

1 Introduction
The FeII–III hydroxycarbonate, that is the carbonated form of green rusts, , was discovered as a corrosion product of steel in an urban water pipe [47]. The role of GRs in the degradation of iron-based materials has been underestimated until recently following the discovery of , which indicated that GRs could form in various environments from various materials. For instance, they were observed as corrosion products of mild steels in boiling chloride or sulphate containing solutions [9] or of cast iron in NaCl, K2SO4 and Na2CO3 solutions at room temperature [24]. They were identified later amongst the products of pitting corrosion processes of stainless steels and related iron alloys [6,7]. And finally, the most severe cases of microbially induced corrosion of carbon steels in seawater, at that time attributed to sulphate reducing bacteria, are somehow associated with the hydroxysulphate [14,31].
The estimation of the standard Gibbs free energy of formation of GRs allowed us to draw potential–pH equilibrium Pourbaix diagrams, where the GR compounds are included [11,13,15,33]. It was the first step towards the full understanding of the importance of GRs in corrosion processes. It predicted that in solutions with moderate concentrations () of Cl−, or , the corresponding GR would form in neutral and slightly alkaline media (pH). A series of experimental studies confirmed this analysis. The corrosion of iron and steel in adequate media, either at open circuit potential or under electrochemical polarisation, led to GRs as predicted. The simplest experiment, that is the immersion of an iron disk in solution, was achieved in 0.1 M NaHCO3 and 0.1 M NaHCO3 + 4 M NaCl electrolytes. Homogeneous layers of were observed in each case [1]. Potentiostatic polarisation was applied to an iron electrode immersed in a 1 M KCl solution of pH about 9. Depending on the applied potential value, the main corrosion product was Fe(OH)2 or GR(Cl−), in agreement with the corresponding Pourbaix diagram [38]. Galvanostatic polarisation applied to iron electrodes immersed in various concrete-simulating electrolytes also produced GRs [16]. And finally, was identified in seawater corrosion problems [40,43]. This is due to the fact that even if the seawater molar ratio is about 19, the affinity of the GR crystal structure for divalent anions is stronger, e.g., , than for monovalent anions, e.g., GR(Cl−) [40].
The oxidation of GRs can lead to the most common constituents of rust, that is goethite, lepidocrocite, akaganeite and magnetite, depending on pH, temperature, oxygen flow, dissolved FeII concentration and, more generally, composition of the electrolyte [10–13,15,28,29,32,38,40]. But FeII cations can also be completely oxidised in situ, by leaving essentially unchanged the initial layered structure and leading to what was called ‘ferric green rust’, as obtained by deprotonation of OH− ions that surround Fe cations [34,41]. This new compound was discovered as the result of the action of hydrogen peroxide upon GRs, but was also obtained by oxidation of a dry GR layer [1] or anodic polarisation of steel [22].
In this article, the mechanisms of formation of GRs from steel are discussed. Two main types of aqueous media are considered, the carbonated media representing freshwaters, and the chloro-sulfated media representing seawater. In the first case, the competition between and FeCO3 siderite is stressed. In the second case, the competition between both types of GRs, and GR(Cl−), is studied. Finally, the conditions that favour the formation of ‘ferric green rusts’ are discussed.
2 Methodology
When a steel electrode is corroding in an aerated electrolyte, the potential, called open-circuit potential (OCP), reaches a value somewhere between the potential of FeII/Fe equilibrium and that of O2/H2O equilibrium. The corrosion process can be accelerated electrochemically by increasing either the potential of the electrode, i.e. the potentiostatic procedure, or the current flowing through the electrode, i.e. the galvanostatic procedure. Such experiments can be devised in the laboratory for monitoring the early stages of the corrosion process. In this article, experiments performed at OCP using E24 steel (98.2% Fe, 0.122% C, 0.206% Si, 0.641% Mn, 0.016% P, 0.131% S, 0.118% Cr, 0.02% Mo, 0.105% Ni and 0.451% Cu) are described. The steel surfaces were polished with silicon carbide (particle size 25 μm), rinsed thoroughly with Milli-Q water and carefully dried. The potential was measured using an Ametek (Princeton Applied Research) 263/A potentiostat system. A saturated calomel electrode (SCE) was used as a reference, but the potential is expressed with respect to the standard hydrogen electrode (SHE) in order to facilitate the comparison with potential–pH equilibrium Pourbaix diagrams.
