



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Instead of being an undesired waste, CO2 can be considered as feedstock to be transformed into sustainable fuels replacing fossil fuels, which could be an interesting alternative to limit both global warming and depletion of fossil fuels. Because of high chemical stability, the photocatalytic reduction of CO2 is thermodynamically unfavorable under direct solar light irradiation. Consequently, CO2 photocatalytic reduction has been considered for a few years as a promising technology for converting CO2 into light-driven C-based compounds, as reported in several reviews summing up the state of the art on that topic [1, 2]. Another important aspect of the solar-driven conversion of CO2 into liquid or gaseous fuels is the high-density storage of solar energy in the form of chemical bonds, mainly C–H bonds. Thus, the non-intermittent energy storage capacity of solar fuels has become a very attractive solution.
The photocatalytic reduction of CO2 can be achieved in gas phase using solar energy at low temperature. During photocatalysis, CO2 and H2O in humified air are utilized as initial reactants allowing to simultaneously reduce CO2 and oxidize H2O to produce renewable fuel products without secondary pollution [3, 4]. Furthermore, water is considered as being the most suitable hydrogen donor (mainly implied as H+ donor in CO2 reduction steps).
Among the different reaction products that could be obtained from CO2 photocatalytic reduction and knowing also that the energy content of oxygenated compounds decreases as the oxygen content increases (due to the relative decrease in C–H bonds storing energy), methane (CH4) is one of the most interesting products, as well as methanol. Nonetheless, as methanol’s photoreactivity is much higher than that of CO2, leading to easy over-oxidation to formaldehyde and formic acid, high selectivity toward methane formation is preferred [5].
Amongst the huge variety of semiconductors and composite materials investigated for CO2 photocatalytic reduction in gas phase, TiO2 often prevails due to its chemical stability, moderate cost and resistance toward corrosion [6, 7, 8]. However, its relative fast recombination rate of electron–hole pairs and limited visible-light harvesting capacity (mainly in the UV region, <5% of the solar spectrum) are considered the main limitations for solar light-driven gas-phase CO2 photocatalytic reduction in presence of water. In recent years, several strategies have been carried out in order to overcome these limitations, including modification of the morphology of TiO2 [9, 10], combination with other semiconductors [11, 12, 13] or elements by means of mono-doping [14, 15] or co-doping approaches [16, 17], or loading with metal nanoparticles (NPs), acting as electron sinks, cocatalyst or inducing surface plasmon (SP) phenomena [8, 18, 19]. Between the different metal NPs used as cocatalysts and exhibiting surface plasmon resonance (SPR) properties in the visible range, gold nanoparticles (Au NPs) can be considered as among the most interesting NPs for driving CH4 production from CO2 gas-phase photocatalytic reduction in presence of water vapor as reducing agent. Indeed, in addition to high inertness in numerous environments, Au NPs exhibit a high quality factor in visible light [20]. Moreover, Pt NPs, because of their large work function (5.64 eV vs. Evacuum), have also been considered as efficient cocatalysts, the deposition of Pt promoting the migration of photoinduced electrons from TiO2 to Pt thus improving the photocatalytic reduction steps [21]. However, finding a cocatalyst with a low price, abundant reserves (typically non-noble metals), and excellent catalytic performance is a crucial concern.
As an alternative, cocatalytic phases, earth-abundant copper-based heterogeneous photocatalysts have received increasing attention for CO2 reduction, since copper is an inexpensive element with multiple oxidation states, meaning that metal and oxide particles can be formed and also contribute positively to the photocatalytic process. Copper oxide (CuO) and cuprous oxide (Cu2O) have narrow band gaps of 1.7 eV [22] and 2.2 eV [23], respectively, and can additionally act as photoactive phases under visible light. Moreover, the introduction of metallic Cu can improve the photocatalytic activity when it is in contact with other semiconductors, acting as CO2 adsorption sites or as catalytic reduction ones, and also achieving limitation of charge carrier recombination by adding electron traps, yielding the formation of a Schottky barrier [24]. Another beneficial impact to be mentioned is the generation of a SPR effect in visible light, knowing that SPR of metallic Cu extends from UV–visible to near-infrared (NIR) regions of the optical spectrum [25]. It is thus necessary to maintain a good balance between the different oxidation states of Cu, the main difficulty consisting in keeping a sufficient proportion of Cu in its metallic state. To do this, the development of approaches designed to mitigate oxidation, such as the formation of bimetallic M–Cu NPs may be a promising solution. Moreover, it is fully admitted that Cu-based bimetallic systems significantly promote activation of the very stable CO2 molecule. Although it is known (by means of electrocatalysis or thermal catalysis) that bimetallic Cu-based systems can efficiently improve CO2 catalytic reduction, and that the second metal plays various roles (morphological, electronic transfer, reactant transfer, generation of supplementary actives sites, intermediates adsorption/desorption) [26, 27], these bimetallic systems have been only scarcely studied in heterogeneous photocatalysis for gas-phase CO2 photoreduction in presence of H2O.
Herein, M–Cu (M = Ag, Pt, Pd, Au)/TiO2 bimetallic samples were prepared by impregnation/chemical reduction methods, characterized and compared toward gas-phase photocatalytic CO2 reduction in humid atmosphere under artificial solar- and visible-light illumination.
2. Results and discussion
2.1. Chemical composition
The real chemical compositions of Pt-, Cu-, Au-, Pd- and Ag-monometallic as well as related bimetallic systems obtained by coreduction are reported in Table 1. First, by considering the TiO2 P25 support, one can observe that on monometallic samples, the deposition yield changed from 73 to 100%. The two noble metals Pd and Pt showed the highest yield among all. Ag deposition was also achieved with a very good yield of 90%. It is worth mentioning that Cu exhibited the lowest yield, close to 70%. Regarding the bimetallic materials, Pt- and Pd-containing materials led to 77–86% yields, the lowest ones being obtained for Au– and Ag–Cu/TiO2 photocatalysts, already identified as more difficult to deposit as monometallics. It is worth noting that the presence of Cu and the difficulty to deposit it onto the TiO2 surface is detrimental to the deposition of a second metal. Nevertheless, even if Cu deposition is more difficult as a monometallic system, the M–Cu molar composition however was close to the expected one, assuming that M–Cu interaction using a codeposition method happened before deposition onto the support.
Determination of total metal content, deposition yield, and bi-metallic composition from ICP-AES measurements
| Sample | Nominal tot. metal wt% | Real total metal wt% | Deposition yield % | Nominal M–Cu mol. composition | Real M–Cu mol. composition |
|---|---|---|---|---|---|
| Pt/TiO2 P25 | 2 | 1.99 | 99 | ||
| Pd/TiO2 P25 | 2 | 2.00 | 100 | ||
| Au/TiO2 P25 | 2 | 1.69 | 85 | ||
| Ag/TiO2 P25 | 2 | 1.81 | 90 | ||
| Cu/TiO2 P25 | 2 | 1.46 | 73 | ||
| Pt–Cu/TiO2 P25 | 2 | 1.72 | 86 | 50–50 | 59–41 |
| Pd–Cu/TiO2 P25 | 2 | 1.69 | 85 | 50–50 | 47–53 |
| Au–Cu/TiO2 P25 | 2 | 1.53 | 77 | 50–50 | 60–40 |
| Ag–Cu/TiO2 P25 | 2 | 1.62 | 81 | 50–50 | 49–51 |
| Pt/TiO2 UV-100 350 °C | 1 | 1.00 | 100 | ||
| Cu/TiO2 UV-100 350 °C | 1 | 0.70 | 70 | ||
| Ptx–Cuy/TiO2 UV-100 350 °C | 1 | 0.99 | 99 | 20–80 | 21–79 |
| 1 | 0.93 | 93 | 50–50 | 53–47 | |
| 1 | 0.97 | 90 | 80–20 | 81–19 | |
| Ptx–Cuy/TiO2 UV-100 350 °C∗ | 1 | 0.90 | 99 | 20–80 | 20–80 |
| 1 | 0.99 | 99 | 50–50 | 57–43 | |
| 1 | 0.99 | 99 | 80–20 | 82–18 |
∗stands for Pt–Cu/TiO2 UV-100 350 °C obtained by successive reduction method. The relative error is estimated at 5%.
