

Avant-propos
Le cycle biogéochimique du fer, c'est-à-dire le schéma décrivant la circulation de cet élément entre les différents compartiments de l'environnement global, doit l'une de ses originalités à deux caractéristiques : l'abondance de cet élément dans la croûte terrestre et les formations superficielles qui en dérivent, et son occurrence naturelle, sous deux états d'oxydation différents. Dans tous les compartiments situés à l'intersection entre géosphère, biosphère, hydrosphère et atmosphère, ces deux particularités vont avoir des conséquences majeures sur son comportement. Si ces compartiments sont ouverts vis-à-vis de l'oxygène atmosphérique, le fer, pour l'essentiel ferreux dans les minéraux primaires, aura tendance à s'oxyder et à s'accumuler dans ces minéraux secondaires que Goldsmith appelait les hydrolysats. En revanche, si la communication entre l'atmosphère et l'hydrosphère est provisoirement interrompue, le fer, quelle que soit son origine, pourra rejoindre la catégorie des cations solubles, où la valeur de son potentiel ionique le situe à proximité immédiate du cation Mg2+.
Dans les sols périodiquement saturés en eau, les réservoirs de nappes phréatiques et certains sédiments récents qui sont tous de bons exemples de ces compartiments situés à l'intersection des quatre sphères mentionnées plus haut, la disponibilité de l'oxydant ultime que constitue l'oxygène atmosphérique est loin d'être assurée d'une façon homogène ou permanente. En effet, la solubilité de l'oxygène gazeux dans l'hydrosphère n'est que de quelques mg l−1. En outre, le coefficient de diffusion de ce gaz dans l'eau est près de dix mille fois inférieur à sa valeur dans l'atmosphère. Dans tous ces milieux, en revanche, les matières organiques, qu'elles soient dissoutes, colloïdales ou particulaires, sont généralement présentes. Il en résulte que, là où l'oxygène est absent, le fer ferrique immobilisé dans les hydrolysats peut devenir un accepteur alternatif d'électrons, pour assurer la “respiration ” de communautés de bactéries anaérobies ou facultativement anaérobies, connues sous le nom de bactéries ferriréductrices.
Comme ces bactéries tirent leur énergie de la dégradation des matières organiques, réduction du fer et oxydation du carbone organique seront des réactions couplées et l'on pourra schématiquement écrire :
| (1) |
La réaction (1) assure donc le passage d'un constituant solide, au sein duquel sont immobilisés des cations ferriques, vers une solution où représente des cations solubles et donc mobiles. Cette réaction n'est pas réversible ; en effet, la conversion du CO2 en CH2O n'est possible que via la photosynthèse et, de ce point de vue, la réaction (1) constitue un des nœuds où le cycle du carbone croise celui du fer. Toutefois, si le milieu vient à s'ouvrir à l'oxygène atmosphérique, on pourra aussi écrire :
| (2) |
Comme le montre la Fig. 1, les Éqs. (1) et (2) délimitent l'enveloppe au sein de laquelle s'inscrit le cycle que décrit le fer entre les éléments de ses hydrolysats et ses solutions ferreuses. Toutefois, comme la réaction (1) donne lieu à un cation mobile, la Fig. 1 doit aussi prendre en compte l'itinéraire que parcourt le fer entre l'endroit où il est mobilisé et celui où il est à nouveau immobilisé. Dans les sols soumis à des alternances saisonnières de conditions réductrices et oxydantes, la distance séparant ces deux types de sites peut varier de quelques millimètres à quelques décimètres. Sur le long terme, cependant, par exemple sur un versant sous lequel circule une nappe phréatique, la répétition des réactions (1) et (2) peut conduire à des transferts de fer atteignant des valeurs kilométriques.
