

1 Introduction
FeII–FeIII hydroxysalts are commonly called green rusts (GR), as first proposed in 1959 by Bernal et al. [3], because they were green compounds found during the process of corrosion of iron. However, they belong to a larger family of di- and trivalent cation compounds, the layered double hydroxides (LDH), and their originality is that the cations come from the same iron element. Therefore, they possess very reactive redox properties and specific precautions must be taken to study them carefully. The purpose of this article is to present the methods that are used to identify and characterise the iron compounds and more specifically the FeII–III hydroxysalts. Special interest will concern the speciation of iron because redox reactions constitute the major part of the studied phases. Not only the crystallographic structure of the compounds must be characterised, but also the degree of oxidation of iron must be distinguished from a quantitative viewpoint. While X-ray diffraction (XRD) studies help to determine the crystallographic structure, Mössbauer spectroscopy completes the speciation. In the first part, the methods and characteristics of iron compounds will be reviewed, with emphasis on synthetic GRs. Then, specific issues relative to synthetic products will be treated before displaying results obtained with the oxyhydroxycarbonate GR mineral, named recently ‘fougerite’ (IMA 2003-057). Three methods for synthesising GRs in the laboratory will be presented separately in following articles: (i) coprecitation of FeII and FeIII ions in solutions containing specific anions [25], (ii) oxidation of Fe(OH)2 in presence of anions [7,11,17,22], (iii) reduction of ferric oxyhydroxides by dissimilatory iron reducing bacteria [4,18].
2 Speciation of Fe and characterisation of iron compounds
A review of all X-ray diffraction patterns encountered in redox reactions involving iron compounds is not intended. We limit ourselves only to the GRs. In contrast, typical Mössbauer spectra of phases usually encountered during the cycle of iron will be presented, with special emphasis on ferrous hydroxide, GRs, ferric oxyhydroxides and magnetite.
2.1 X-ray diffraction
XRD was first extensively utilised by Bernal et al. [3], who recognised two types of patterns, one called green rust one, GR1, and the other one green rust two, GR2. Within the frame of LDH, their crystal structure is described as brucite-like layers that alternate with interlayers made of anions (, , Cl−...) and water molecules. As a general rule, GR1 forms in the presence of spherical or planar anions such as Cl− and , whereas GR2 forms with three-dimensional anions such as . Space group of GR1 is and patterns resemble those of hydrotalcite [1] or of pyroaurite [2]. No specific position for anions and water molecules in interlayers was at that time proposed. In contrast, the structure of GR2 was definitively determined only recently and in the case of hydroxysulphate the formula is precisely and the structure was determined to be trigonal of space group by Rietveld analysis [26]. XRD patterns of hydroxycarbonate and hydroxysulphate are shown in Fig. 1.
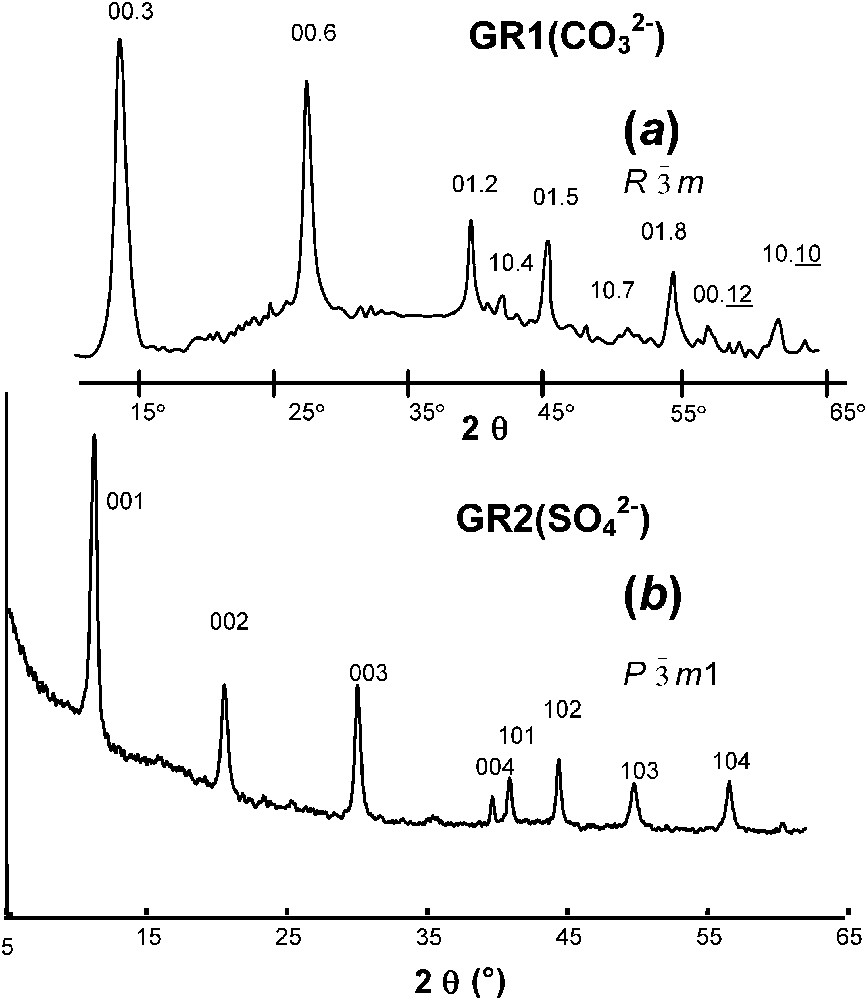
XRD patterns of GR1 versus GR2 (radiation Co K with λ=0.1789 nm): pattern of (a) hydroxycarbonate; (b) hydroxysulphate.
Clichés de diffraction de rayons X de rouilles vertes 1 et 2 (rayonnement Co K, avec λ=0,1789 nm) : Cliché (a) de l'hydroxycarbonate ; (b) de l'hydroxysulfate.
2.2 Mössbauer spectroscopy
The recoilless emission and absorption of γ-rays in solids was discovered by R. Mössbauer in 1958, who was rewarded by the Nobel Prize. The obtained information concerns the nuclear levels of the probed elements that are the object of the hyperfine interactions between nuclei and surrounding electric charges, electrons and ions as well. It strongly resembles nuclear magnetic resonance (NMR), but is limited to elements where exist adequate radioactive sources, e.g., Fe, Sn, Sb and rare earths. Fe constitutes about 90% of studies where the source is usually embedded in an Rh matrix and the resonant isotope is , i.e. only 2% of natural iron. The energy scale in a spectrum, E, is expressed in term of velocity v in mm s−1 since the Doppler effect is used to modulate the emitted γ-ray; where c is the velocity of light, the transition energy, i.e. 14.4 keV for and v the relative velocity between source and absorber as obtained from the electronic drive. Each component is essentially constituted of absorption lines, assembled into singlets, quadrupole doublets or magnetically split sextets. Each is attributed to an iron environment within a specific iron compound, at a crystal site and with its degree of oxidation, i.e. Fe0 metallic iron, FeII ferrous and FeIII ferric states in this case [14,15].