Globally, except for the Cu monometallic sample, the total deposition yield onto the TiO2 UV-100 350 °C support was very important, higher than 90%. As well as what was observed above for the TiO2 P25 support, in terms of bimetallic molar composition, the real compositions are close to the targeted ones, meaning that despite the weaker interaction of Cu with TiO2 UV-100 350 °C when alone, this metal takes advantage of the simultaneous presence of Pt.
2.2. Photocatalytic activity under simulated solar light
From the photocatalytic activity of the M0.5–Cu0.5/TiO2 P25 bimetallic systems, one can underline that the association with Pt resulted in the best performance, considering both the reaction rate and selectivity toward C-based products, namely CH4 formation (more than 40 μmol⋅h−1⋅g−1) (Figure 1). With a production rate for CH4 formation of ca. 23 μmol⋅h−1⋅g−1, and despite a selectivity close to 100%, the Pd0.5–Cu0.5/TiO2 P25 photocatalyst exhibited a reaction rate almost twice lower than the Pt–Cu-based one. The activity on Au0.5–Cu0.5/TiO2 P25, although accompanied by almost total selectivity toward CH4, was decreased to less than 15 μmol⋅h−1⋅g−1. The lowest performance was observed when associating Ag and Cu, in parallel with a drastic decrease in selectivity. Among all the M–Cu bimetallic systems, one has to underline that, except for Pt0.5–Cu0.5/TiO2 P25, enhanced activity was obtained compared to the monometallic counterparts, even if the monometallic Cu/TiO2 reference resulted in poor activity. Beside this observation, we can note that the Ag/TiO2 and Cu/TiO2 monometallics led to very low activity and selectivity towards C-containing molecules compared to the noble-metal monometallic systems.
Gas-phase photocatalytic CO2 reduction on (top left) 2 wt% Ag0.5–Cu0.5/TiO2 P25 (top right) 2 wt% Au0.5–Cu0.5/TiO2 P25 (bottom left) 2 wt% Pt0.5–Cu0.5/TiO2 P25 (bottom right) 2 wt% Pd0.5–Cu0.5/TiO2 P25. Comparison with the monometallic counterparts.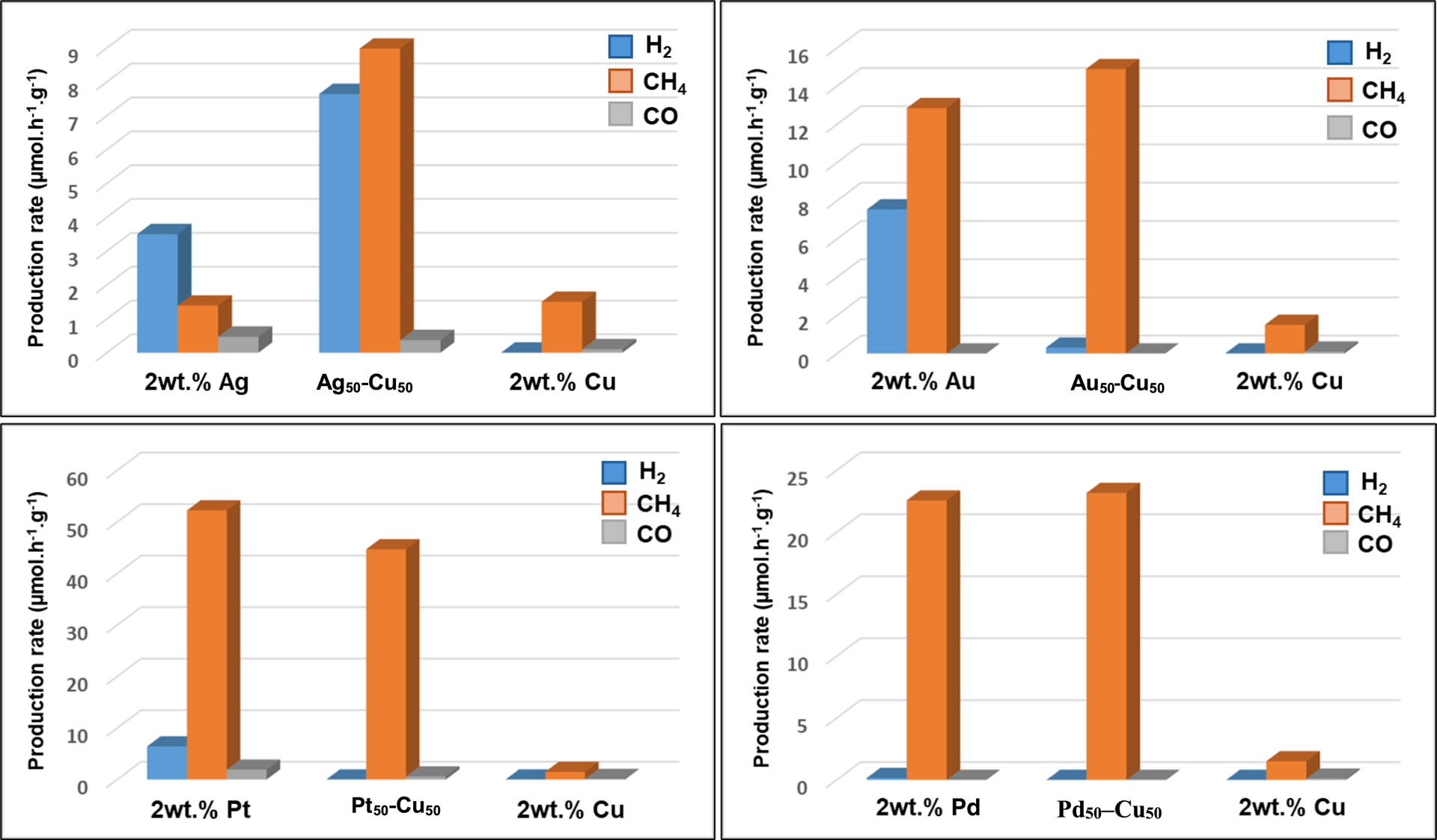
The two 2 wt% Pd0.5–Cu0.5/TiO2 P25 and 2 wt% Pt0.5–Cu0.5/TiO2 P25 samples previously identified as the most active in terms of production rate and selectivity toward CH4 were further chosen to study the influence of total metal content, molar composition, and TiO2 support. Looking at the effect of metal content varying from 1 to 3 wt%, a common general trend emerged, while keeping the electronic selectivity close to 100% (Figure 2). It was observed that doubling total metal loading from 1 to 2 wt% resulted in doubling the active sites that contribute the most strongly to the rate-determining steps, without impacting the reaction routes. However, increasing further to 3 wt% led to a deviation with a non-proportional increase in the reaction rate but, above all, detrimental to selectivity toward the formation of C-based compounds.