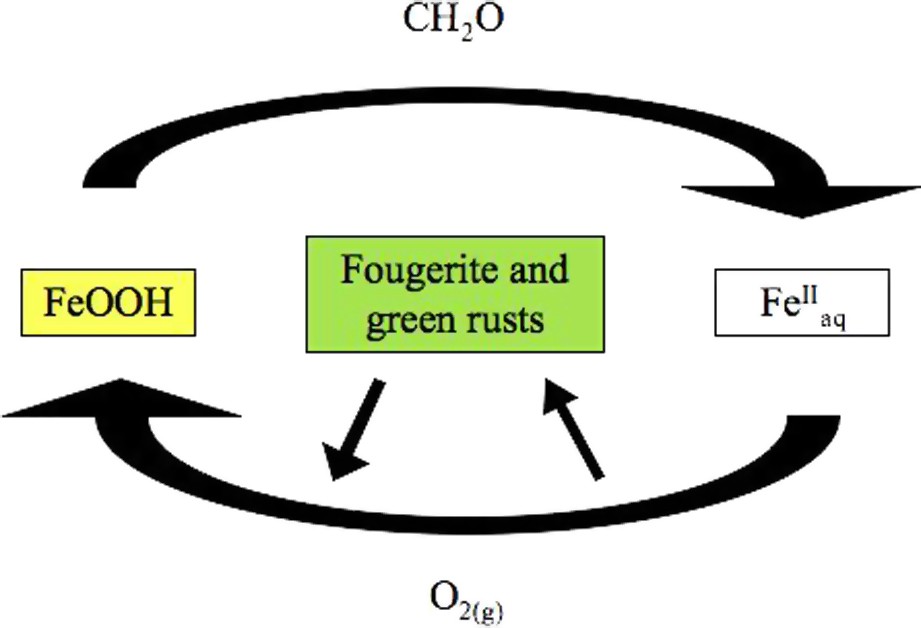
Sketch of the part of the biogeochemical cycle of iron taking place between ferric oxyhydroxides and ferrous solutions.
Schéma décrivant la partie du cycle biogéochimique du fer se déroulant entre les oxydes et oxyhydroxydes ferriques et les solutions ferreuses.
Par ailleurs, il est bien établi que la nature cristallographique des solides FeOOH qui apparaissent comme réactif dans (1) et comme produit de réaction dans (2) peut différer. Les bactéries ferriréductrices mobilisent en effet préférentiellement le fer occlus dans les oxydes de fer les moins cristallins [17]. En revanche, la nature cristallographique de la ou des phase(s) ferrique(s) restituée(s) par la réaction (2) dépend des conditions physicochimiques locales [5]. La répétition du cycle schématisé par la Fig. 1 conduit donc fréquemment à modifier la composition minéralogique du compartiment des hydrolysats.
Dans de nombreux sites ou microsites visités par des solutions ferreuses, la disponibilité en oxygène (le PO2 qui apparaîtrait dans l'expression décrivant les conditions d'équilibre de la réaction (2)) n'est pas toujours suffisante et la précipitation de FeOOH n'est donc pas observée. Dans ces cas, particulièrement là où les conditions de pH s'approchent de la neutralité, les cations FeIIaq peuvent néanmoins donner lieu à la formation de précipités en suivant une autre voie.
Selon que les anions qui assurent l'électro-neutralité de la solution ferreuse initiale sont ou non impliqués dans le phénomène, deux types de réactions alternatives ont été envisagés. Utilisant le même formalisme que précédemment, elles s'écrivent:
| (3a) |
| (3b) |
Les solides qui résulteraient de la réaction (3a) sont connus dans la littérature sous le nom d'hydroxydes ferrosiques [1]. Parmi ces différents précipités, l'un d'entre eux, à savoir Fe3(OH)8 (ou (FeII FeIII2(OH)8)), a connu une certaine célébrité parmi les auteurs décrivant le devenir du fer, tant dans les sols périodiquement saturés en eau (entre autres, [14,15,23,30]), que dans les réservoirs de nappes phréatiques [13]. Précédemment, en effet, Ponnamperuma et al. [18] avaient observé que, pendant la plus grande partie de la phase de submersion des sols de rizières, l'activité des ions Fe(II)aq correspondait sensiblement bien au produit de solubilité de Fe3(OH)8 tel que l'avait déterminé Arden [1]. Toutefois, ni Ponnamperuma et al. [18], ni les auteurs qui suivirent ne purent mettre directement en évidence la présence d'un tel hydroxyde et ceci conduisit certains (entre autres, [11,13,16,29]) à douter de son existence. Ainsi, Langmuir [13] remarque que dans un diagramme –pH construit en tenant compte de la composition chimique habituelle des eaux continentales, le domaine de stabilité de Fe3(OH)8 serait tellement étendu qu'il est probable que la valeur de l'énergie libre standard de formation attribuée traditionnellement à cet hydroxyde ferrosique doive être révisée. De même, tout en soulignant le rôle important que joue la précipitation de constituants ferrosiques dans les sols périodiquement submergés, Kirk [12] reconnaît aussi clairement que leur attribuer une formule d'hydroxyde n'est qu'une approximation, rendant mal compte de leur identité et de leur composition chimique réelles.
