, 128.3, 126.7, 123.2, 121.4, 118.8, 118.6, 118.4, 55.5 (CH2), 39.0 (N–CH3), 21.9 (2s, CH3 mes ortho), 21.7 (CH3 mes para) ppm. UV–visible (CH2Cl2): 𝜆max(log𝜀) = 418 (5.65), 514 (4.28), 548 (3.80), 590 (3.75), 647 (3.55) nm. MALDI-TOF+MS: calcd for C64H55Au4N12Cl4: 1066.38, found 1066.40.
4.2.13. Pyropheophorbide a 12
The synthesis and characterization data of pyropheophorbide a 12 were reported in [63, 64, 65, 66].
4.2.14. Chlorin 13
Pyropheophorbide a12 (62 mg, 0.12 mmol, 1.0 eq), 1-(3-aminopropyl)imidazole (18.6 μL, 0.16 mmol, 1.4 eq) and 4-DMAP (18.4 mg, 0.15 mmol, 1.3 eq) were dissolved in dry CH2Cl2 (4.0 mL). DCC (31.1 mg, 0.15 mmol, 1.3 eq) was portionwise added at 0 °C under argon atmosphere. The reaction mixture was stirred at room temperature for 3 h in the dark. The reaction was monitored by TLC. Once finished, H2O (4.0 mL) was added and the mixture was stirred for 15 min. Then, organic phase was extracted with CH2Cl2, washed (distilled H2O), dried (MgSO4) and concentrated. The residue was dissolved in a minimum amount of CH2Cl2 and refrigerated overnight. The crude was then filtered, washed with cold CH2Cl2, and the filtrate was concentrated (the operation was reiterated twice). Recrystallization from CH2Cl2/n-hexane afforded the chlorin 13 as a dark green solid in 46% yield (34 mg). 1H NMR (400 MHz, CD2Cl2, 298 K): 𝛿 9.44, 9.41, 8.64 (each s, each 1H, 5, 10 and 20-H), 8.04 (dd, 3 JH–H = 17.8, 11.6 Hz, 1H, 31), 7.18 (br dd, 1H, Hd), 6.81 (br dd, 3 JH–H = 1.3 Hz and 4 JH–H = 1.3 Hz, 1H, Hf), 6.62 (br dd, 3 JH–H = 1.3 Hz and 4 JH–H = 1.3 Hz, 1H, He), 6.34–6.16 (m, 2H, 32), 5.24 and 5.06 (2d AB system, 2 JH–H = 19.8 Hz, each 1H, 132), 4.58–4.51, 4.39–4.36 (each m, each 1H, 17 and 18), 3.75–3.69 (m, 2H, 81), 3.59–3.49 (m, 2H, Hc), 3.65, 3.43, 3.26 (each s, each 3H, 21, 71 and 121), 2.89–2.72, 2.66–2.42, 2.25–2.17 and 1.95–1.80 (each m, each 2H, 171, 172, Ha and Hb), 1.81 (d, 3 JH–H = 7.3 Hz, 3H, 181), 1.67 (t, 3 JH–H = 7.6 Hz, 4H, 82), −1.79 (s, 2H, NH) ppm. UV–visible(CH2Cl2): 𝜆max(log𝜀) = 412 (4.57), 508 (3.88), 538 (3.82), 609 (3.75), 667 (4.13) nm. MALDI-TOF+MS: calcd for C39H43N7O2 642.35, found 642.30.