Gas-phase photocatalytic CO2 reduction on (left) Pd0.5–Cu0.5/TiO2 P25 and (right) Pt0.5–Cu0.5/TiO2 P25 for 1, 2, and 3 wt%.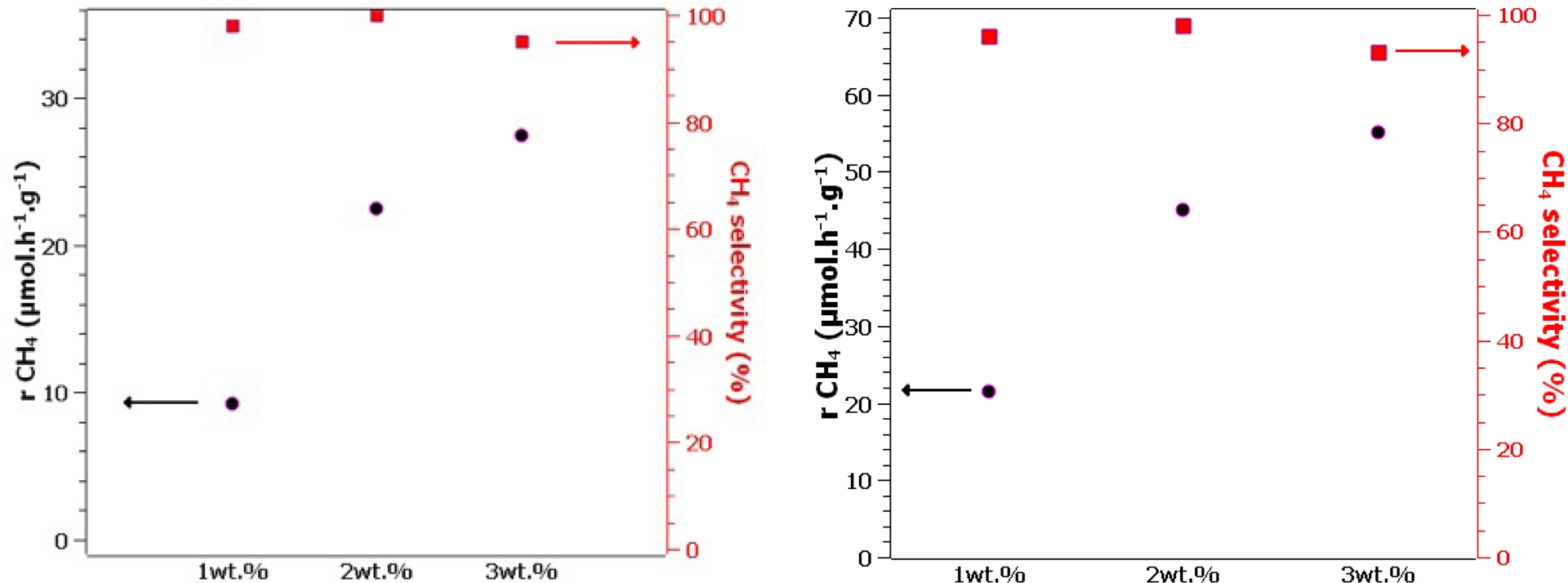
The evolution of both CH4 production rate and selectivity as a function of molar composition (keeping the metal content constant at 2 wt%) showed an optimum close to 80–20 between Pd (or Pt) and Cu (Figure 3). For this optimal composition, the resulting performance regarding CH4 production rate exceeded the one of the corresponding Pd (or Pt) activity. Increasing further Cu content yielded a strong decrease in activity, without impacting the selectivity, however.
Gas-phase photocatalytic CO2 reduction on (left) 2 wt% Pdx–Cuy/TiO2 P25 and (right) 2 wt% Ptx–Cuy/TiO2 P25, x = 0.2, 0.5, and 0.8.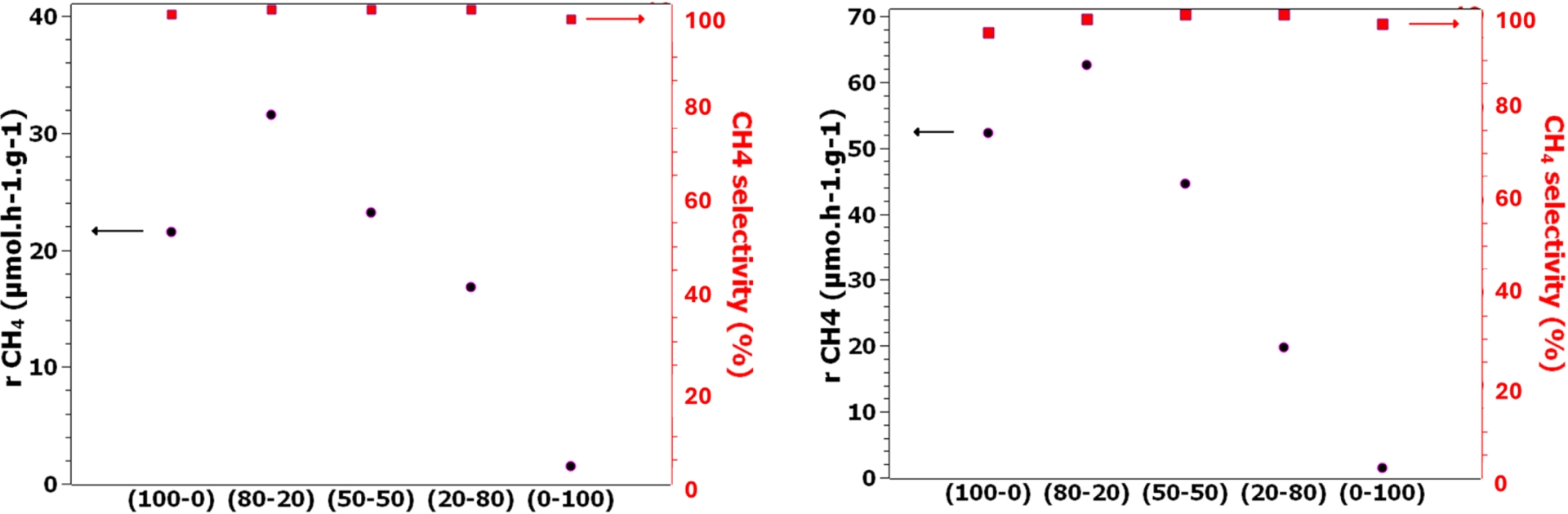
Whether for Pd–Cu or Pt–Cu bimetallic system, a UV-100 support resulted in slightly higher activity, and lower selectivity than for TiO2 P25. This first feature can probably be attributed to the larger surface area of the UV-100 photocatalyst, compared to the P25 one. Moreover, it is worth mentioning that the CH4 reaction rate notably increased with the UV-100 350 °C substrate, accompanied by full selectivity. Knowing the lower surface area (ca. 140 m2⋅g−1) of TiO2 UV-100 350 °C, compared to 315 m2⋅g−1 for bare UV-100, this important enhancement in activity is thus not correlated to this parameter, but probably to the better crystallinity of TiO2, which might be beneficial for charge carrier dynamics (Figure 4).
Gas-phase photocatalytic CO2 reduction on (left) 2 wt% Pd0.5–Cu0.5/TiO2 P25 and (right) 2 wt% Pt0.5–Cu0.5/TiO2, using various TiO2 supports (x-axes).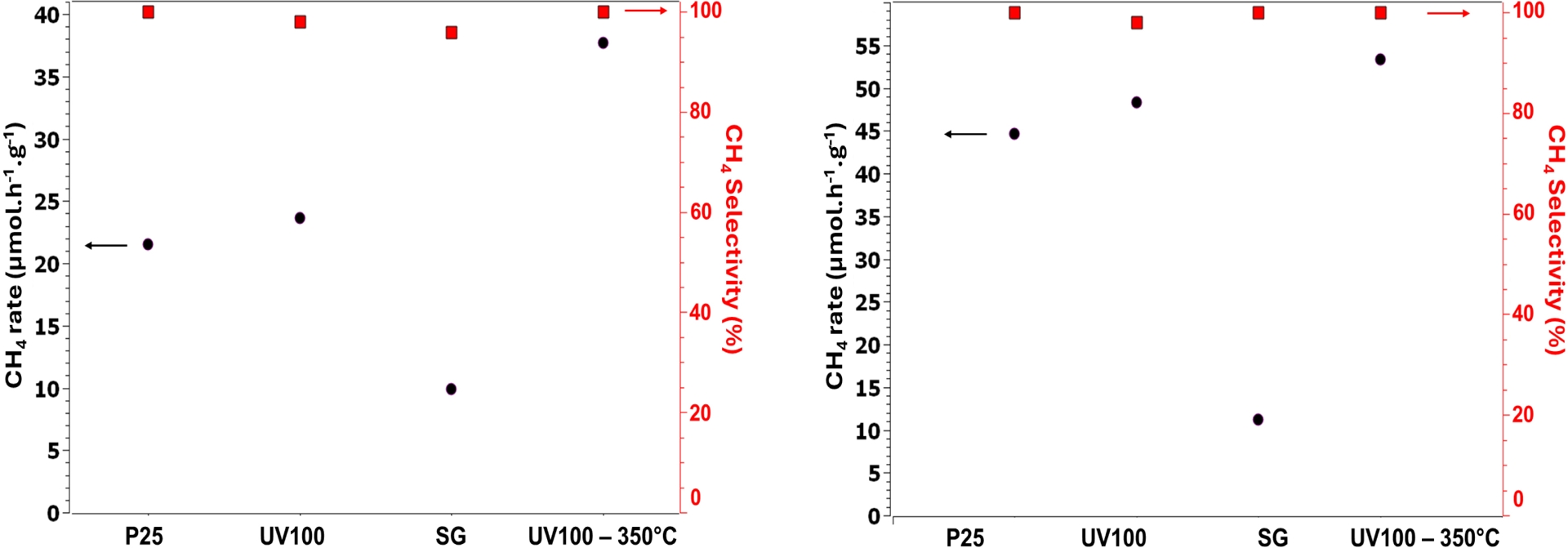
2.3. Photocatalytic activity under simulated visible light on 1 wt% PtxCuy/TiO2 UV-100 350 °C
Subsequently, given that amongst the studied materials, 2 wt% Pt0.5–Cu0.5/TiO2 UV-100 350 °C sample yielded the best photocatalytic activity and selectivity for CH4 formation, further investigations were undertaken with 1 wt% of total metal content, analyzing in more details the PtxCuy molar composition, i.e., Pt0.8–Cu0.2, Pt0.5–Cu0.5, and Pt0.2–Cu0.8, the deposition method, and targeting visible-light illumination (Figure 5).