Les solides résultant de la réaction (3b) sont connus dans la littérature sous le nom de rouilles vertes. Cette appellation découle de leur occurrence fréquente parmi les produits de corrosion du fer métallique et de leur couleur bleu-vert [10]. Contrairement aux hypothétiques hydroxydes ferrosiques des sols de rizières, qui étaient considérés comme des solides amorphes, les rouilles vertes sont des solides présentant une organisation cristalline. Initiée par les études de Feitknecht et Keller [6] et de Bernal et al. [2], la détermination de leur structure et de leur cristallochimie a fait l'objet d'un nombre considérable de travaux. Les derniers développements sur ce sujet seront présentés en détail par ailleurs [8]. Ici, il suffit de rappeler que leur architecture cristalline résulte de l'empilement régulier de couches brucitiques abritant les cations FeII et FeIII et d'espaces interlamellaires occupés par les anions et des molécules d'eau. Ces particularités structurales rangent les rouilles vertes dans la grande famille des hydroxydes doubles lamellaires, famille à laquelle appartiennent également des minéraux hydroxycarbonatés, comme l'hydrotalcite et la pyroaurite.
En dépit de leurs différences notables en termes de structure et de composition chimique, rouilles vertes et hydroxydes ferrosiques ont en commun de développer une couleur bleu-vert très caractéristique. En outre, lorsque ces solides sont exposés à l'air, cette couleur disparaît très rapidement. Pour les pédologues, ces deux propriétés ne manquaient pas de rappeler des observations faites dans certains horizons de sols longuement saturés en eau. Il était, dès lors, légitime, de suspecter qu'il existait dans ces horizons des pigments, c'est-à-dire des minéraux inconnus, responsables, tant de cette couleur, que de son caractère fugace.
Dans le début des années 1980, Taylor [25] et Taylor et McKenzie [27] ont largement contribué à répandre l'idée que la couleur bleu-vert de certains sols saturés en eau, plutôt que d'être due à la présence d'hydroxydes ferrosiques, résultait vraisemblablement de la précipitation de rouilles vertes carbonatées. Toutefois, même après avoir surmonté la difficulté liée à l'instabilité de ces solides, Taylor [26] ne put le démontrer. Plus de 15 ans plus tard, c'est à Trolard et al. [28] et à Génin et al. [7] que l'on doit d'avoir montré définitivement que les pigments colorant en bleu-vert les horizons d'un Réductisol de la forêt de Fougères possédaient des caractéristiques spectrales et chimiques les rattachant clairement aux rouilles vertes. Dans le même temps, ces auteurs proposaient que ce nouveau minéral reçoive le nom de fougérite.
Cette proposition ayant entretemps été entérinée par l'Association internationale de minéralogie (IMA-2003-057), il est maintenant justifié d'attribuer à la fougérite la place qu'occupaient autrefois les hydroxydes ferrosiques et les rouilles vertes dans le cycle du fer. Comme l'illustre la Fig. 1, ce minéral y joue le rôle de plaque tournante. D'une part, il y est le précurseur de la plupart des oxyhydroxydes ferriques connus. D'autre part, sa solubilité, plus élevée que celles des oxydes ferriques, le désigne comme le meilleur candidat pour contrôler l'activité des espèces fer présentes en solution et, par là, influer sur le devenir des espèces solubles ou potentiellement solubles d'un grand intérêt environnemental, comme par exemple les phosphates, les nitrates et les métaux lourds.