The centre of gravity of a spectral component determines the isomer shift δ expressed in mm s−1 with respect to a reference, e.g., that of metallic α iron measured at room temperature; it is due to the Coulombic interaction between the valence s electrons and the nucleus considered as a point charge. The distance Δ in mm s−1 between the two peaks within a doublet characterises the quadrupole splitting due to the interaction between the nuclear quadrupole moment and the electric-field gradient at the resonant nucleus, which comes from the shape of the electron shells and the symmetry of the ion distribution within the lattice as well. Finally, the magnetic sextet comes from the coupling between the hyperfine field H, expressed in kOe, and the magnetic moment of the nucleus in the often encountered case where the quadrupole shift ε can be considered only as a perturbation of the Zeeman splitting. Therefore, all spectral components are characterised by or ε, and H. Most interesting features are that values for δ and Δ are within definite ranges for the various degrees of oxidation of iron and constitute a signature for speciation, i.e. 0.7–1.5 and 0.25–0.50 mm s−1 for high spin ferrous and ferric states, respectively, where metallic α iron with a field of 331 kOe at room temperature is taken for reference of δ. Each magnetic phase has also characteristic hyperfine magnetic fields (several Fe sites can exist in a phase) and the temperature at which magnetism vanishes when increasing it, Curie or Néel points, is also a way for characterisation. In that respect, a phenomenon called superparamagnetism may render the interpretation more difficult. Curie or Néel temperature of large and bulk samples is sharp. However, a doublet may still exist below the usual magnetic ordering, due to the small size of the crystal particles. Therefore, a paramagnetic behaviour lasts as long as the bulk magnetic energy is counterbalanced by the thermal agitation. Such a superparamagnetic behaviour is a good indication of small particle size. Note also that amorphous materials give rise to Mössbauer spectra displaying a change of magnetic ordering that lies within a broad temperature range. Most of the time, computer fitting of Mössbauer spectra with Lorentzian-shape lines is sufficient. However, Voigt profile analysis can be more relevant and components are then the convolution of a Gaussian distribution with the Lorentzian transition. Each component is thus characterised by its quadrupole splitting Δ or hyperfine field H along with their respective standard deviation δΔ or δH.
Fig. 2 exhibits Mössbauer spectra measured at 77 K for the principal ferric oxyhydroxides that are encountered in Nature or as corrosion end-products of iron. These are goethite α-FeOOH, akaganeite β-FeOOH, lepidocrocite γ-FeOOH, feroxyhyte δ-FeOOH, and ferrihydrite to which magnetite Fe3O4 is added. They are different for all phases, even though measuring spectra at several temperatures is sometimes necessary to strengthen the identification; for instance, a temperature chosen between those of the magnetic ordering of two phases can distinguish them unambiguously.
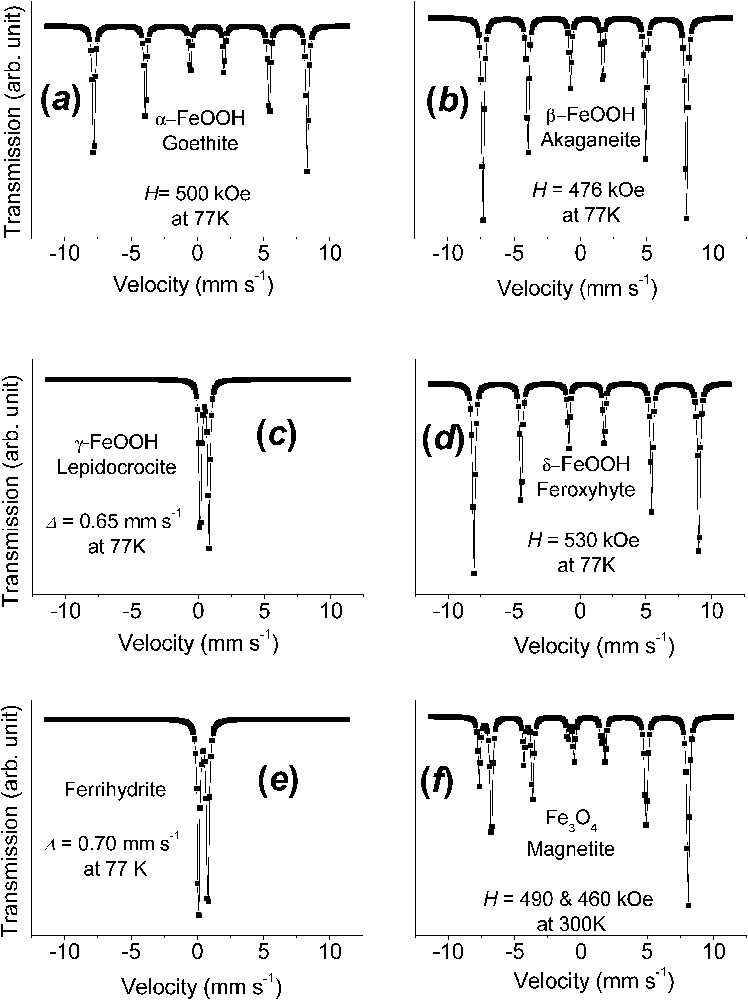
Mössbauer spectra measured at 77 K of the principal ferric oxyhydroxides, the common rusts and minerals, and of magnetite at 300 K: (a) α-FeOOH goethite; (b) β-FeOOH akaganeite; (c) γ-FeOOH lepidocrocite; (d) δ-FeOOH feroxyhyte; (e) ferrihydrite; (f) Fe3O4 magnetite.
Spectres Mössbauer mesurés à 77 K des principaux oxyhydroxydes ferriques, les rouilles ordinaires et les minéraux, et de la magnétite à 300 K : (a) α-FeOOH, la goethite ; (b) β-FeOOH, l'akaganéite ; (c) γ-FeOOH, la lépidocrocite ; (d) δ-FeOOH, la feroxyhyte ; (e) la ferrihydrite ; (f) Fe3O4, la magnétite.
The spectrum of α-FeOOH goethite, which is brownish ochre, is characterised by asymmetric lines of the magnetic sextets due to a field distribution, up to 500 kOe at 77 K, and negative quadrupole shift (Fig. 2a). The Néel point is at 120 °C. β-FeOOH akaganeite, which is exclusively encountered in chloride-containing solutions, has several Fe sites, probably due to the presence of Cl− ions in the solid (Fig. 2b). γ-FeOOH lepidocrocite, which is bright orange, is commonly encountered as corrosion product in slightly chloride containing media. It is paramagnetic at room temperature with and at 77 K . Its Néel point is around of 70 K (Fig. 2c). δ-FeOOH feroxyhyte is a less common phase presenting some amorphous character with a field of 530 kOe at 77 K (Fig. 2d). Ferrihydrite, which is a most common reddish-brown compound of oxidation is badly crystallised and is designated as 2-line or 6-line ferrihydrite, depending on the appearance of the XRD pattern. Paramagnetic at 77 K with Δ of about 0.70 mm s−1 (Fig. 2e), a Mössbauer spectrum with a field around 500 kOe at 4 K can characterise it without difficulty.
A last phase often encountered as a corrosion product when the presence of oxygen is insufficient, but also common in Nature, is black magnetite and its more oxidised form is maghemite γ-Fe2O3. Mössbauer studies of magnetite is a topic by itself, since it has raised many questions. When at stoichiometry Fe3O4, the spectrum is constituted of two sextets of 491 and about 460 kOe at 300 K, respectively [6,28]. Its inverse spinel structure possesses two crystal sites A and B, which are tetrahedral and octahedral, respectively. At room temperature, the larger field that corresponds to the tetrahedral site A has half the intensity of the smaller field of B. This is due to electron hopping in the octahedral site between FeII and FeIII ions, which are in fact in some intermediate Fe2.5+ configuration above the Verwey temperature of 120 K for Fe3O4. Below it, e.g., 82 K, there exist five sextets: one FeIII in A site at 511 kOe, two FeIII in B site at 533 and 516 kOe and two FeII in B site at 473 and 374 kOe [6,28]. It often occurs also that the intensity ratio between A and B sextets is no longer equal to 2 because of vacant FeII ions in the octahedral sites. This ratio allows to determine the departure from the ideal formula Fe3O4 and maghemite γ-Fe2O3 is merely the same phase once all FeII ions have been removed; there exists only one sextet at 490 kOe at 77 K [28].