Gas-phase photocatalytic CO2 reduction on 1 wt% Ptx–Cuy/TiO2 UV-100 350 °C, x = 0.2, 0.5, and 0.8. Effect of (A) simultaneous- and (B) successive-reduction deposition methods.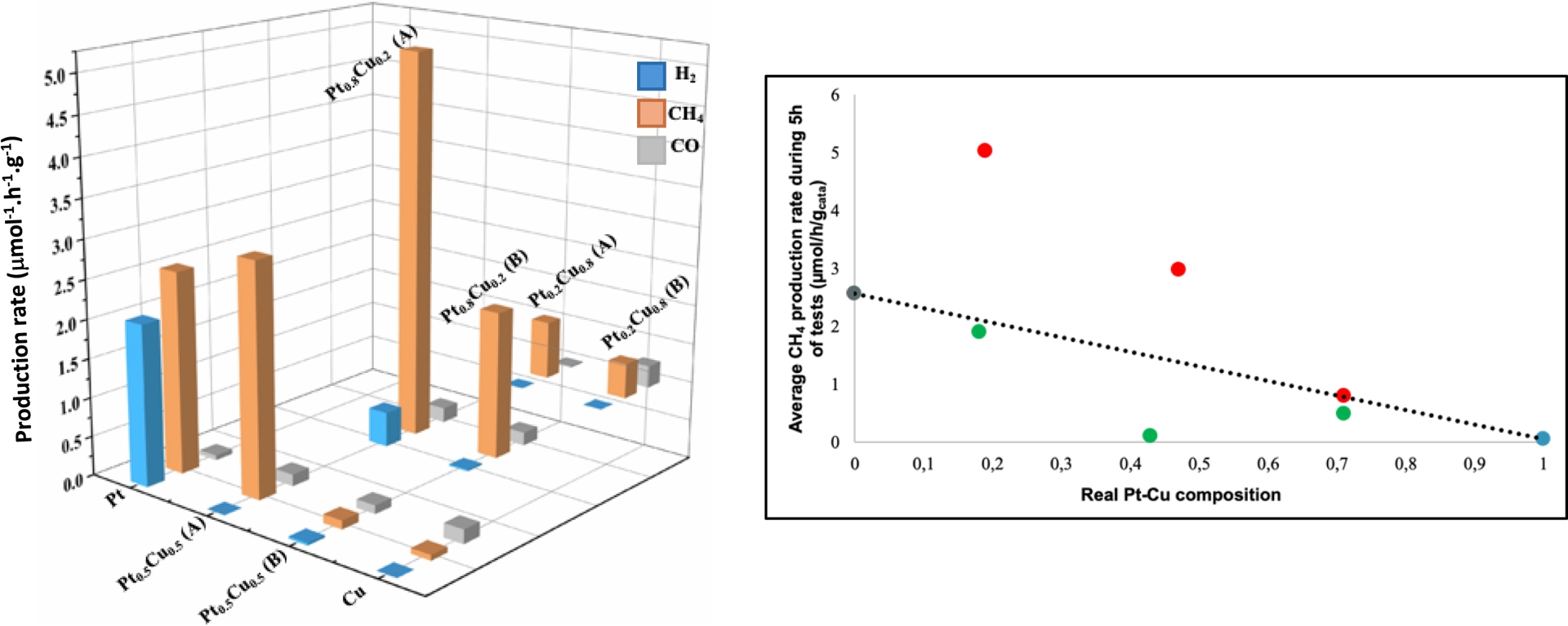
Comparing the effect of the addition order of the metal precursors revealed the superiority of the coreduction method (labeled A) over the successive one (labeled B) (Figure 5). It must also be underlined that all the synthesized Pt–Cu bimetallic samples showed improved activity compared to their monometallic counterparts under visible light activation. Looking at the best 1 wt% coreduced bimetallics, the Pt0.8–Cu0.2 material showed by far the highest performance, amongst all compositions.
Looking further into the experimental CH4 production rate in relation with the weighted combination of the respective activities of the monometallic counterparts (Figure 5-right), it must be highlighted that the Pt–Cu bimetallics exhibited a real synergy provided that the composition was not too rich in Cu. In addition, it should also be noted that on those Pt–Cu/TiO2 UV-100 350 °C materials, electronic selectivity higher than 90% was obtained under visible-light activation, unlike their monometallic counterparts (83% for 1 wt% Pt/TiO2 and 61% for 1 wt% Cu/TiO2, with the production of non-negligible CO amounts).
2.4. Optical properties
2.4.1. On the TiO2 P25 support
From the UV–Vis absorption spectra of the 2% M0.5Cu0.5/TiO2 P25 bimetallic samples (Figure 6), one can observe that beside the contribution in the UV region due to anatase TiO2, additional absorption occurred in the visible spectrum, extending from 400 nm to NIR, whose intensity and extent depend on the metal associated to Cu.
UV–Vis absorption spectra of (left) 2 wt% M0.5–Cu0.5/TiO2 P25 samples and (right) corresponding monometallic references.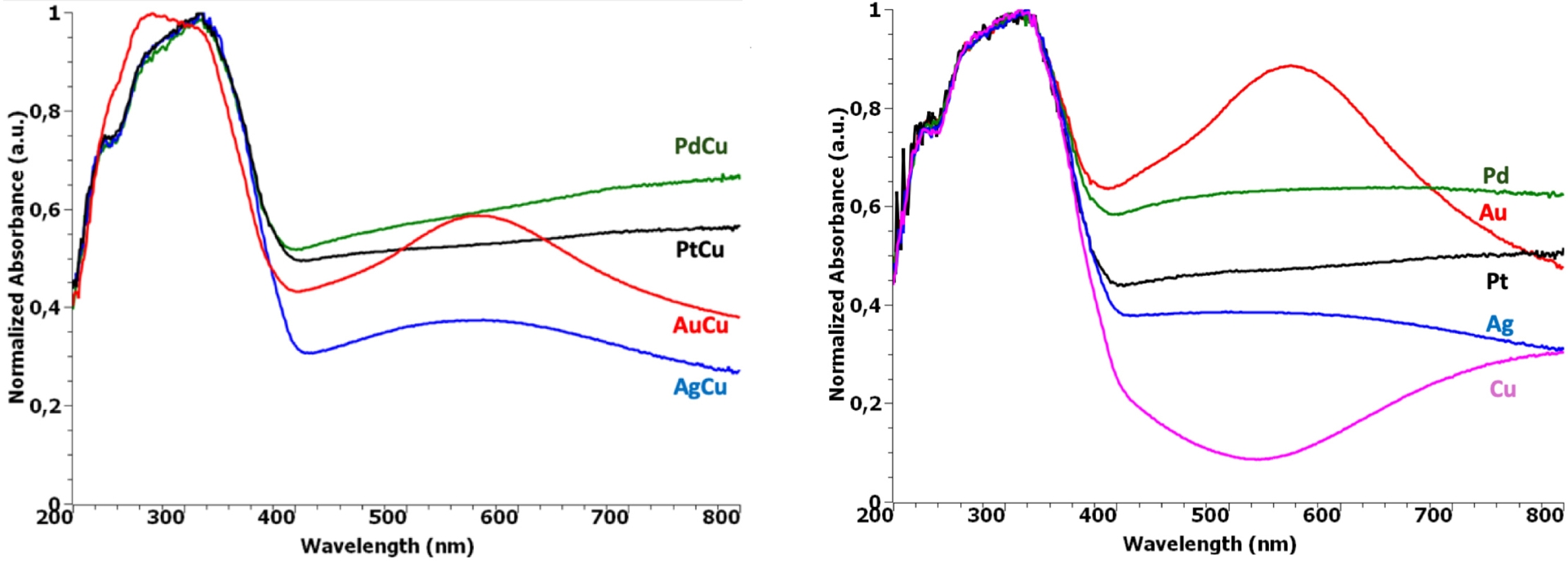
The two noble-metal-based bimetallic systems exhibited a continuous increase in the visible spectrum as also observed on the monometallic Pt and Pd references (Figure 6-right), which can be attributed mainly to their intraband absorption [28]. Nevertheless, by comparing with the Cu/TiO2 counterpart, this broad signal might also include contributions from Cu species, namely from plasmonic Cu(0) inter- and intraband transitions located at ca. 380–450 and 580 nm, respectively [29]. Moreover, knowing the low resistance to oxidation of Cu(0), CuOx species probably also contribute to this signal. Indeed, it is admitted that Cu2O [30] and CuO [31] semiconductor NPs show absorption from ca. 600 and 700 nm corresponding to their band-edge position, a feature that becomes more prominent in NP forms.