Bien que cette place centrale soit aujourd'hui bien reconnue (voir par exemple la localisation des rouilles vertes dans le schéma plus détaillé que proposaient récemment Bigham et al. [4]), de nombreuses questions restent en suspens. Dans ce numéro thématique des C. R. Geoscience, ce sont celles d'entre elles qui ont trait, tant à la structure et la cristallochimie précises de ce minéral et de ses équivalents synthétiques, qu'aux mécanismes chimiques et biochimiques conduisant à leur formation qui seront développées.
Dans le premier article de ce fascicule, Génin et al. [8] utilisent l'information obtenue par spectroscopie Mössbauer pour décrire les différences structurales survenant dans les cristaux de rouilles vertes (RV) lorsque ces solides incorporent des anions possédant des charges électriques et des encombrements différents tels que par exemple Cl− ou , ou encore et . Ils montrent aussi que des RV carbonatées préparées à leur stœchiométrie (c'est-à-dire avec un rapport FeIII/Fetotal = 0,33) peuvent subir, après séchage à l'air, une oxydation progressive et ce, sans modification structurale autre qu'une déprotonation équivalente de leurs anions hydroxyles structuraux. Comme tous les échantillons de fougérite examinés jusqu'à présent sont caractérisés par des rapports FeIII/Fetotal compris entre 0,33 et 0,66, ce comportement particulier permet donc de comprendre comment ce minéral peut, en conditions naturelles, ajuster sa cristallochimie aux conditions rédox prévalant dans le site où il se forme et évolue.
L'étude des RV a de tous temps fait appel à leur obtention par voie de synthèse in vitro. Dans ce numéro thématique, les résultats provenant de deux méthodes de synthèse différentes sont exposés et comparés. En procédant à la coprécipitation d'ions FeII et FeIII en conditions strictement contrôlées, Ruby et al. [22] décrivent comment un précurseur FeOOH se transforme en une RV stœchiométrique lorsque sont réalisées en solution les conditions appropriées. Cet article présente aussi dans le détail un diagramme “bilan de matière ” permettant de visualiser l'itinéraire suivi par toute réaction survenant dans ce système et de prédire la nature de la phase ou des phases solide(s) qui s'y forme(nt).
En faisant usage de ce même diagramme, Génin et al. [9] réexaminent les résultats des nombreuses expériences dans lesquelles Fe(OH)2 avait été choisi comme matériau de départ pour l'obtention, tant de RV, que d'autres phases ferrosiques et ferriques. Cet article explique pourquoi, lors de l'oxydation spontanée de Fe(OH)2, la synthèse de la RV seule n'est possible qu'en présence d'un excès d'ions FeII en solution. En leur absence, en effet, Fe(OH)2 s'oxyde en livrant la magnétite qui, plus tard, se transformera en maghémite. Les phases solides résultant de l'oxydation des RV sont également identifiées. Ici, il est notamment montré que l'obtention de la lépidocrocite n'est observée que lorsque des ions FeII sont encore présents en solution. Ainsi, et contrairement à ce qui a quelquefois été rapporté, la synthèse de cet oxyhydroxyde métastable ne dépend aucunement de la nature de l'anion incorporé dans la RV initiale.
Ensuite, trois articles traitent du rôle joué par les bactéries ferriréductrices dans le cycle biogéochimique du fer. Dans le premier, Berthelin et al. [3] discutent de l'abondance de ces bactéries dans les différents horizons du Réductisol de Fougères, où le minéral fougérite fut découvert et montrent que, là comme ailleurs, ces bactéries coexistent fréquemment avec des populations de bactéries fermentatives qui fournissent aux bactéries ferriréductrices les substrats organiques favorisant leur développement. Ces auteurs décrivent ensuite la séquence de réactions suivant laquelle l'activité d'une souche particulière de bactéries ferriréductrices donne lieu à la dissolution de la lépidocrocite et, finalement, à la biosynthèse d'une RV carbonatée possédant un rapport FeIII/Fetotal semblable à celui fréquemment observé dans la fougérite naturelle.