A typical spectrum of ferrous state is illustrated by that of ferrous hydroxide Fe(OH)2 (Fig. 3) [11,24]. At 78 K, it is a quadrupole doublet with large negative quadrupole splitting Δ of about (Fig. 3a). At 4 K, the spectrum is more complex (Fig. 3b). It is below the Néel point of about 15 K, but since Δ is very large compared to the hyperfine field H, it cannot be treated any longer as a perturbation of the Zeeman splitting and the spectrum comprises eight lines that have to be computed fully by solving the eigenvalues of the Hamiltonian. Each line corresponds to a mixture of quantum states as established in Fig. 3 with computed Clebsch–Gordon coefficients and the best fitting is obtained with a magnetic field of 220 kOe with perpendicular to the axis of symmetry of the electric field gradient, i.e. lying within the brucite-like layer [11]. A spectrum below 10 K is sometimes necessary to recognise Fe(OH)2 without any ambiguity.
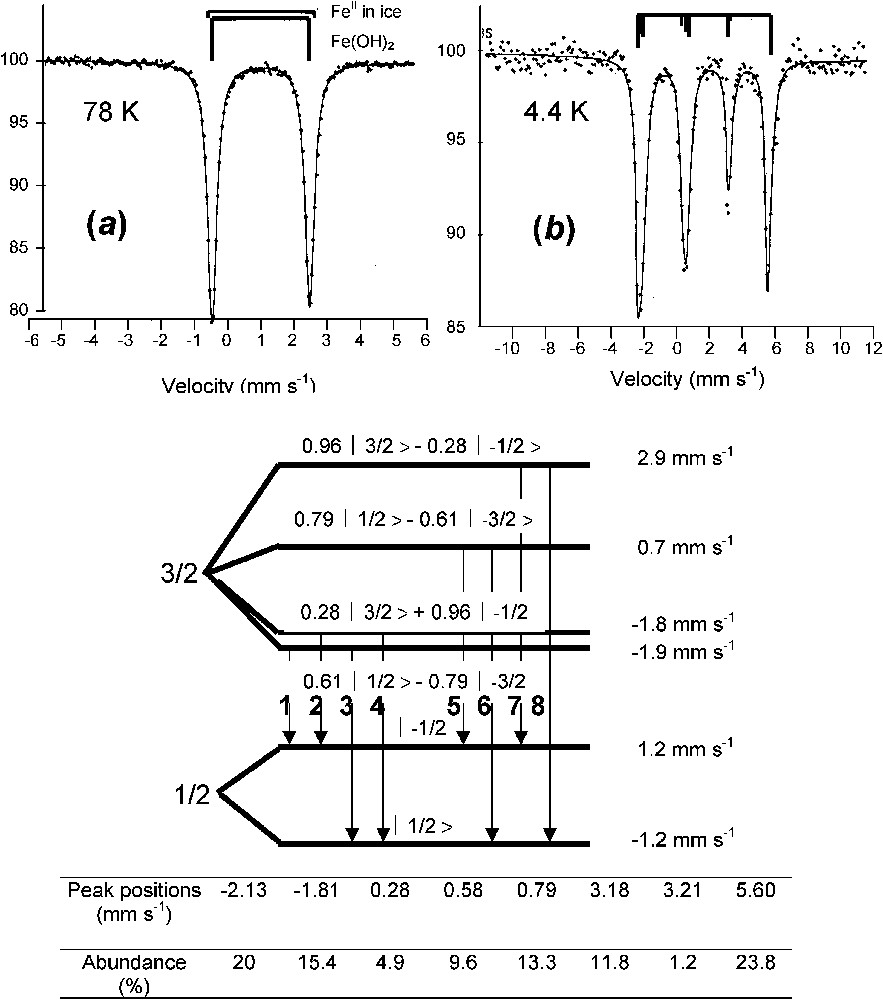
Mössbauer spectra of ferrous hydroxide Fe(OH)2 measured at (a) 78 K and (b) 4 K. The spectrum at 4 K comprises eight absorption peaks, an unusual situation where the quadrupole effect cannot be considered as only a perturbation of the magnetic Zeeman splitting [11,24].
Spectres Mössbauer de l'hydroxyde ferreux Fe(OH)2 mesuré (a) à 78 K et (b) à 4 K. Le spectre à 4 K comprend huit pics d'absorption, car l'effet quadripolaire n'est plus considéré comme perturbation de l'effet Zeeman nucléaire.
Spectra of green rusts GRs measured at 78 K are presented in Fig. 4 [12]. They all have common features (Table 1). One or two quadrupole doublets and with large values of Δ are distinguished along with one doublet with a small Δ value. The large values of Δ come from FeII state, whereas the small one comes from FeIII. Besides the relative intensity ratios, it is not possible to see differences on the sole basis of the values taken by the hyperfine parameters. The structural model that will be presented later will have to explain the Fe environment as seen in the spectra for all GRs (Fig. 4) [12]. A spectrum measured at 4 K for hydroxychloride GR1(Cl−) displays how components that are distinguished in the paramagnetic state become [20]. The ferric doublet becomes the usual sextet, whereas the two ferrous doublets become eight lines each the way the quadrupole doublet did in the case of Fe(OH)2. Again, the magnetic field is perpendicular to the three-fold axis of the structure and there exists a mixture of quantum states.
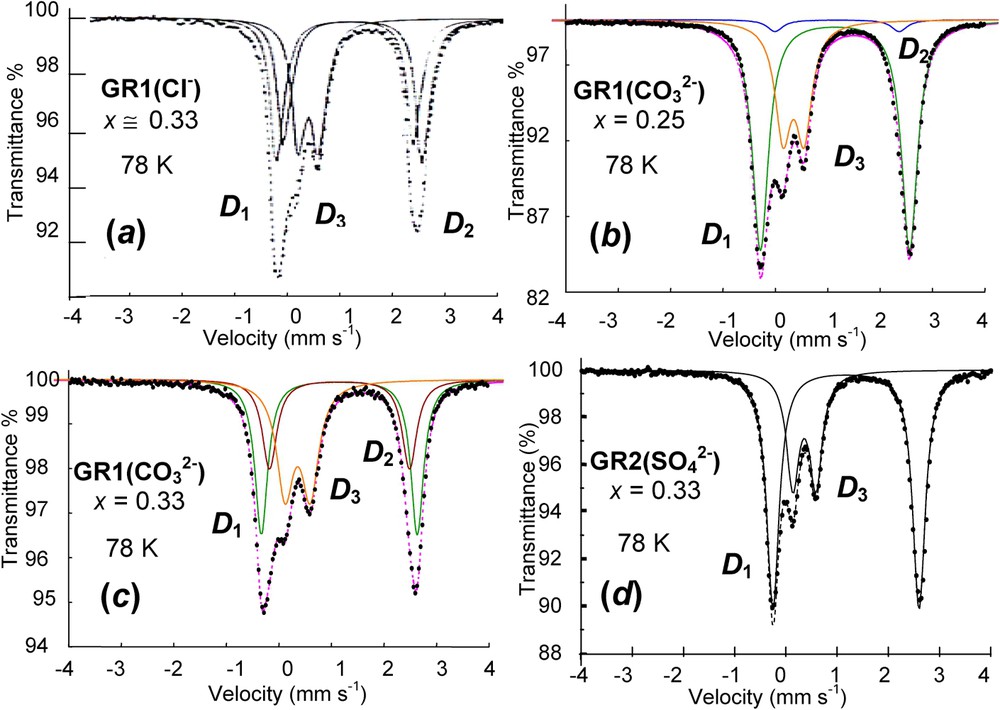
Mössbauer spectra measured at 78 K of four various green rusts: (a) GR1(Cl−) at ; (b) at x=0.25; (c) at x=0.33 and (d) at x=0.33. and quadrupole doublets are attributed to ferrous FeII(H2O) and FeII(anion), respectively, whereas is ferric FeIII state.