The kinetics of degradation of steel tends to decrease as the metal is covered by a rust layer and the behaviour of a steel structure dipped in water for years is governed by the properties of this several-millimetre-thick layer. The understanding of the mechanisms at such late stages of the corrosion process requires a detailed analysis of the morphology and composition of the rust layers. The case of a steel structure left for 25 years in the Atlantic Ocean is detailed. The identification of transient unstable phases such as GRs is only made possible if the samples are sheltered immediately from oxygen. Therefore, the corrosion layers, about 10–15-mm thick, scrapped from the metal in the permanently immersed zone, 1 m above the mud line, were immediately placed in acetone, preventing any chemical evolution of the samples for 3–4 months. They were coated with epoxy resin (Struers Epofix®), sawed up into 2-mm-thick slices, and again coated with resin. Cuts were achieved so that slices were perpendicular to the steel/rust layer interface. More details concerning this procedure can be found elsewhere [25].
Additional information about the mechanisms of formation and transformation of rust can be obtained via the study of the oxidation of a Fe(II) precipitate in aqueous suspension [10–13,15,33,38,40]. The oxidation processes involved in chloro-sulfated media were then studied using the following methodology. FeCl2⋅4 H2O and FeSO4⋅7 H2O were dissolved in a 100-ml flask of milliQ water. The ratio was set at 1/12. NaOH was dissolved in another 100-ml flask and two ratios were considered, 1 and 0.58. The NaOH concentration was set at 0.4 M, and the temperature at 25 °C. The solutions were mixed, leading to the precipitation of a Fe(II) compound. Magnetic stirring (∼500 rpm) in the open air ensured a progressive homogeneous oxidation of the precipitate and a thermostat controlled the temperature, which was kept at . Reactions were monitored by recording the pH, measured via a glass electrode, and the potential E of a platinum electrode immersed in solution, using the saturated calomel electrode as a reference (but all potentials in the following refer to the standard hydrogen electrode, SHE).
3 Rust layer characterisation
Different characterisation methods must be combined to identify unambiguously the various components of a rust layer. X-ray diffraction (XRD) can be used along with Raman and/or Mössbauer spectroscopy. Here, analyses performed by XRD, conversion electron Mössbauer spectroscopy (CEMS) and micro-Raman spectroscopy are presented.
3.1 CEMS
The decay of the nuclear excited state of 57Fe leads to the emission and subsequent resonant absorption of 14.4-keV photons. These are the photons that are not detected in the Mössbauer effect transmission measurements and correspond to the usual negative peaks of the Mössbauer spectra. The principle of CEMS is based upon the detection of the internally converted electrons emitted after the resonant absorption of γ-rays has taken place. This emission of K-conversion electrons with 7.3-keV energy is detected in backscattering experiments and is adequate for a non-destructive surface analysis. The analysed depth is about 300 nm. A room-temperature spectrum presented here was recorded using a gas flow proportional counter. The constant-acceleration Mössbauer spectrometer was calibrated with an α-Fe disk. The spectra were computer-fitted with a sum of Lorentzian shape lines. Errors on the Mössbauer parameters are about for isomer shift δ and quadrupole splitting Δ and ±2 kOe for hyperfine field H.
3.2 XRD
Products were also analysed by XRD using Co K wavelength () in Bragg–Brentano geometry. Reactive compounds such as GRs are coated with glycerol to avoid any oxidation [18].
3.3 Raman spectroscopy
The Raman study of the marine corrosion products of steel was performed using a multichannel DILOR OMARS 89 spectrometer®. The apparatus is fitted with a diode array detection system, which enables a 500-cm−1 broad spectrum to be analysed for a typical acquisition time of 30 s. Excitation of the samples was carried out with 514.5-nm radiation from a Spectra Physics 2017® argon ion laser. The power of the source was 20 mW and the spectral resolution was .
4 Fresh water and
E–pH Pourbaix diagrams are maps that summarise the thermodynamic information describing the possible routes followed during the corrosion of steels in carbonated aqueous media (Fig. 1). Values of the standard Gibbs energy of formation that are retained for the various species are listed in Table 1 and the equilibrium equations are reported in Table 2. For Fe species, the values were taken from [5,20], or re-computed from solubility products using the value of Table 1. This allowed us to have a complete set of consistent values and equations. The value of FeCO3 was computed from the solubility product () value of −10.80 given by [8], but values ranging from −10.43 to −11.20 are reported [19]. The other values were taken from Wagman et al. [48]. The diagrams were drawn using an activity of carbonate species equal to 0.1. A first diagram was drawn considering , but ignoring FeCO3. It is represented in solid lines. The second diagram was drawn considering both phases. Equilibrium reactions involving FeCO3 are represented in dotted lines. The domain of stability of is totally included inside that of FeCO3, which is delimited by lines (15), (16), (17) and (18). This demonstrates that the hydroxycarbonate is metastable with respect to FeCO3.