La contribution suivante traite de la cinétique du phénomène de dissolution des oxydes ferriques à l'intervention des bactéries ferriréductrices. Observant que la réduction de ces oxydes par voie enzymatique n'obéit pas aux modèles cinétiques décrivant leur dissolution en conditions abiotiques, Roden [20] démontre que la densité de cellules bactériennes et l'étendue de la surface “disponible ” des oxydes ferriques sont les deux paramètres-clés contrôlant, tant la vitesse initiale, que l'importance, sur le long terme, de leur réduction. Ainsi donc, il apparaît que, lorsque la réduction des oxydes ferriques est causée par une activité bactérienne, les propriétés thermodynamiques de ces derniers n'exercent qu'une importance secondaire sur la cinétique de leur dissolution.
On sait que les silicates ferriques également sont susceptibles d'être dissous à l'intervention des bactéries ferriréductrices. Néanmoins, lorsque de tels silicates sont des smectites, c'est-à-dire des phyllosilicates développant des surfaces spécifiques considérables, il a été observé que la réduction in situ de leur FeIII structural précède et peut entrer favorablement en compétition avec leur mise en solution. Dans leur article, Stucki et Kotska [24] décrivent les conséquences des changements survenant dans l'état de valence des cations Fe structuraux sur les propriétés physiques et chimiques des smectites qui les subissent, ainsi que sur leur réactivité vis-à-vis de différentes substances organiques. Ils mettent dès lors en évidence le fait que, dans les sols et les sédiments, les smectites également peuvent ajuster rapidement leur cristallochimie aux conditions rédox de leur environnement et qu'en conséquence, il convient de reconsidérer le point de vue selon lequel ces minéraux argileux seraient partout et toujours doués de propriétés de surface stables.
Les RV apparaissent aussi comme des phases transitoires lors de la corrosion de l'acier en milieu aqueux, et ce sujet est couvert ici par l'article de Refait et al. [19]. Deux types de RV sont observés au cours de ce processus: la en présence d'eau douce et la en présence d'eau de mer. En présence d'eau douce, il existe une compétition entre la formation de la et celle de la sidérite, FeCO3. En milieu marin et en dépit d'un concentration très élevée en ions Cl−, la formation de la prend cependant rapidement le pas sur celle de la RV(Cl−). Dans ces conditions, en raison de l'intervention des bactéries sulfato-réductrices qui utilisent les comme source de sulfates, le cycle du fer est alors couplé à celui du soufre.
La précipitation d'hydroxydes ferreux ainsi que celle d'oxyhydroxydes ferriques, d'hydroxysels ou d'oxydes ferrosiques résultent toutes de phénomènes de condensation impliquant des ions fer initialement présents en solution. Dans la dernière contribution de ce numéro thématique, Jolivet et al. [11] décrivent les différentes étapes conduisant à ces précipitations et montrent comment la structure des différentes espèces polymériques présentes en solution oriente la nucléation et la croissance des nombreuses phases solides susceptibles d'apparaître dans le système FeO–Fe2O3–H2O. Cette mise au point jette donc un pont entre la chimie du fer en solution et en phase solide et, à ce titre, se révèle être d'une importance primordiale pour comprendre l'itinéraire de cet élément dans tous les environnements mitoyens entre géosphère, biosphère, hydrosphère et atmosphère.
Foreword
The biogeochemical cycle of iron, i.e. the scheme that describes the circulation of this element between the various compartments of the global environment, owes one of its originalities to two features: its abundance in the Earth's crust and the superficial formations that ensue on the one hand, and, on the other hand, its natural occurrence under the two different states of oxidation, ferrous and ferric. In all compartments lying at the intersection between geosphere, biosphere, hydrosphere, and atmosphere, these two features have major consequences on its behaviour. If these compartments are open to atmospheric oxygen, iron, which is essentially ferrous in primary minerals, would tend to get oxidised and stored in these secondary minerals that Goldsmith used to call hydrolysates. In contrast, if the communication between atmosphere and hydrosphere is momentarily interrupted, iron, whatever its origin, will tend to join the category of soluble cations where the value of its ionic potential ranks in close vicinity of that of Mg2+ cation.