Spectres Mössbauer mesurés à 78 K de quatre diverses rouilles vertes : (a) GR1(Cl−) à ; (b) à x=0,25 ; (c) à x=0,33 et (d) à x=0,33. Les doublets quadrupolaires et sont respectivement attribués aux ions ferreux FeII(H2O) et FeII(anion), alors que est l'état ferrique FeIII.
Hyperfine parameters of Mössbauer spectra of GRs measured at 78 K as illustrated in Fig. 4 and selected from the literature; δ (mm s−1): isomer shift (α-iron as reference at ambient); Δ (mm s−1): quadrupole splitting; RA (%): relative abundance
Paramètres hyperfins des spectres Mössbauer de GRs mesurés à 78 K et illustrés sur la Fig. 4 à partir d'une sélection trouvée dans la littérature ; δ (mm s−1) : déplacement isomérique (référence fer α à l'ambiante) ; Δ (mm s−1) : éclatement quadrupolaire ; RA (%) : abondance relative
| x | GR1(Cl−) | GR1(Cl−) | |||||||||||||
| 0.25 | 0.33 | 0.25 | 0.33 | 0.25 | |||||||||||
| Ref. | Not shown | Fig. 4a | Fig. 4b | Fig. 4c | Fig. 4d | ||||||||||
| [11] | [20] | [12] | [4] | [26] | |||||||||||
| δΔ (mm s−1) | RA (%) | δΔ (mm s−1) | RA (%) | δΔ (mm s−1) | RA (%) | δΔ (mm s−1) | RA (%) | δΔ (mm s−1) | RA (%) | ||||||
| 1.26 | 2.88 | 48 | 1.27 | 2.89 | 37 | 1.28 | 2.97 | 62 | 1.27 | 2.93 | 51 | 1.27 | 2.88 | 66 | |
| 1.25 | 2.60 | 24 | 1.25 | 2.60 | 32 | 1.28 | 2.55 | 12 | 1.28 | 2.64 | 15 | ||||
| 0.47 | 0.41 | 24 | 0.47 | 0.41 | 31 | 0.47 | 0.43 | 26 | 0.47 | 0.42 | 34 | 0.47 | 0.44 | 34 | |
| W | 1.37 | 3.36 | 4 |
2.3 Miscellaneous methods
Besides its deep green colour when synthesised and the celadon porcelain shades of fougerite in the field, GRs display thin hexagonal crystals as seen (Fig. 5) by transmission electron microscopy (TEM). Their size depends strongly upon the method of preparation from coprecipitation ( diameter) (Fig. 5a) to bioreduction ( diameter) [4,5,18]. Infrared spectra have also been used essentially for characterising the incorporated anion within interlayers [16]. XAS study was also done at the K absorption edge of Fe to compare synthetic GRs and fougerite mineral [23]. It is a Fe selective method the way Mössbauer spectroscopy is and shows that the pavement of iron is similar to that of brucite. However, the presence of anions was ignored and no indication about their exact nature ever obtained.
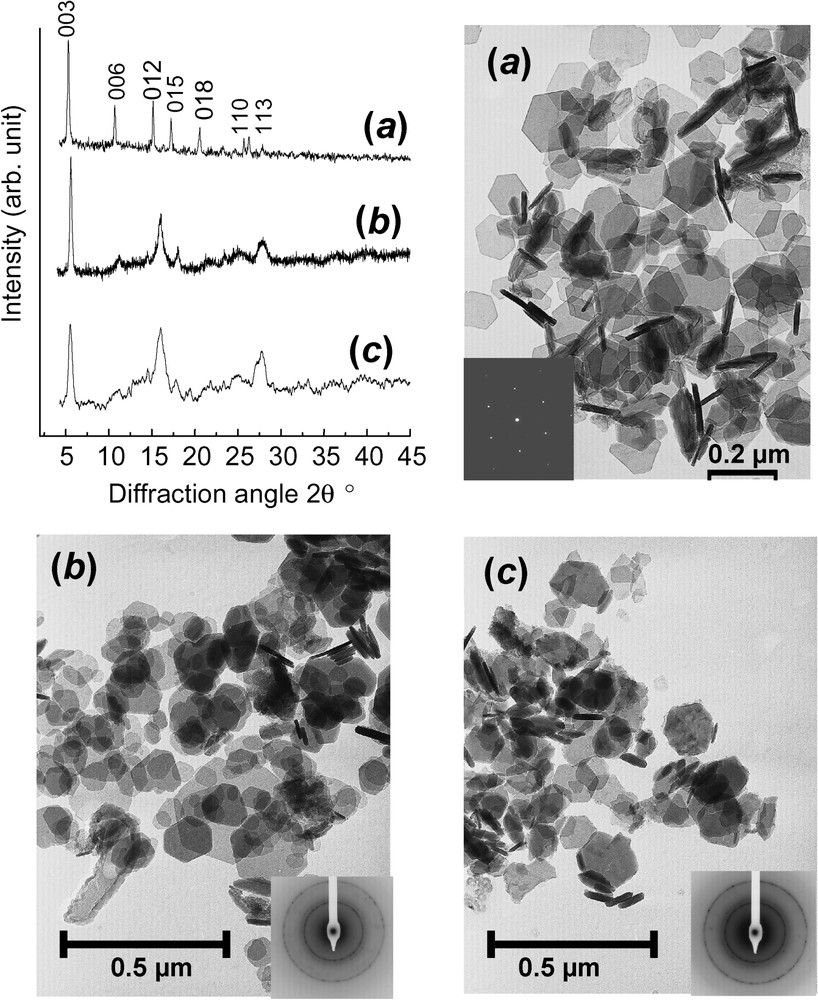
XRD patterns using Mo K radiation with λ=0.070930 nm and corresponding transmission electron micrographs of: (a) FeII–III hydroxycarbonate green rust at stoichiometry, i.e. ; (b) ferric green rust obtained by aerial oxidation of dried ; (c) ferric green rust obtained by fast oxidation of using H2O2.
Clichés de diffraction X utilisant le rayonnement Mo K,où λ=0,070930 nm et micrographies électroniques par transmission de : (a) la rouille verte hydroxycarbonate FeII–III à la stœchiométrie, c'est-à-dire ; (b) la rouille verte ferrique , obtenue par oxydation à l'air de séchée ; (c) la rouille verte ferrique obtenue par oxydation rapide de traitée par H2O2.
3 Structure of FeII–III hydroxysalt green rusts
Models of crystal structures of GRs and fougerite must be consistent with both XRD and Mössbauer spectroscopy. Starting from cation layer stacking determined by XRD in Fe(OH)2 and GRs, cation and anion distributions in layers and interlayers can been derived [12].
3.1 Stacking sequences from XRD (Fig. 6)
GRs are issued from Fe(OH)2 by intercalating anions that are balanced by FeIII inside brucite-like layers while replacing FeII. If or , and c represent the three sites in a hexagonal pavement, Fe(OH)2 structure consists of hexagonal close-packed layers of OH− ions separated by where the sequence along the three-fold c axis is designated by letters ... with parameter [3] (Fig. 6a). Octahedrons sharing edges are occupied by FeII ions at c positions between OH− layers at A and B positions. FeII ions constitute a hexagonal layer with also a cation distance a. The overall stacking sequence is ... if V represents vacant sites and the lattice is trigonal with space group [3].