With regard to the Au–Cu samples, in addition to Cu species absorption, d–d interband transitions from Au NPs were observed centered approximatively around 570 nm, characteristic of SPR fingerprint of Au NPs as confirmed with the monometallic reference. It is currently admitted that the intensity and position of this SPR strongly depend on the Au NP size, shape, and interaction with the support [32, 33].
Looking at the very broad and extended Ag–Cu/TiO2 visible-light signal and comparing with the monometallic counterparts, one can suppose that it encompasses optical behaviors of the different (above-mentioned) Cu and Ag species. It should indeed be noted that, depending on the size and shape of Ag(0) NPs, the corresponding SPR peak position can be extended from 390 to 460 nm, in addition to the presence of contributions from the Ag2O semiconductor (maximum absorption at ca. 550 nm that can extend up to 800 nm) [34], as Ag(0) is likely to easily undergo surface oxidation.
2.4.2. On the TiO2 UV-100 350 °C support
Focusing on the Pt–Cu bimetallic system immobilized onto the TiO2 UV-100 350 °C support, optical properties were investigated by simultaneously varying the molar composition between the two metals and the addition order of the precursors (coreduction or successive reduction) (Figure 7). The general shape of the absorption curve was similar to that already noted for the Pt0.5–Cu0.5 system deposited onto the TiO2 P25 support, with the relative intensity increasing with Pt content in case of coreduction (Figure 7-left). However, for successive reduction (Figure 7-right), the trend was reversed for molar content in Pt greater than 50%.
UV–Vis absorption spectra of 1 wt% Ptx–Cuy/TiO2 UV-100 350 °C (x = 0.2, 0.5, 0.8) samples prepared by (left) coreduction and (right) successive reduction.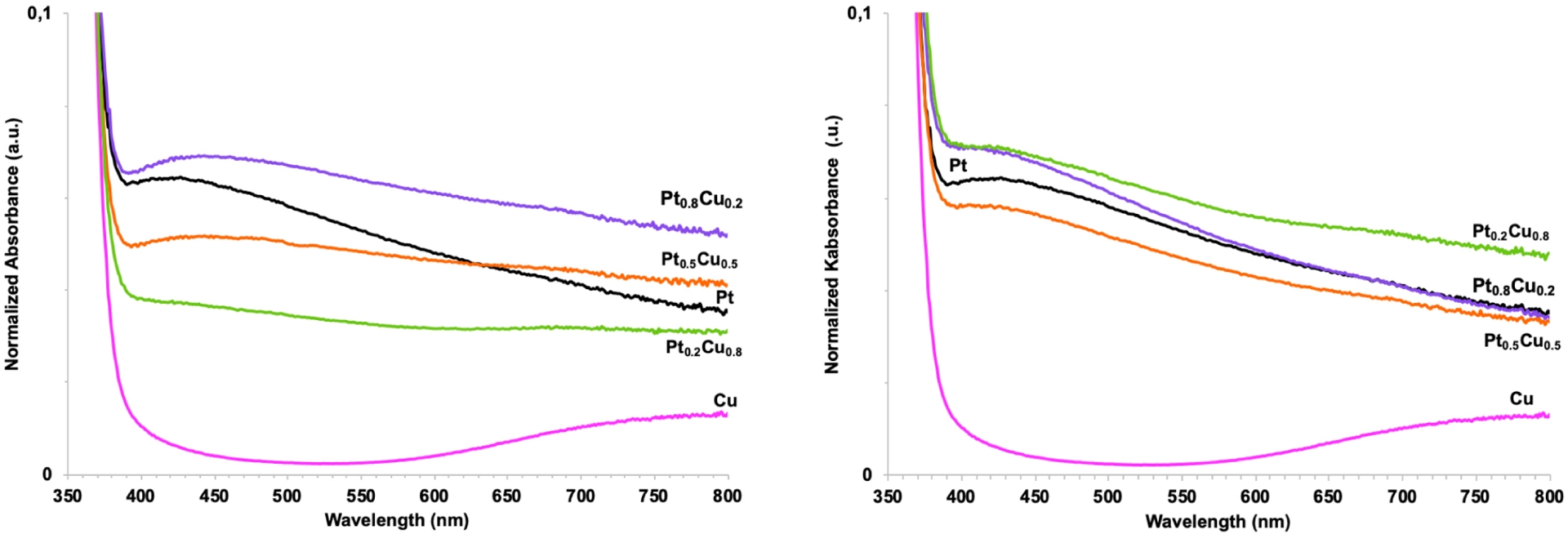
2.5. Transmission electronic microscopy (TEM) of PtxCuyTiO2 UV-100 350 °C
We consequently performed TEM analyses on the 1 wt% Ptx–Cuy/TiO2 UV-100 350 °C (coreduction) series (Figure 8). Independently of the mono- or bimetallic material, the NPs appeared to be rather homogeneously deposited onto the TiO2 UV-100 350 °C substrate. Looking first at the monometallic references, one can observe that Cu NPs exhibited a small and narrowly distributed average particle size of 1.6 nm, whereas Pt exhibited a broader distribution centered around 2.7 nm, both with moderate standard deviation. On bimetallic systems, it must be underlined that regardless of the molar composition and addition order of the metallic precursors, their average particle size was always greater than that of the monometallic counterparts, also accompanied by larger standard deviation. Thus, one can assume that the differences observed in optical properties and photocatalytic CO2 reduction activity do not originate either from discrepancies in metal loading or composition or from particle size and/or dispersion disparities (Figure 8).