In numerous locations situated at the intersection of the above-mentioned spheres, e.g., in periodically waterlogged soils, ground waters and recent sediments, the availability of atmospheric oxygen is far to be insured permanently or homogeneously. The solubility of oxygen in the hydrosphere is in fact only a few mg l−1. Moreover, its diffusion coefficient in water is about ten thousands times smaller than its value in the atmosphere. However, in all these locations, organic matter, either dissolved or colloidal or as particles, is generally present. It results that wherever oxygen is lacking, ferric iron stored in hydrolysates may become an alternative electron acceptor to insure the respiration of communities of anaerobic or facultative anaerobic bacteria designated as dissimilatory iron-reducing bacteria.
Since these bacteria extract their energy from the decay of organic matter, the reduction of iron and the oxidation of organic carbon are coupled reactions that could be schematically described as follows:
| (1) |
Reaction (1) thus insures the passage of a solid constituent within which ferric cations are immobilised towards a solution containing soluble cations that are mobile. This reaction is irreversible, since a conversion of CO2 into CH2O is only made possible via photosynthesis, and consequently, reaction (1) constitutes one knot where the cycle of carbon meets the cycle of iron.
However, if the medium occurs to be open to atmospheric oxygen, one can also write:
| (2) |
As shown in Fig. 1, Eqs. (1) and (2) are the limits within which the cycle of iron is inscribed between the elements of its hydrolysates and ferrous solutions. However, since reaction (1) gives rise to a mobile cation, Fig. 1 must take into account the path followed by iron from the location where it was mobilised and that where it will be immobilised again. In soils that undergo alternative seasoning of reducing and oxidising conditions, the distance separating these two types of sites may vary from few to few hundreds of millimetres. However, on the long term, for instance on a slope where a groundwater flows down, the repetition of reactions (1) and (2) can lead to a transfer of iron reaching kilometres.
Otherwise, it makes sense that the crystallographic nature of FeOOH solid compounds appearing as a reactant in (1) and as a product in (2) may differ. Iron-reducing bacteria mobilise indeed the iron preferentially contained in poorly crystallised iron oxides [17]. In contrast, the crystallographic nature of the ferric phases that are delivered back by reaction (2) depends upon the local physicochemical conditions [5]. The repetition of the cycle as schematised in Fig. 1 leads often to a modification of the mineralogical composition within the pool of hydrolysates.
In many sites or microsites that are explored by ferrous solutions, the availability of oxygen, as measured by its partial pressure that would appear in the expression describing the equilibrium conditions of reaction (2), is not always sufficient and the precipitation of FeOOH is consequently not observed. In these cases, particularly where the pH conditions are close to neutrality, cations can give rise to the formation of precipitates by following another route.
Two types of alternative reactions have been considered, depending upon whether the anions that insure the electroneutrality of the initial ferrous solution are or are not involved in the phenomenon. By using a formalism similar to that used previously, one can write:
| (3a) |
| (3b) |
The solid compounds that would result from reaction (3a) are known in the literature under the name of ferrosic hydroxides [1]. Among all these various precipitates, one of them, Fe3(OH)8 or , has known some fame among authors who have described the fate of iron in periodically waterlogged soils (e.g., [14,21,23,30]) and in groundwaters as well [13]. In fact, Ponnamperuma et al. [18] had observed previously that during most of the submersion period of paddy soils, the activity of ions in solution would rather correspond consistently with the solubility product of the assumed Fe3(OH)8 as determined by [1]. Neither Ponnamperuma et al. [18] nor authors that followed were ever able to show directly the evidence of the presence of such a hydroxide and this led some authors [12,16,29] to cast doubt on its very existence. For instance, though emphasizing the role played by the precipitation of mixed Fe(II)–Fe(III) compounds in submerged soils, Kirk [12] clearly recognizes that taking their formula as Fe3(OH)8 is only an approximation hiding the many uncertainties left concerning their identities and their actual chemical compositions. Similarly, Langmuir [13] noticed that the domain of stability of Fe3(OH)8 would be so large within a –pH diagram built by taking into account the usual chemical composition of continental waters that the value of the standard Gibbs free energy of formation usually attributed to this ferrosic hydroxide should be likely revisited.