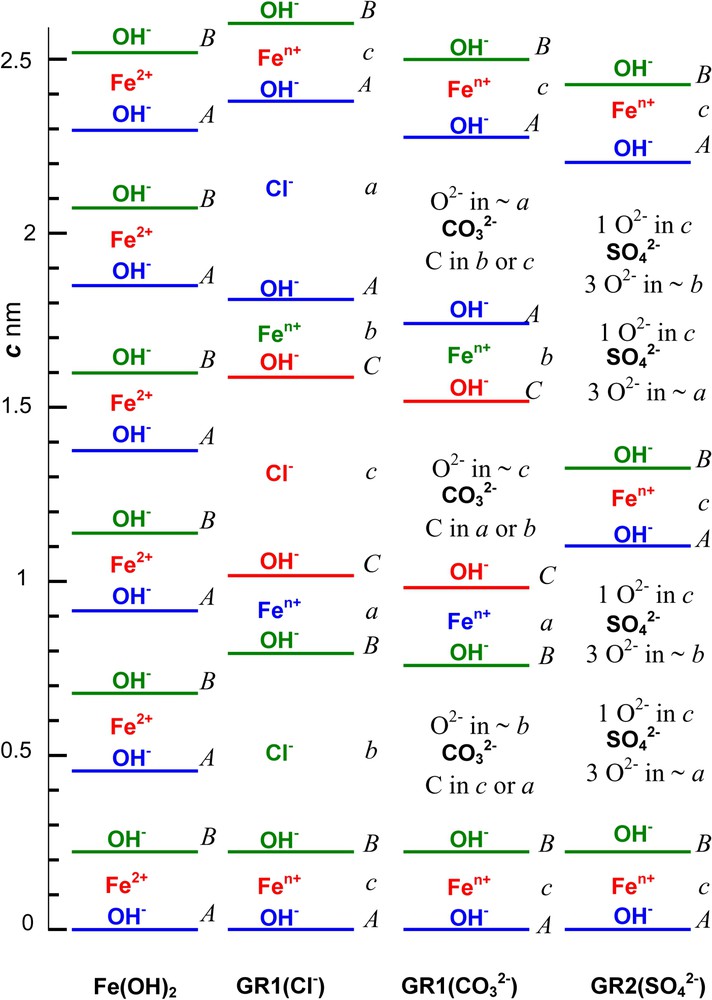
Stacking sequence along the 3-fold axis at scale of Fe cation layers, OH− ion layers and anion interlayers in compounds: (a) Fe(OH)2; (b) GR1(Cl−); (c) ; (d) . A,B,C and a,b,c positions represent the sites in the hexagonal pavement of ions for OH− ions and other ions, respectively.
Séquence d'empilement représentée à l'échelle le long de l'axe ternaire des couches cationiques de fer, des couches d'ions OH− et d'intercouches anioniques dans les composés (a) Fe(OH)2 ; (b) GR1(Cl−) ; (c) ; (d) . Les positions A,B,C et a,b,c représentent respectivement les sites dans le pavage hexagonal des ions OH− et des autres ions.
Within a space group, the stacking sequence of OH− layers for GR1s is ... Lattice parameters are and 0.31605 nm and and 2.2455 nm for GR1(Cl−) hydroxychloride (Fig. 6b) and GR1() hydroxycarbonate (Fig. 6c), respectively [7,22]. The overall sequence is ... if are anion interlayers where anion positions depend on their shape. Trigonal space group of GR2 keeps the OH− ion layer sequence of ... as in Fe(OH)2 [26]. The overall sequence ... displays 2 anion interlayers (Fig. 6d). For , , parameters are , , and [26]. GR2 has a long-range ordered structure with correlated anion and cation positions. In contrast, XRD does not reveal ordering in GR1.
3.2 Cation and anion distributions from Mössbauer spectroscopy
The models we propose concern only the GR1s since the Rietveld analysis has completely settled the structure of GR2 [26]. It is consistent with the Mössbauer spectrum, since the order of anions generates only one type of FeIII site (Fig. 4, Table 1). Two GR1 are treated: those of hydroxychloride GR1(Cl−) and hydroxycarbonate . Experimentally, it was observed that the ratio cannot exceed in both cases [12,22]. It is a general rule found in LDH: 2 FeIII cations cannot be first nearest neighbours in a layer due to electrostatic repulsion. Consequently, corresponds to a long-range order of FeIII cations with , where is the distance between cations, but this order is not detected by XRD. In contrast, x can be smaller than , since the repulsion is still compatible and ordering still exists down to , leading to a range for x that extends over . Enough anions are necessary to shift from hydrogen bonds in Fe(OH)2 to ionic bonds in GR1 and from ... stacking to ... stacking.
Let us first take the example of GR1(Cl−) for , i.e. where long-range order is met [12]. All FeIII ions are surrounded by six FeII ions within a layer. The overall stacking sequence is precisely ... as illustrated in Fig. 6b and since a Cl− anion is spherical, it lies in register with the OH− planes below and above it. For instance, if Cl− is in a b position, the closest FeIII ions are in a c position below and a position above. Therefore, each anion has adjacent cations above and below and vice-versa. Chains of anions and cations are thus constituted. The projections perpendicularly to the c axis of the hexagonal layers and interlayers are drawn in Fig. 7. In particular, the superimposition of one Fe layer in the way between two Cl− interlayers allows us to visualise the environments determined by Mössbauer spectroscopy (Fig. 7d). Each anion is close to 2 FeIII cations, which are both shared by 2 anions. Moreover, two types of FeII ions exist, those close to 2 Cl− ions also shared by 2 FeII ions, and those far from any anion. The first ones are called FeII(Cl−), whereas the other ones are FeII(H2O), since water molecules can fill sites which are not occupied by Cl−. The abundance for the three sites FeIII, FeII(Cl−) and FeII(H2O) is the same and equal to , as observed in the spectrum (Fig. 4a, Table 1). If , repulsion among FeIII ions still exists, so that 2 FeIII cannot be first-nearest neighbours and site abundance for FeIII, FeII(Cl−) and FeII(H2O) is thus and , respectively. Moreover, it has been shown recently that the range of composition for x is met in most LDHs and corresponds in fact to crystals composed of domains with two types of long range order, one with periodicity of and the other one of matched epitaxially, as drawn in Fig. 8. This result obtained from Mössbauer spectroscopy for GRs can be extended to other LDHs, such as pyroaurite or hydrotalcite.

(001) Projections perpendicular to the c axis of the GR structure of, from left to right, GR1(Cl−), and , as obtained for with long range order. OH−-ion layers are not taken into account; (a) one anion interlayer, (b) a Fe layer and (c) the next anion interlayer; (d) superimposition of (a), (b), (c) where only one Fe layer in the way between two interlayers is represented; (e) three adjacent interlayers in GR1s to have a complete repeat c showing the anion pillars that maintain the stacking of the structure.
Projections (001) perpendiculaires à l'axe c de la structure GR, de gauche à droite, GR1(Cl−), et obtenues à avec ordre à grande distance. Les couches d'ions OH− ne sont pas prises en compte ; (a) intercouche anionique, (b) couche de fer et (c) intercouche anionique suivante ; (d) superposition de (a), (b) et (c), où seule une couche de fer à mi-chemin entre deux intercouches est représentée ; (e) trois intercouches adjacentes dans GR1s afin d'avoir un motif complet se répétant le long de c et montrant ainsi les piliers d'anions qui soutiennent l'empilement de structure .