Transmission electronic microscopy micrographs and particle size distribution of (top left) 1 wt% Pt/TiO2 UV-100 350 °C (top right) 1 wt% Cu/TiO2 UV-100 350 °C (middle) 1 wt% Pt0.2–Cu0.8/TiO2 UV-100 350 °C, 1 wt% Pt0.5–Cu0.5/TiO2 UV-100 350 °C, and 1 wt% Pt0.8–Cu0.2/TiO2 UV-100 350 °C obtained by coreduction (top left) 1 wt% Pt0.2–Cu0.8/TiO2 UV-100 350 °C and 1 wt% Pt05–Cu0.5/TiO2 UV-100 350 °C obtained by successive reduction.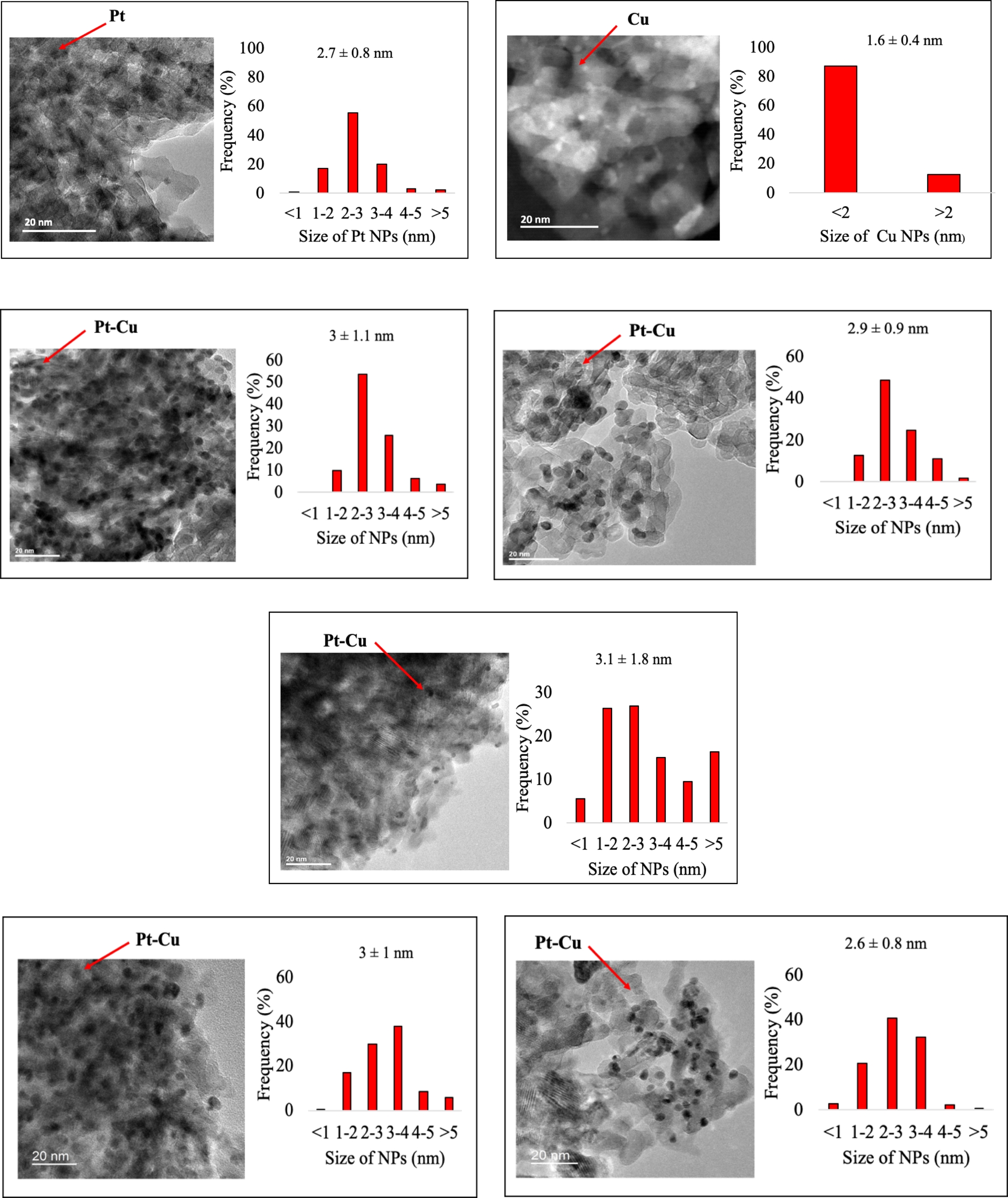
2.6. XPS analysis on the TiO2 UV-100 350 °C support
The surface and bulk Cu/(Cu+Pt) atomic ratios of the 1 wt% PtxCuy/TiO2 UV-100 350 °C samples varying in their molar composition and deposition method A (coreduction) or B (successive reduction) displayed two main features (Figure 9). Regarding samples resulting from coreduction and regardless of the Cu–Pt molar composition, the Cu/(Cu+Pt) surface atomic ratio was larger than the total one, consistent with a surface enrichment in Cu, all the more important as the composition in Cu increased. Nevertheless, it seems that this behavior did not happen in the case of successive reduction, even if the measurements were performed only on one Pt–Cu composition. From these observations, it may be assumed that the addition order of the Pt and Cu precursors impacts the surface enrichment in Cu particles. More precisely, the photocatalytic materials exhibiting the best performances seems to be those showing an optimal surface enrichment in Cu, without being too important however. Complementary detailed XPS analysis (to be published elsewhere) revealed that Cu(0) and/or Cu(+1) surface species are the predominant Cu oxidation states, apparently excluding the presence of other Cu oxidation states.
Cu/(Cu+Pt) surface (determined from XPS) and bulk (determined from ICP-AES) atomic ratios as a function of molar composition and deposition method (A and B stand for coreduction and successive reduction deposition, respectively) for 1 wt% PtxCuy TiO2 UV-100 350 °C samples.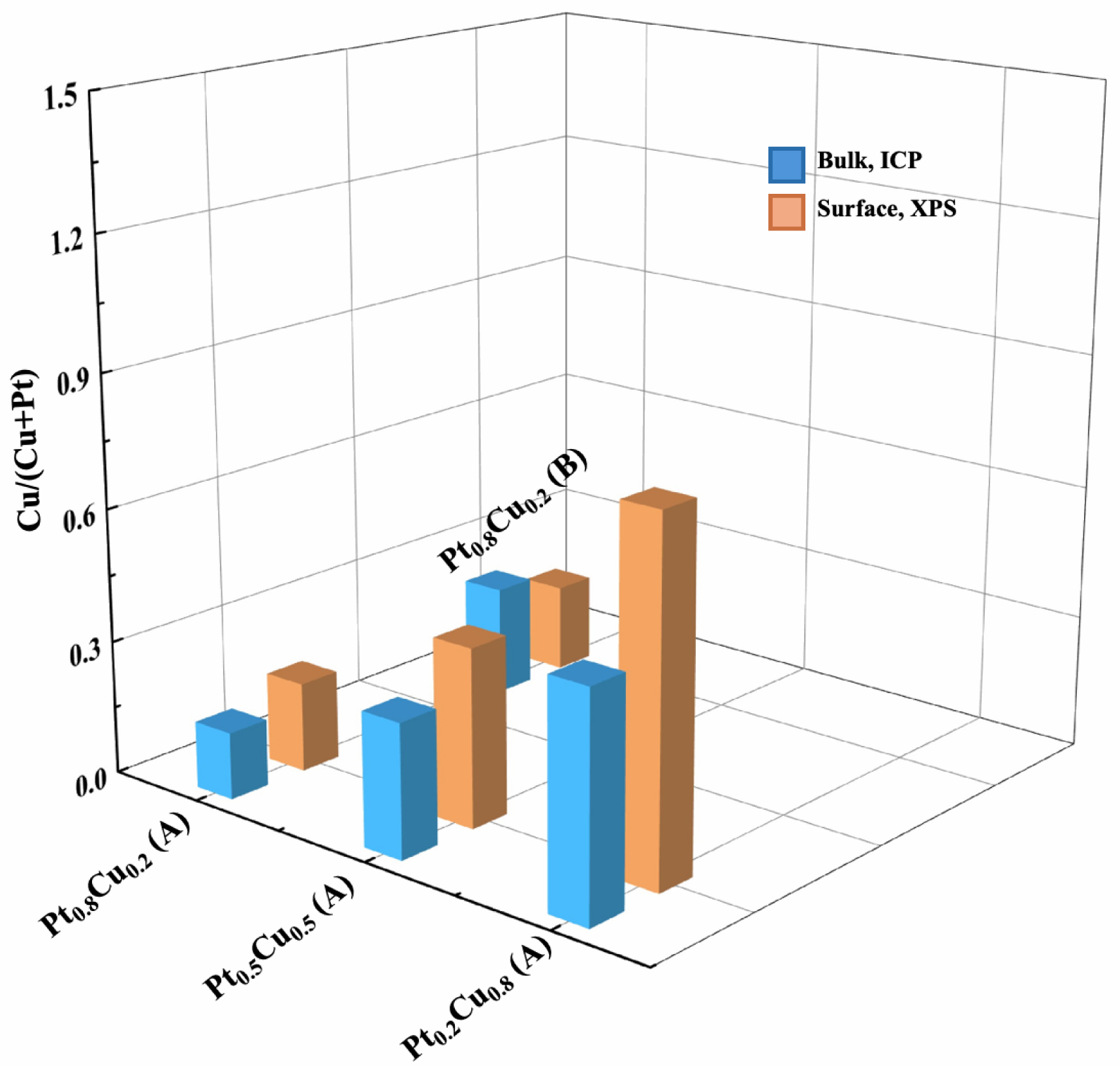
It is well known that noble-metal NPs, such as Au, Pt, Pd are able to absorb visible light thanks to surface plasmonic resonance (SPR), which may contribute to visible light activation of TiO2. In addition to this beneficial SPR feature that would help extend the response of TiO2 from UV to visible light, the presence of a noble metal would also result in improving charge carrier separation thanks to the formation of a Schottky barrier at the metal/semiconductor (SC) interface and also be helpful in supplying cocatalytic function. Because of their large work function (5.64 eV vs. Evacuum), Pt NPs have been considered an efficient cocatalyst for TiO2. However, more recently, non-noble plasmonic metals such as copper Cu, Bi, Ni, etc. have been proposed as alternatives to plasmonic noble metals, also considering their massive Earth abundance, low cost, and promising SPR tunable from UV to NIR [35, 36]. Non-metal-based plasmonic materials often exhibit plasmonic properties comparable to those of noble metals, but they can outperform their noble-metal counterparts in some cases. There are several recent studies showing that Cu is used to replace Au, which is considered as the most widely researched metal in the plasmonic field. Moreover, as the work function of Cu (4.65 eV) is smaller than that of Au (5.1 eV), it can form a lower Schottky barrier with TiO2. It is usually admitted that intraband transitions are characterized by the creation of hot electrons above the Fermi level more suitable for reductive catalytic pathways, whereas interband transitions mainly lead to the generation of hot d-band holes below the Fermi level, thus better for oxidative catalytic pathways. However, as the interband transition energy level of Cu is also lower than that of Au, it is possible to produce hot electrons with higher energy, thus allowing hot electrons excited from both intraband and interband transitions to be extracted more efficiently [30]. As CO2 is also a very stable linear molecule, CO2 activation is a key step for its further reduction. For this purpose, a Cu-based metallic catalyst is considered to significantly promote CO2 activation. Indeed, it has already been demonstrated that the presence of Cu atoms on the surface of TiO2 significantly stabilizes the adsorption of curved CO2, meaning that Cu can effectively stabilize the CO2 adsorbed on the catalyst [37]. Thus, loading Cu onto TiO2 leads to increase CO2 adsorption but also to inhibit the competitive H2 evolution [38]. In addition, as a conductive substance, Cu can be associated with TiO2 to form a metal–semiconductor Schottky junction allowing effective inhibition of the charge carrier recombination.