Solid compounds that result from reaction (3b) are better known in the literature under the name of green rusts, GR(), where is the corresponding anion in the chemical formula. This appellation comes from their bluish green colour [10] and their frequent occurrence among the corrosion products of metallic iron. In contrast with the hypothetical ferrosic hydroxides of waterlogged soils, which were considered as amorphous solid phases, green rusts are compounds that present a crystalline organisation. Initiated by the studies of [6] and [2], the determination of their structure and crystal chemistry has been the object of many intensive studies. The last developments about this topic will be presented later into details [8]. It is sufficient here to recall that their crystalline architecture results from the regular stacking of brucite-like layers containing FeII and FeIII cations with interlayers occupied by anions and some water molecules. These structural features rank the green rusts among the wide family of double layered hydroxides and several carbonate-bearing minerals, such as hydrotalcite and pyroaurite, belong to this family.
In spite of notable differences concerning their structure and chemical composition, green rusts and hypothetical ferrosic hydroxides have in common a characteristic bluish-green colour. Moreover, when these compounds would be exposed to the air, this colour would vanish very quickly. For soil scientists, these two properties reminded the observations done in some horizons of water-saturated soils often denominated gleys. Thus, it became sensible to suspect that there exist in these horizons pigments, i.e. unknown minerals responsible for this colour and its fugacious character as well.
In the early 1980s, Taylor [25] and Taylor and McKenzie [27] have widely contributed to advocate the idea that the bluish-green colour of some waterlogged soils would result most likely from the precipitation of carbonated green rusts instead of being due to the presence of a hypothetical ferrosic hydroxide. However, even after having been able to overcome the difficulty inherent to these unstable compounds, Taylor [26] was never able to demonstrate it. One had to wait for 1996 to definitively identify by using essentially 57Fe Mössbauer spectroscopy that the pigments, which tint in bluish green the subsurface horizons sampled from a Gleysol of the forest of Fougères (Brittany, France), had spectral and chemical characters relating them clearly to the green rust family [7,28]. Meanwhile, these authors proposed to give the name fougerite to this new mineral.
As this proposal has recently been endorsed by the International Mineralogical Association (IMA-2003-057), it is now justified to assign to the fougerite the role attributed previously to the ferrosic hydroxides. As illustrated in Fig. 1, in the biogeochemical cycle of iron, these hydroxysalts, which are highly unstable and for this reason always difficult to detect, play the role of a turntable. On the one hand, they are precursors of most ferric oxides or hydroxides that are known. On the other hand, their solubility that is always larger than that of ferric oxides designates them as serious candidates to control the activity of iron species present in the hydrosphere and consequently to influence the fate of most soluble or potentially soluble species that have an eminent agronomical and environmental interest such as, e.g., nitrates, phosphates and heavy metals.
Although the central location of fougerite in this part of the cycle of iron is now clearly identified (see also the more detailed sketch presented recently by Bigham et al. [4]), many questions remain to be clarified. It is thus the purpose of the present thematic issue to report on those dealing more particularly with the crystal chemistry and precise structure of fougerite and its synthetic counterparts along with the description of the chemical and biochemical mechanisms leading to its formation.
In the first paper of this issue, Génin et al. [8] make use of the information obtained from X-ray diffraction and Mössbauer spectroscopy to depict the structural differences occurring in the green rusts (GR) crystals when they incorporate either planar and spherical anions, e.g., and Cl− or tetrahedral anions, e.g., and . They show also that a carbonated GR prepared at stoichiometry, i.e., with a {FeIII/Fetotal} ratio at 0.33, experiences, once carefully dried, a progressive oxidation in the air suffering in situ structural changes that only concern an equivalent deprotonation of its structural hydroxyl groups, thus conserving the stacking of the brucite-like layers and anion interlayers. Since all fougerite samples identified so far in the field are characterised by a {FeIII/Fetotal} ratio comprised between 0.33 and 0.66, this finding is a key observation to explain how this mineral can adjust its crystal chemistry to the environmental redox conditions prevailing in the natural sites where it forms and evolves.