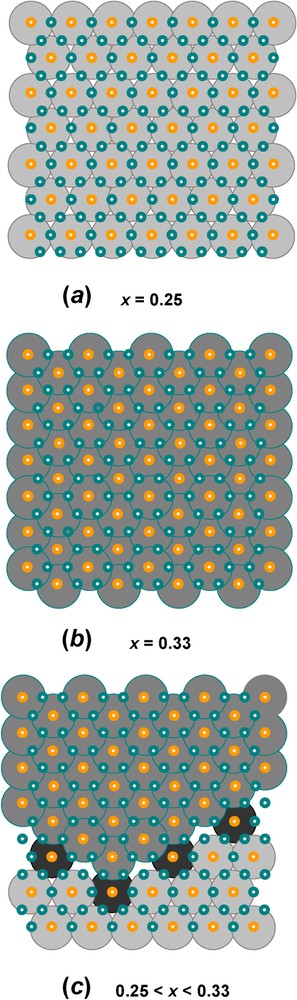
Domains with long-range order of FeIII cations in the hexagonal pavement of brucite-like layers found in FeII–III hydroxysalt green rusts: (a) long-range order at x=0.25 with periodicity ; (b) long-range order at x=0.33 with periodicity : (c) ordered domains that can adjust any value of x in the [0.25,0.33] range. These domains can be extended to any LDH.
Domaines avec ordre à grande distance des cations FeIII dans le pavage hexagonal des couches de type brucitique que l'on trouve dans les rouilles vertes, hydroxysels ferreux–ferriques : (a) ordre à grande distance pour=0,25 de périodicité ; (b) ordre à grande distance pour x=0,33, de périodicité ; (c) domaines ordonnés qui peuvent s'ajuster pour n'importe quelle valeur de x dans l'intervalle [0,25;0,33]. Ces domaines peuvent s'étendre à tout hydroxyde double lamellaire.
Let us take now for example, the case of GR1() at [12]. The cation pavement is that already seen for GR2(), with a long-range order corresponding to . Since each ion occupies three sites for two charges, 2 FeII and one FeIII ions are associated with it. Within its pavement, a anion can lie in a Δ or a ∇ configuration. The centre of gravity of the anion is a carbon atom, which is in register with the layer either above or below it. Fig. 7a shows that an ordered array of anions with a periodicity can solely meet this requirement, that is 12 times the original unit cell of the pavement, for sharing 2 FeIII cations on one side and 1 FeII() on the other side. As a whole, when looking at the superimposition of one Fe layer (Fig. 7b) in the way between two interlayers (Fig. 7a and c), the abundance for FeIII, FeII() and FeII(H2O) is (), (1/6) and (), respectively (Fig. 7d). This result is in perfect agreement with the abundances observed for the three sites by Mössbauer spectroscopy (Fig. 4c, Table 1). Finally, the superimposition of three successive anion interlayers is displayed in Fig. 7e. This shows the anion pillars with the 3-fold screw axis that maintain the stacking within the structure of GR1. These projections illustrate completely the distribution of cations and anions inside the periodicity along the c axis. If , since the repulsion among FeIII cations also exists, the site abundance for FeIII, FeII() and FeII(H2O) is now and leading to 0.25, 0.125 and 0.625 for (Fig. 4b, Table 1).
Finally, if comparing all cases of GR1(Cl−), GR1() and GR2(), Mössbauer spectra reveal one or two FeII doublets (Fig. 4) (Table 1). In all cases, the doublet that corresponds to the largest quadrupole splitting Δ around 2.90 mm s−1 must be attributed to the FeII cations that are far from any inserted anion. These Fe environments are indeed alike, whatever the GR.
4 Deprotonation of green rusts
Aerial oxidation of GRs within the aqueous solution as observed during the corrosion process of iron has been extensively studied and will be developed later on. However, one most interesting phenomenon is the difference that is observed if the GR sample has been dried before slow aerial oxidation giving (Fig. 5b) or if the oxidation is fast as it is by pouring hydrogen peroxide giving (Fig. 5c). Instead of obtaining the usual products that are obtained by aerial oxidation inside the solution such as goethite, lepidocrocite or ferrihydrite, the fast or dried oxidation gives rise to hexagonal crystals identical to those of the initial GR as illustrated in electron micrographs (Fig. 5a–c), but the colour turns from green to orange. This compound, which is called ‘ferric green rust’ , , conserves its initial crystal lattice keeping the c parameter, due to the anion intercalation even though some differences after oxidising are noticeable in the XRD patterns (Fig. 5). A sample obtained by air oxidation of dried green rust maintains a prominent peak attributed to (003) planes, whereas other peaks are broadened to a great extent, but conserving their essential features (Fig. 5b). In contrast, the XRD pattern for the sample obtained by H2O2 fast oxidation displays much broader peaks (Fig. 5c). All this suggests that the crystal structure depends crucially on the preparation method. The oxidation of a dried by atmospheric oxygen giving is a slow process providing enough time to readjust crystalline distortions. Lattice parameters are thus and , whereas those of were initially and testifying of a shrinkage of the unit cell. In contrast, oxidation by H2O2 distorts the structure to a greater extent than expected, since it is a very strong oxidant allowing no local relaxation, as by quenching.
Mössbauer spectra are fitted with Voigt profiles. They demonstrate that all Fe cations are ferric since only quadrupole splitting with low Δ value or magnetic sextets are displayed (ferrous states give rise to octets) (Fig. 9, Table 2). Measurements for and spectra are done below and above the Néel temperatures, which have been determined to be about 75 and 50 K, respectively. At 16 K, they exhibit magnetic sextets with three components , and for (Fig. 9f), but only two, and for (Fig. 9b), whereas they present quadrupole doublets in the paramagnetic state, two doublets and for (Fig. 9h) but one, D, for (Fig. 9d). Attribution of components is as follows: (i) ferric sextets , and of at 16 K correspond to doublets , and of (Fig. 9f); (ii) the FeIII site due to deprotonation, doublet , can only be distinguished in paramagnetic state from the FeIII site due to anions, doublet (Fig. 9h); (iii) FeIII sites due to deprotonation are only distinguished, whether they are far from or close to an anion for (Fig. 9f and h), but not for (Fig. 9b and d) (sextets and , versus only ). These observations confirm the XRD interpretation. Fast oxidation leading to quenches the GR structure where the long-range order among anions is conserved. All sites of the initial remain distinguishable the way they were and the only change is that each FeII gets oxidised. In contrast, slow aerial oxidation of a dried sample allows a lattice relaxation, probably along with diffusion in interlayers and some water molecules are likely missing. All ferric sites are also equivalent in the paramagnetic state and no FeIII ion can be attributed to a specific anion; this explains the only doublet D. But, since magnetic interactions are of a longer range than quadrupole effect, the two FeIII sites are distinguished at 16 K (Fig. 9b). Mössbauer spectroscopy gives indeed a deeper insight than XRD.