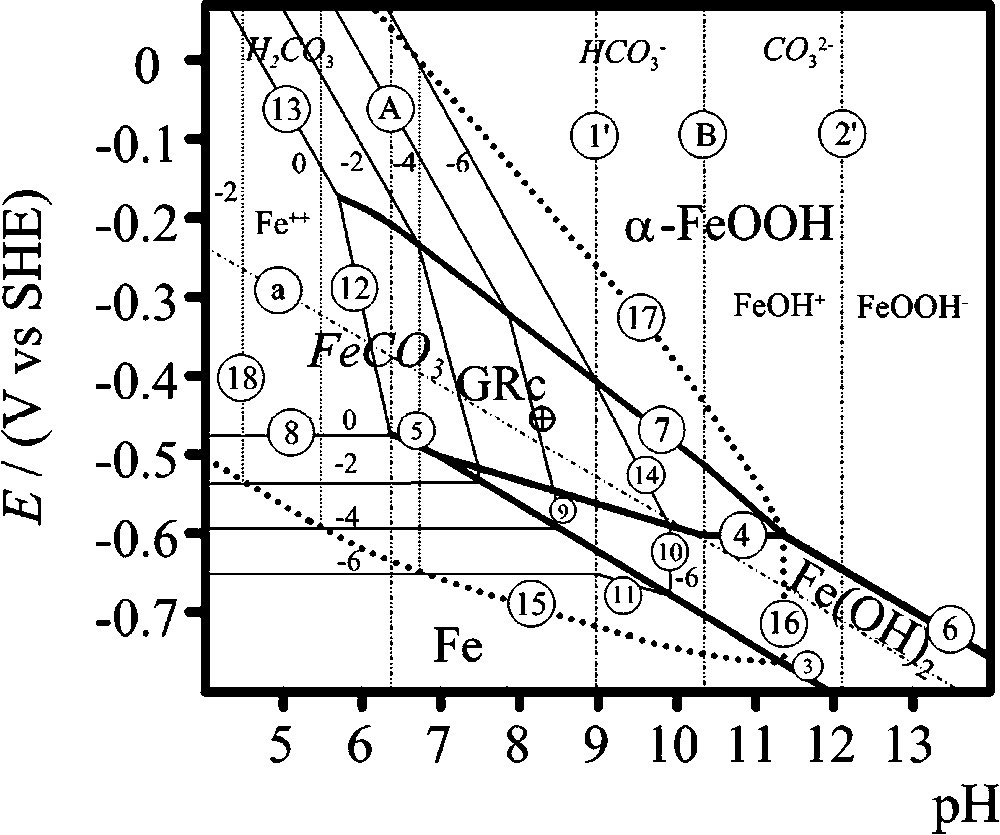
Superimposition of Pourbaix diagrams of iron in carbonate-containing aqueous solution at 25 °C, for an activity of carbonate species of 0.1. GRc designates . The diagram in solid line was drawn omitting FeCO3, the diagram in dotted lines was drawn considering each solid phase.
Superposition des diagrammes de Pourbaix du fer en solution aqueuse carbonatée à 25 °C, pour une activité des espèces carbonate de 0,1. GRc désigne . Le diagramme en ligne pleine est tracé en omettant FeCO3, le diagramme en pointillé est tracé en considérant chaque phase solide.