However, despite their low cost and Earth abundance, Cu NPs are easily oxidized and can exist in different oxidation states where metallic Cu and CuOx can be present at the same time. More precisely, copper oxide (CuO) and cuprous oxide (Cu2O) can be formed exhibiting narrow band gaps of 1.7 eV and 2.2 eV, respectively, which may permit visible-light absorption. It is worth mentioning that, in addition to their band-gap energy smaller than that of TiO2, they also exhibit higher conduction band values facilitating CO2 reduction. Many works have reported the use of CuOx with different morphologies [39, 40] for photocatalytic reduction of CO2, overcoming the rapid charge carrier recombination by formation of (hetero)junction with another material. However, for longer-term stability, keeping an optimal balance between the different Cu oxidation states remains a tricky concern, especially in the case where O2 production occurs as a result of the first H2O oxidation step (essential for providing H+ to further advance CO2 reduction) and can thus be considered as one of the key-point that can be satisfied by designing M–Cu bimetallic systems.
Cu–Pt/TiO2 bimetallic photocatalysts with optimized Cu–Pt molar composition were identified as the best materials for CO2 reduction due to the synergistic effects where each metal complements the role of the other one. Indeed, Cu is an excellent cocatalyst for CO2 activation by lowering the energy barrier required to break the stable linear C=O bond. Pt enhances transfer of the photogenerated electrons from the TiO2 semiconductor to the CO2 molecule, a crucial step in the reduction pathway. Moreover, optimized interface between Cu and Pt domains can lead to the formation of specific active sites promoting CO2 reduction and driving toward the desired products. Within the Cu–Pt bimetallic system, specific electronic interactions between the two metals result in preventing too important an oxidation of Cu(0) NPs, allowing stabilization of the optimal balance between Cu(0) and Cu(+1) (Cu2O) oxidation species, also resulting both in the formation of TiO2/Cu2O heterojunctions for increased charge carrier separation and visible light photosensitization of the TiO2 semiconductor. In addition to all those positive effects deriving from the optimal association between Pt and Cu, it is worth mentioning the ones related to SPR effects. Hot electrons from Cu can be injected into Pt thus lowering the energy barrier for the kinetically slow steps of CO2 reduction, thus accelerating the reaction. This electron injection can also influence the reaction pathway. For instance, an increased concentration of electrons at the active Pt sites can favor the reduction of specific intermediates, thereby inhibiting competing pathways like hydrogen evolution.
3. Conclusion
Tuning and elaboration of M–Cu bimetallic phases supported on TiO2 semiconductors can be considered an interesting strategy considering that photocatalysts based on non-noble plasmonic metals may be a promising alternative to noble-metal-based ones due to their advantages like Earth abundance, effectiveness, and thus large-scale application possibility. However, despite their low cost and Earth abundance, Cu NPs are easily oxidized. As a consequence, the development of approaches to mitigate oxidation represents a key point. Amongst them, the intimate association, in an optimized way, of Cu and noble metals can result in synergistic effects on photocatalytic properties compared to the monometallic catalysts. Overall, Pt–Cu bimetallic catalysts—benefiting from catalytic synergy, plasmonic enhancement, tailored electronic interactions, and optimized balance between the different Cu species—represent a promising direction for the development of efficient, stable, and solar-driven systems for CO2 conversion.
4. Experimental section
4.1. Materials
Commercial TiO2 P25 (surface area of 50 m2⋅g−1) and UV100 Hombikat (315 m2⋅g−1) were purchased from Evonik and Sachtleben Chemie, respectively. UV 100 is used either as received or after calcination in air at 350 °C for 4 h (named UV-100 350 °C) in a muffle furnace to obtain a pure-phase anatase, nanocrystalline, without amorphous phase and with well-defined crystallites of 13 nm, exhibiting a surface area of 140 m2⋅g−1. H2PtCl6⋅xH2O, Na2PdCl6, HAuCl4⋅3H2O, AgNO3, and NaBH4 were purchased from Sigma-Aldrich and used without further purification. Cu(NO3)2⋅xH2O was purchased from Alfa Aesar & used without further purification.
4.2. Photocatalyst preparation
The TiO2 support (1 g) was dispersed and stirred at 1000 rpm for 5 min in distilled water (400 mL). For all bi-metallic M–Cu systems, the codeposition/chemical reduction protocol was carried out. For this, two aqueous solutions of metal (Pt, Pd, Au, or Ag) precursor, and Cu(NO3)2⋅xH2O (0.25 M) were added simultaneously to the suspended TiO2 support under vigorous stirring for 45 min. A freshly prepared solution of NaBH4(n(NaBH4)/nmetal = 5) was added and left to react for 15 min. The resulting powder in suspension was isolated by filtration under vacuum, washed with distilled water (1 L), and dried under air for 24 h at 100 °C. The final photocatalyst was recovered after a calcination at 200 °C for 2 h.
Nevertheless, in the case of UV-100 350 °C support, sequential chemical reduction was also applied in order to assess whether there was an influence of the addition order of the metal precursors. To do this, in a first step, the aqueous solution of the metal precursor was added to the medium, and the synthesis was then pursued as described above to obtain the M/TiO2 catalyst. Secondly, the aqueous solution of Cu(NO3)2⋅xH2O was added, and the same synthesis was performed yielding M–Cu/TiO2 catalyst.
Different series of M–Cu/TiO2 materials were targeted. Series of 1, 2, and 3 wt% PdxCuy/TiO2 P25, PtxCuy/TiO2 P25, AuxCuy/TiO2 P25 and AgxCuy/TiO2 P25 (with y = nominal content of Cu = 0, 0.5, 1; x = 1 − y), and of 2 wt% AuxCuy/TiO2 P25, 2 wt% AuxCuy/TiO2 P25 (with y = 0, 0.2, 0.5, 0.8, 1) were prepared in order to investigate the impact of bimetallic composition and total metal loading. Additionally, a series of 1 wt% PtxCuy/TiO2 UV-100 350 °C (with y = 0, 0.2, 0.5, 0.8, 1) was also investigated in order to study the influence of the TiO2 support.