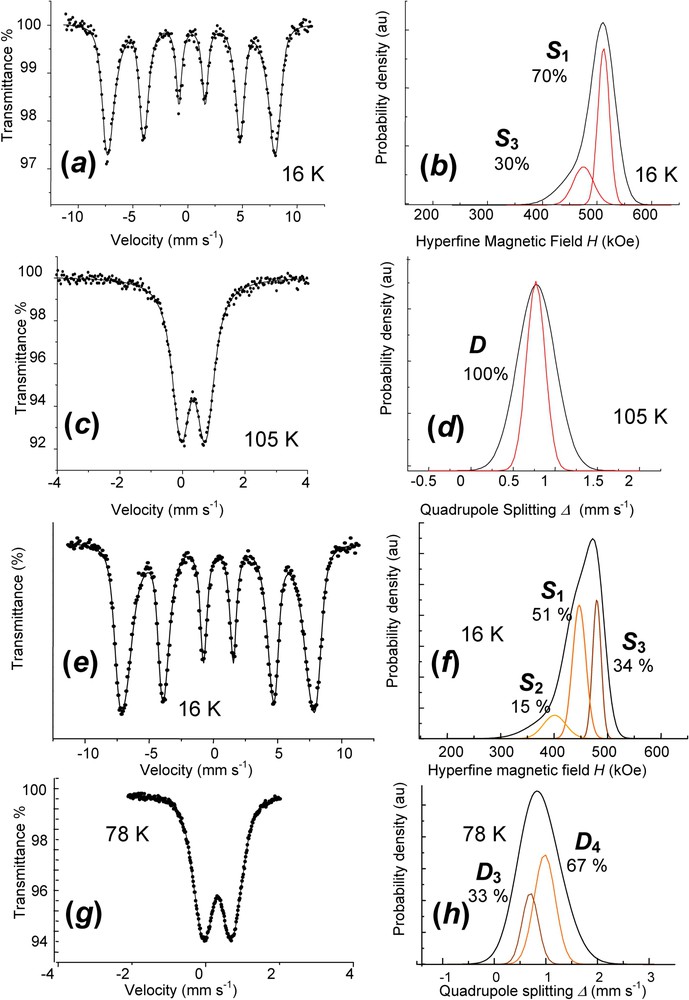
Mössbauer spectra and Voigt profile analysis of fully deprotonated ferric : (a) and (b) ferric green rust obtained by aerial oxidation of dried . Spectrum measured at 16 K; (c) and (d) ferric green rust obtained by aerial oxidation of dried . Spectrum measured at 105 K; (e) and (f) ferric green rust obtained by fast oxidation of using H2O2. Spectrum measured at 16 K; (g) and (h) ferric green rust obtained by fast oxidation of using H2O2. Spectrum measured at 78 K.
Spectres Mössbauer avec leur analyse en utilisant des profils de Voigt pour ferrique totalement déprotonée : (a) et (b) rouille verte ferrique obtenue par oxydation à l'air de séchée. Spectre mesuré à 16 K ; (c) et (d) rouille verte ferrique obtenue par oxydation à l'air de séchée. Spectre mesuré à 105 K ; (e) et (f) rouille verte ferrique obtenue par oxydation rapide de avec H2O2. Spectre mesuré à 16 K ; (g) et (h) rouille verte ferrique obtenue par oxydation rapide de avec H2O2. Spectre mesuré à 78 K.
Mössbauer parameters for FeII–III hydroxycarbonate green rust and its ferric forms from Voigt profile analysis or classical Lorentzian shape lines (Fig. 9)
Paramètres Mössbauer pour la rouille verte, hydroxycarbonate FeII–III et ses formes ferriques à partir d'analyse de profil de Voigt ou raies classiques de forme lorenztienne (Fig. 9)
| Sample | Temp. | δ (mm s−1) | or δ (mm s−1) | (mm s−1) | (kOe) | (kOe) | RA (%) | |
| [4] | 12 K | 1.18 | 3.00 | 50 | ||||
| 1.18 | 2.59 | 17 | ||||||
| 0.50 | 0.45 | 33 | ||||||
| 16 K | 0.48 | ∼0.00 | 477 | 21 | 70 | |||
| dry oxidation | 0.48 | ∼0.00 | 440 | 37 | 30 | |||
| 105 K | D | 0.47 | 0.77 | 0.22 | 100 | |||
| 16 K | 0.48 | ∼0.00 | 447 | 25 | 51 | |||
| fast oxidation | 401 | |||||||
| 0.48 | ∼0.00 | 42 | 15 | |||||
| 0.48 | ∼0.00 | 480 | 16 | 34 | ||||
| 77 K | 0.47 | 0.88 | 0.41 | 67 | ||||
| 0.47 | 0.60 | 0.30 | 33 |
Finally, a continuous deprotonation can be observed for the overall range of x between 0.33 and 1. Samples of have been treated by the exact amount of H2O2 necessary to aim at a given value of x, according to the reaction for full deprotonation:
Obtained values were 0.33, 0.50, 0.63, 0.78 and 1 and the corresponding spectra measured at 78 K are displayed in Fig. 10 (Table 3). The reference for displays the three doublets comprising the two ferrous and and the ferric in the ():():() ratio. Other spectra are fitted by Voigt profile analysis. A new ferric component appears, whereas the two ferrous components decrease when x increases (Fig. 10). Components and remain in the 3:1 ratio, meaning that the deprotonation affects equally both types of FeII cations. The formula is thus for the continuous . In the case of the air oxidation of a dried , the same phenomenon occurs, but only one ferric doublet is observed, increasing with x, and all FeIII cations become banalised. The deprotonation goes along a lattice relaxation and diffusion may proceed along with missing water molecules.
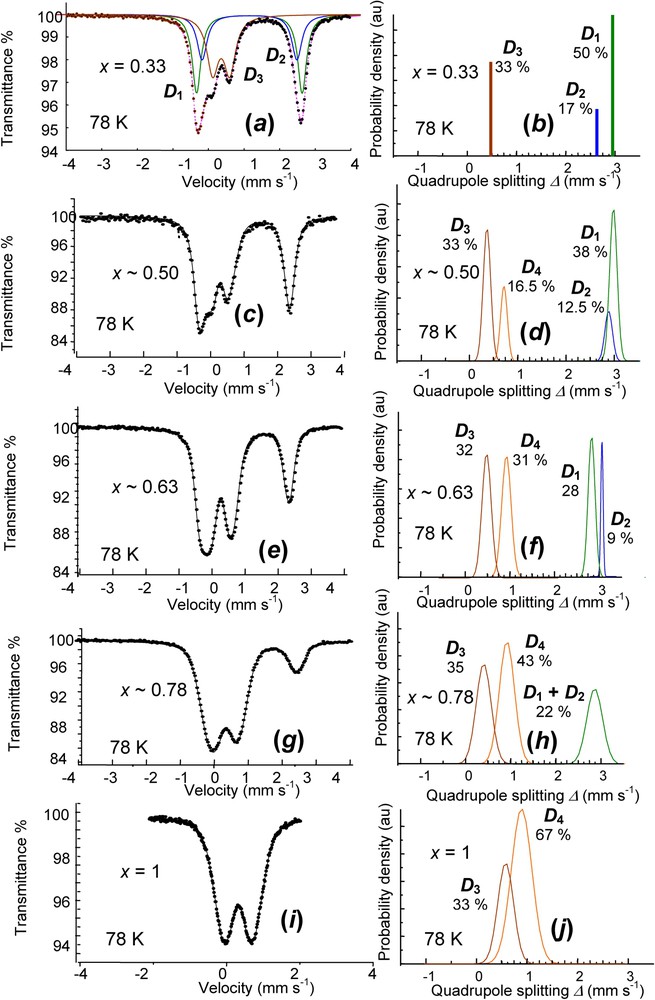
Mössbauer spectra measured at 78 K of a set of partially deprotonated hydroxycarbonate green rust samples using H2O2 for values of / ranging from 0.33 to 1. The values experimentally obtained are 0.33, 0.50, 0.63, 0.78 and 1. On right, Gaussian distributions of quadrupole splitting for fitting Voigt profile doublets.
Spectres Mössbauer mesurés à 78 K d'un ensemble d'échantillons de rouille verte hydroxycarbonate partiellement déprotonée en utilisant H2O2 pour des valeurs de / allant de 0,33 à 1. Les valeurs obtenues expérimentalement sont 0,33, 0,50, 0,63, 0,78 et 1. À droite, distributions gaussiennes d'éclatement quadrupolaire pour les doublets de profil de Voigt.