4.2.15. Chlorin 14
Chlorin 13 (30 mg, 0.047 mmol, 1.0 eq) was dissolved in dry THF (6.0 mL) and CH3I (290 μL, 4.7 mmol, 100 eq) was added. The solution was stirred at 40 °C for two days under argon atmosphere and protected from light. The reaction was monitored by TLC. After evaporation of the solvent, recrystallization from CH2Cl2/n-hexane afforded the monocationic chlorin 14 as a black solid in 52% yield (19 mg). 1H NMR (400 MHz, (CD2Cl2/CD3OD 9:1 (v:v), 298 K): 𝛿 9.50, 9.37, 8.72 (each s, each 1H, 5, 10 and 20-H), 9.26 (br s, 1H, Hd), 8.04 (dd, 3 JH–H = 17.9,11.5 Hz, 1H, 31), 7.04, 6.98 (each br s, each 1H, He and Hf ), 6.31 and 6.18 (2d AB system, 2 JH–H = 19.8 Hz, each 1H, 32), 5.32 and 5.07 (2 d AB system, 2 JH–H = 19.2 Hz, each 1H, 132), 4.70–4.64 and 4.33–4.28 (each m, each 1H, 17 and 18), 3.79 (s, 3H, Hg), 3.62 and 3.23 (each s, each 3H), 3.00–2.91 (m, 2H, Ha), 2.71–2.56 and 2.43–2.32 (each m, each 3H, 171, 172 and Hb), 1.81 (d, 3 JH–H = 7.4 Hz, 1H, 181), 1.69 (t, 3 JH–H = 7.6 Hz, 4H, 82), −1.75 (s, 2H, NH) ppm. 13C{1H} NMR (150.9 MHz, (CD2Cl2/CD3OD 9:1 (v:v), 298 K): 𝛿 197.1 (C=O), 174.4, 173.1, 161.8, 155.6, 151.1, 149.4, 145.6, 142.1, 138.1, 137.3, 136.7 (2s), 136.1, 132.8, 130.8, 129.6, 128.6, 123.5, 122.9, 122.7, 104.4, 97.1, 94.5, 52.5, 50.5, 49.6, 48.8, 47.7, 37.0, 35.7, 34.4, 33.6, 31.0, 29.9, 26.2, 25.6, 23.5, 19.9, 17.7, 12.5, 12.2, 11.5 ppm. UV–visible (CH2Cl2): 𝜆max(log𝜀) = 413 (4.67), 509 (3.75), 540 (3.67), 610 (3.61), 668 (4.28) nm. HR ESI-MS (positive mode): calcd for
4.3. Biological studies
Cell culture. Human breast cancer cells (MCF-7) were purchased from ATCC (American Type Culture Collection, Manassas, VA). Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM-F12) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin. Cells were allowed to grow in humidified atmosphere at 37 °C under 5% CO2.
4.3.1. Cytotoxicity study in the dark
MCF-7 cells were seeded into 96-well plates at 5000 cells per well in 200 μL culture medium and allowed to grow for 24 h. Increasing concentrations (from 0 to 2 μM) of compounds were incubated in culture medium of MCF-7 cells during 72 h. Then, a MTT assay was performed to evaluate the toxicity. Briefly, cells were incubated for 4 h with 0.5 mg⋅mL−1 of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Promega) in media. The MTT/media solution was then removed, and the precipitated crystals were dissolved in EtOH/DMSO (1:1). The solution absorbance was read with a microplate reader at 540 nm.
4.3.2. PDT experiments
For 450 nm and 545 nm in vitro phototoxicity, 1000 cells by well were plated in a 384-well plates, in 50 μL of culture medium. Twelve hours after seeding, compounds were added on cells at a concentration of 0.5 μM for 24 h. After this incubation, cells were submitted (or not) to laser irradiation for 10 min with the Leica DM IRB at 450 nm (4.6 J⋅cm−2) or 545 nm (19.5 J⋅cm−2). The laser beam was focussed by a microscope objective lens (magnification × 4). Two days after irradiation, an MTT assay was performed to measure the cell viability. For 650 nm in vitro phototoxicity, 5000 cells by well were plated in a 96-well plates, in 100 μL of culture medium. Twelve hours after seeding, compounds were added on cells at a concentration of 0.5 μM for 24 h. Then, cells were submitted (or not) to laser irradiation with a red laser at 650 nm for 10 min (18.75 J⋅cm−2). Two days after irradiation, an MTT assay was performed to evaluate the cell viability.
Acknowledgments
The authors are grateful to the University of Montpellier, the CNRS and the French Ministry of Research for financial support. SR is also grateful for financial support from the Région Languedoc-Roussillon (Research Grant Chercheur(se)s d’Avenir – 2015-005984) and the FEDER Program (Fonds Européen de Développement Régional).
Supplementary data
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.98 or from the author.
1The values of photo-induced cell death are related to the non-irradiated cells in the same experiment to fairly describe the PDT efficiency, i.e. the cell death induced by light excitation only.