4.3. Material characterization
Elemental analyses of the composites were performed using inductively coupled plasma atomic emission spectroscopy (ICP-AES). The detection threshold of the instrument is 0.1 mg⋅L−1 for Au, Cu, Pd, Pd and Ag. Analysis of the metal component in the composites allowed us to calculate deposition yields, which are defined as the ratio between the real deposited mass of Au (deduced from ICP-AES analysis) and the mass of Au introduced during the deposition step. High-resolution transmission electron microscopy (HRTEM) was performed on a Jeol 2100F microscope equipped with a lanthanum hexaboride cathode (LaB6) operating at 200 kV. The sample was sonicated in EtOH, and then a drop of the suspension was deposited on a carbon membrane placed on a copper grid. The size of M NPs was determined by HRTEM, and consequently the size distribution was obtained by measuring particle size for more than 100 particles using the ImageJ software. UV–Vis absorption spectra were recorded on a Perkin Elmer 950 spectrophotometer fitted with a Labsphere RSA ASSY 100 nm integrating sphere. The spectra were acquired in reflection mode (diffuse reflectance).
The surface of the catalysts was studied by X-ray photoelectron spectroscopy (XPS). The spectrometer used was an ultrahigh vacuum (UHV) one with an incident X-ray source such as a dual anode Al K𝛼 (1486.6 eV). The electron analyzer was a VSW Class WA hemi-sphere. The spectra, survey and high resolution, were recorded in constant pass energy mode, 90 eV for survey scan and 20 eV for high resolution spectra. The % surface atomic ratios were calculated using the corresponding core level peaks, which were normalized to the photoemission cross-section, and assuming a homogeneous distribution arrangement.
4.4. Photocatalytic evaluation
Photocatalytic setup has already been described in detail elsewhere [41]. During the test, the continuous reaction mixture (CO2+H2O, CO2/H2O = 97/43) flow (0.3 mL⋅min−1) passes through a light-transparent photoreactor (6 mL) equipped with a ventilator-cooled Hg lamp simulating artificial solar light (150 W ceramic metal-halide Hg lamp). Note that, to do the tests under visible light, a Schott GG 400 optical filter (Edmund Optics) was placed between the photoreactor and the lamp to cut all wavelengths below 400 nm. Total irradiance was 4317 W/m2 for the whole lamp spectrum (from 300–900 nm) and 4271 W/m2 with the 400 nm optical filter (simulated visible light) (Figure 10). The irradiated surface was 19.65 cm2. For photocatalytic tests, materials (50 mg) were dispersed in EtOH and deposited on a 50 mm diameter glass disk by evaporation at 100 °C. The surface concentration of the photocatalyst on the glass disc was 25 g⋅m−2. The glass disk was then placed in the reactor and the setup was purged for 5 min with CO2 flow (>100 mL⋅min−1) to remove air and other gas impurities. As a blank test, the setup was run without CO2 gas flow under illumination to control the absence of gaseous products that may result from conversion of carbon-based contaminants [42]. Products from the reactor were analyzed by an online micro-GC (Agilent 3000A SRA instrument). Finally, the lamp was switched on and the duration of test went from 5 h to 10 h.
Emission spectra of the 150 W ceramic metal-halide Hg lamp with and without Schott GG 400 optical filter.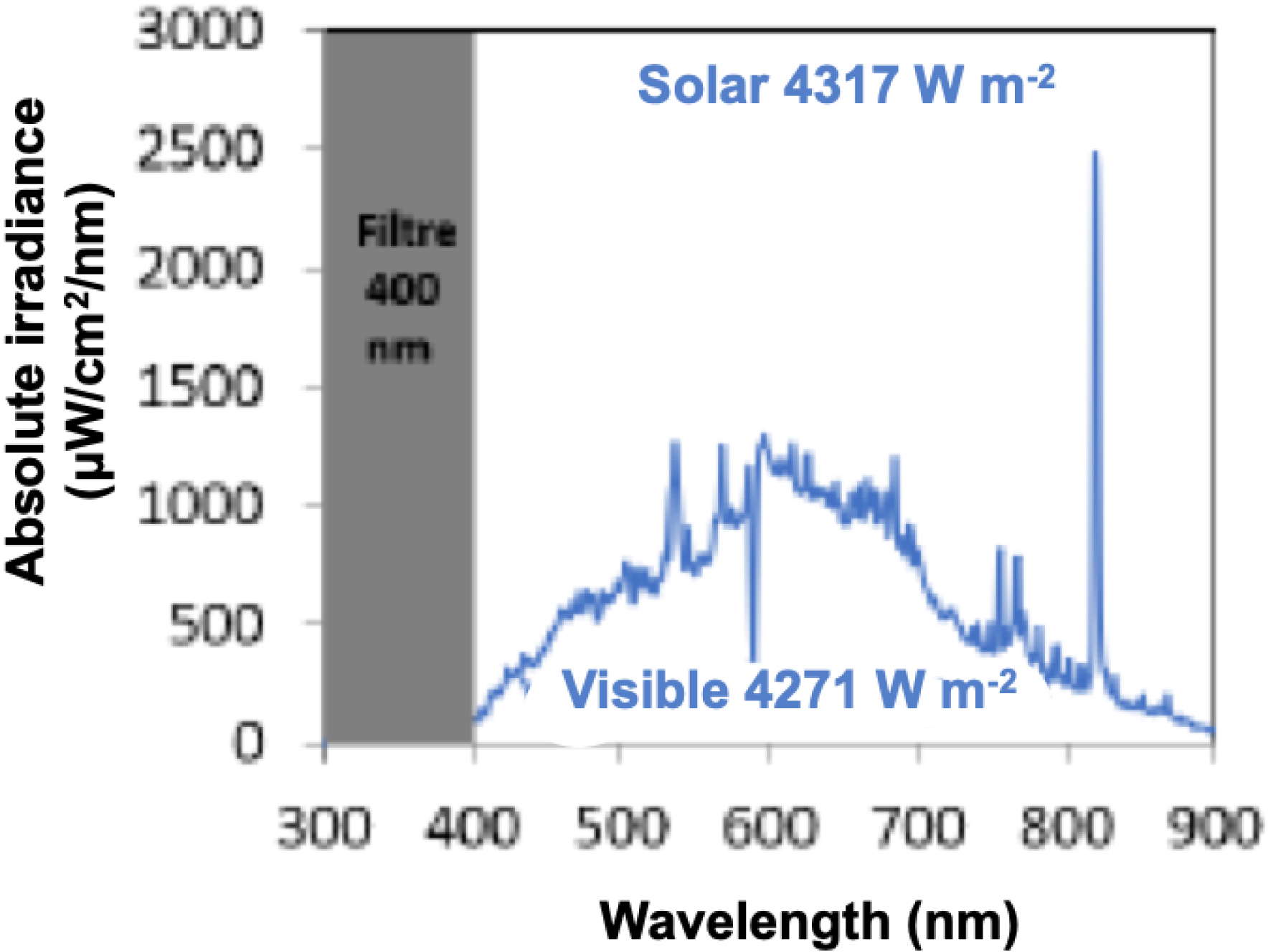
Production rates were calculated according to the following equation:
| \begin {equation*} r_{X}(\mathrm {mol}{\cdot }\mathrm {h}^{-1}{\cdot }\mathrm {g}^{-1}) = \dfrac {[X] \times 10^{-6} \times (\mathrm {flow~rate}) \times 60} {V_{m} \times m_{\mathrm {photocat}}} \end {equation*} |
Electronic selectivity was calculated by these equations:
| \begin {eqnarray} \mathrm {CH}_{4}\;\mathrm {Selectivity} &=& \dfrac {8[\mathrm {CH}_{4}]} {8[\mathrm {CH}_{4}] + 2[\mathrm {H}_{2}]} \label {eq1} \end {eqnarray} | (1) |
| \begin {eqnarray} \mathrm {H}_{2}\;\mathrm {Selectivity} &=& 1 - \mathrm {CH}_{4}\; \mathrm {Selectivity} \label {eq2} \end {eqnarray} | (2) |
Acknowledgments
Vasiliki Papaefthimiou (ICPEES, Strasbourg France) is acknowledged for XPS analyses and discussion.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
The authors acknowledge PEPR PowerCO2 and ECOCHEM projects for financial support of Eliane Khoury’s PhD and Clément Marchal’s postdoctoral fellowship.