Since the early works devoted to GRs, their in vitro synthesis was the privileged way to study their nature as well as the mechanisms of their formation. In the present issue, the results obtained from two different preparation methods are reviewed and compared. Using the coprecipitation of FeII and FeIII cations under strictly controlled and pH conditions, Ruby et al. [22] describe how a FeOOH precursor transforms into a stoichiometric GR if the appropriate solution conditions are fulfilled. This paper presents also in detail a mass-balance diagram permitting to describe the pathways followed by all types of reaction susceptible to take place and to predict the nature of the solid phase or mixture of solid phases that forms.
Making use of the same diagram, Génin et al. [9] revisit the results of the numerous experiments in which Fe(OH)2 was chosen as the starting compound for the synthesis of GR and other ferrosic and ferric compounds. It is explained why along the pathway followed during the oxidation of Fe(OH)2, GR alone forms only if an excess of FeII cations is initially present in solution. In contrast, Fe(OH)2 alone gives rise to the precipitation of magnetite, which under further oxidation evolves towards maghemite. The products of oxidation of GRs are also identified and it is shown that along their oxidation pathways, the occurrence of the metastable lepidocrocite compound is caused by the presence of FeII cations within solution and not, as it was sometimes reported, by the nature of the anion initially incorporated in the GR.
Next, three papers deal with the role played by the dissimilatory iron reducing bacteria (DIRB) in the cycle of iron. Berthelin et al. [3] report on their abundance in the Gleysol of Fougères where the mineral fougerite was first discovered and show that in this soil as in others, they are frequently associated with populations of fermentative bacteria providing the DIRB with the types of organic substrate favourable to their development. Then, they describe the sequence of reactions by which the activity of a DIRB strain dissolves lepidocrocite and gives rise to the microbiologically-induced precipitation of a carbonated GR whose FeIII/FeII ratio is within the range of those of natural fougerite samples.
The following paper deals with the kinetics of Fe(III) oxides dissolution by DIRB bringing a new light on this topic. Observing that the enzymatic reduction of these oxides does not follow the kinetic models describing their abiotic dissolution, Roden [20] demonstrates that the bacterial cell density as well as the ‘available’ oxide surface are the two key parameters controlling both the initial rate and the long term extent of their reduction. Therefore, the solubility product of the involved oxides appears to play a very secondary role in this biotic dissolution process.
FeIII-bearing silicates also may experience dissolution as a result of the activity of DIRB. However, when the large surface area iron-bearing smectites are concerned, the solid-state reduction of their structural FeIII cations competes favourably with their dissolution. In their paper, Stucki and Kostka [24] describe the consequences of this microbially induced solid-state reaction on the physical and chemical properties of smectites and the reactivity of these clay minerals towards various organic pollutants. Thus they bring to the fore that in sediments and soils, the crystal chemistry of smectites can adjust itself rapidly to the environmental conditions and consequently the view considering these clay minerals as having everywhere stable surface properties should be reconsidered.
Green rusts are also transitory intermediate products during the wet corrosion of steel and this topic is covered here by the paper of Refait et al. [19]. Two types of GR are forming during this process: , the hydroxycarbonate in freshwater, and , the hydroxysulphate in seawater. In freshwater, a competition exists between the formation of and that of siderite FeCO3. In seawater, it is explained why, in spite of the very large concentration of chloride, the sulphated GR takes rapidly precedence over the chlorinated GR(Cl−). The authors show also that where forms as a transitory corrosion product, the cycle of iron becomes coupled to the cycle of sulphur due to the intervention of the sulphate-reducing bacteria that use the GR as a sulphate reservoir.
The precipitation of ferrous hydroxides, FeIII oxides and oxihydroxides, and ferrosic hydroxysalts and oxides as well results from the condensation phenomena of iron cations initially present in solution. In the last paper of this issue, Jolivet et al. [11] describe the different steps involved in these precipitation processes and show how the structure of the various polymeric precursors orientates the nucleation and growth of the numerous crystalline solid phases found in the FeO–Fe2O3–H2O system. This review is thus building a bridge between the solution and solid-state chemistry of iron and it proves to be of primary importance to understand the manifold routes that this element can take in all environments that are situated at the interface between geosphere, biosphere, hydrosphere, and atmosphere.