Hyperfine parameters of a set of partially deprotonated hydroxycarbonate green rust samples for values of / ranging from 0.33 to 1 (Fig. 10)
Paramètres hyperfins d'un ensemble d'échantillons d'hydroxycarbonate partiellement déprotonés pour des valeurs de / allant de 0,33 à 1 (Fig. 10)
| Quadrupole doublets | δ (mm s−1) | (mm s−1) | (mm s−1) | RA (%) | |
| x=0.33 | |||||
| FeII | 1.25 | 2.92 | 0 | 50 | |
| 1.25 | 2.63 | 0 | 17 | ||
| FeIII | 0.48 | 0.47 | 0 | 33 | |
| x∼0.50 | |||||
| FeII | 1.21 | 2.98 | 0.14 | 38 | |
| 1.21 | 2.72 | 0.16 | 12.5 | ||
| FeIII | 0.49 | 0.40 | 0.15 | 33 | |
| 0.49 | 0.70 | 0.28 | 16.5 | ||
| x∼0.63 | |||||
| FeII | 1.24 | 2.80 | 0.15 | 28 | |
| 1.24 | 3.05 | 0.05 | 9 | ||
| FeIII | 0.48 | 0.49 | 0.20 | 32 | |
| 0.48 | 0.90 | 0.21 | 31 | ||
| x∼0.78 | |||||
| FeII | 1.21 | 2.89 | 0.31 | 22 | |
| FeIII | 0.47 | 0.45 | 0.32 | 35 | |
| 0.47 | 0.95 | 0.34 | 43 | ||
| x=1 | |||||
| FeIII | 0.47 | 0.60 | 0.30 | 33 | |
| 0.47 | 0.88 | 0.41 | 67 |
5 Cation substitution in green rusts
Studies were devoted to the substitution in GRs of FeII and FeIII by di- and trivalent cations proving that Mössbauer spectroscopy is the most proficient method. For instance, replacing FeII by NiII allowed us to stabilise GR(Cl−) to measure carefully lattice parameters [21]. An XAS study for understanding the structure of fougerite and the {[FeII]/[FeIII]} variation suggested that peak intensities could vary if some FeII were substituted by MgII ions, even reaching pyroaurite [23] and Mössbauer studies were equally successful by replacing FeII with MgII or FeIII with AlIII [12]. A continuous solid solution exists by substituting FeII by MII divalent cations or FeIII by MIII trivalent cations in GRs. However, the stoichiometry of {[MII]/[MIII]} = 2 is easier to reach with Fe than with all other cations, since diffusion in a solid phase is unnecessary and electron exchange simply does it.
6 FeII–III oxyhydroxycarbonate fougerite
A GR mineral sampled in the Gr horizon of a gley soil was first identified in 1996 in the forest of Fougères (Brittany) by Mössbauer spectroscopy and thus christened ‘fougerite’ (Fig. 11) [13,27]. However, the first Mössbauer spectrum of fougerite ever published was reported in 1985 by the Geological Survey of Denmark and Greenland [8], but it was attributed at that time to the clay minerals with which it was then mixed (Fig. 11c–f) [8,9]. Anyhow, all arguments lead now to the evidence that fougerite is the FeII–III oxyhydroxycarbonate, even though there is no direct identification yet of the inserted anions that lie in the interlayers. Following an old tradition [19], Fe(OH)(2+x) was for some time proposed essentially because of its variation of composition, whereas at that time all synthetic GRs had a well-established stoichiometry [13]. Since then, arguments have been devoted to cope with this challenge [12].

Mössbauer spectra of fougerite mineral compared to that of synthetic sample. (a) Synthetic with x=0.50. (b) Original sample from the forest of Fougères (Brittany) [13,27]. (c)–(f) Samples extracted around Syv Creek (Denmark) [8,12]: (c) sample from the completely oxidised zone at a depth of 2 m, (d) sample from the partially oxidised zone at a depth of 2.5 m, (e) sample from the completely reduced zone at a depth of 3 m and (f) histograms from the computer fittings of reduced and oxidised zones samples [12]. Spectrum of Fig. 11e is measured in a velocity range, since the sample is completely paramagnetic and thus computed with two ferrous and two ferric doublets. One couple is attributed to fougerite (80%) and the other one to clay minerals (20%) [12].
Spectres Mössbauer de minéral fougérite comparés, à celui d'un échantillon synthétique (a) de synthèse avec x=0,50. (b) Premier échantillon extrait de la forêt de Fougères à l'origine du nom fougérite (Bretagne) [13,27]. (c)–(f) Échantillons extraits aux alentours de Syv Creek (Danemark) [8,12] : (c) échantillon de la zone complètement oxydée à une profondeur de 2 m, (d) échantillon de la zone partiellement oxydée à une profondeur de 2,5 m, (e) échantillon de la zone complètement réduite à une profondeur de 3 m et (f) histogrammes issus des ajustements à l'ordinateur pour les échantillons des zones réduites et oxydées [12]. Le spectre de la Fig. 11e est mesuré dans un domaine de vitesse , puisque l'échantillon est entièrement paramagnétique et donc calculé avec deux doublets ferreux et deux doublets ferriques. Une paire est attribuée à la fougérite (80%) et l'autre paire aux minéraux argileux (20%) [12].
The GR mineral called fougerite is therefore based on the FeII–III hydroxycarbonate , . Moreover, it is the partially deprotonated , formed by bioreduction of ferric oxyhydroxides, which keeps some unreduced FeIII(OOH)3 octahedrons where x lies within the interval, as later explained [12]. Cation substitution, e.g., by MgII and AlIII, may lead to a slight variation of x, relating fougerite with isomorphic hydroxycarbonate minerals, pyroaurite and hydrotalcite. However, a major difference exists between fougerite and other LDHs, e.g., pyroaurite or hydrotalcite. The {[MIII]/[MII]} ratio cannot change by deprotonation, as it does in . They are most stable phases that do not evolve in oxidising conditions.
7 Conclusion
XRD is the most reliable method for characterising crystalline compounds. However, amorphous or nanocrystals are hardly detectable and it is often badly quantitative. Therefore, XRD is of no help to detect fougerite in the field, because this mineral is competing with many well-crystallised phases. In contrast, Mössbauer spectroscopy of occurs to be the most sensitive method for characterising iron-containing phases, since it eliminates all other compounds and cannot be matched by any other method for field monitoring. It was even possible to follow in situ the status of iron with time and depth, without removing anything, by using the miniaturised MIMOS spectrometer that has been devised to characterise iron oxides on planet Mars, once the apparatus set in a pit dug in the ground [10]. The presence of fougerite would have not been successfully identified without Mössbauer effect. Moreover, it was possible to separate the contribution of iron within clay minerals from that of hydroxycarbonate fougerite [12].
All synthetic GRs have a stoichiometry for a ratio , as observed by Mössbauer spectroscopy. However, x can be smaller than this value with ordered cation domains that still ensure that FeIII ions are all surrounded by 6 FeII ions within cation layers. In contrast, a fast or dry oxidation deprotonates a GR, keeping the initial LDH structure. Mineral fougerite, which forms by bioreduction of ferric FeOOH due to DIRB in soils, is partially deprotonated , i.e. , which keeps some unreduced FeIII(OOH)3 octahedrons where x remains within the [] interval. It is isomorphous with pyroaurite and hydrotalcite of space group. Anyhow, an old tradition proposing a mythic Fe(OH)(2+x) compound as being the bluish green mineral found in reduced gley soils and never produced in a laboratory whatsoever is clearly outdated. The outstanding redox properties and unmatchable reactivity of GRs, more specifically its carbonated form, will be the object of other articles in this issue.