Gibbs free energies of formation used for calculations in standard temperature and pressure conditions
Enthalpies libres de formation utilisées pour les calculs dans les conditions standard de température et de pression
| Species | Average oxidation number of Fe | (kJ mol−1) | References |
| Solid species | |||
| α-Fe | 0 | 0 | |
| Fe(OH)2(s) | +2 | −492 | [5,20] |
| FeCO3(s) | +2 | −681 | Computed from [8] |
| , that is [12]: | +7/3 | −4076 | Computed from [13] using of Fe(OH)2(s) given above |
| α-FeOOH(s) | +3 | −485.3 | [5] |
| Liquid and dissolved species | |||
| H2O | — | −237.18 | [5,20] |
| +2 | −91.5 | [5,20] | |
| FeOH+ | +2 | −277.4 | [5,20] |
| FeOOH− | +2 | −376.4 | [5,20] |
| H2CO3 | — | −623.2 | [48] |
| — | −586.8 | [48] | |
| — | −527.9 | [48] |
Equilibrium equations of E–pH Pourbaix diagrams drawn in Fig. 1
Équations d'équilibre des diagrammes de Pourbaix E–pH de la Fig. 1
| Water and carbonate system |
|
| Fe–H2O system |
|
|
However, it was reported that homogeneous layers could form on iron disks dipped in 0.1 M NaHCO3 solutions [1]. This experiment was performed again, using a 30-mm diameter E24 steel disk. The OCP of the disk was measured during the 24 h of immersion. It decreased from about down to about , while the initial Fe2O3 film formed in air upon the steel surface dissolved. It stabilised then at as the corrosion of iron proceeded. The green rust layer obtained after 24 h was analysed by CEMS at room temperature in the inert atmosphere of the gas flow proportional counter. The spectrum (Fig. 2) is composed of three spectral components, two doublets due to a paramagnetic compound and one sextet due to a magnetically ordered compound. The hyperfine parameters of this sextet prove to be typical of α-Fe at room temperature. This is the signal coming from the substrate, detected because the rust layer is porous and does not cover completely the metal. The hyperfine parameters of the doublets ( and ) and ( and ) are characteristic of FeII and FeIII atoms in a GR compound [38]. Since only carbonate species were present in the solution, this GR can only be the hydroxycarbonate of chemical formula . The area ratio, which is practically identical to the FeII/FeIII ratio within the GR, is measured at 1.8 (≡35.7% FeIII), very close indeed to the awaited value of 2 (≡33.3% FeIII). Moreover, a slight discrepancy due to the small emission percent comes from ignoring a second ferrous doublet , the intensity of which should be (1/3) that of . Consequently, the value that is computed must lie between 1.5 and 2. The measured value of the OCP is thus reported in the Pourbaix diagram (Fig. 1). The pH of the 0.1 M NaHCO3 solution is buffered at about 8.3 by the hydrogenocarbonate ions and the corresponding point is represented by a ringed cross, which is located at the centre of the domain of stability of .
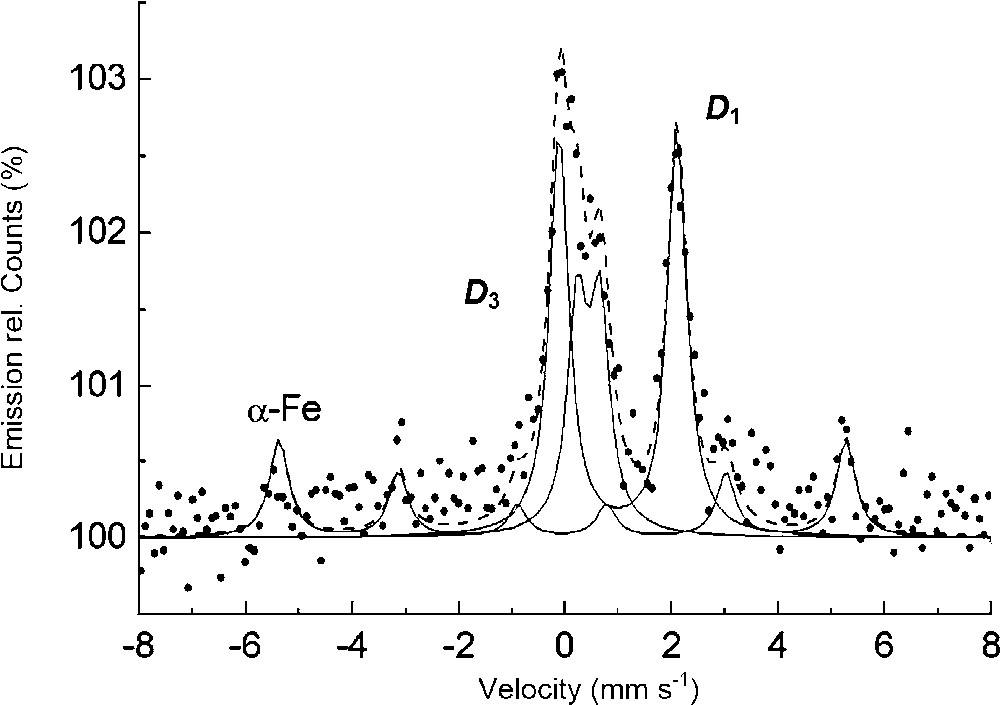
CEMS spectrum at room temperature of a steel disk dipped during 24 h in a 0.1 M NaHCO3 solution. ⋅⋅⋅⋅⋅⋅⋅: Experimental curve, - - - - -: global computed curve, ------: components of the spectra.
Spectre Mössbauer en électron de conversion (CEMS) à l'ambiante d'un disque d'acier trempé 24 h dans une solution de NaHCO3 à . ⋅⋅⋅⋅⋅⋅⋅ : courbe expérimentale, - - - - - : courbe globale calculée, ------ : composantes du spectre.
When NaCl is added to the 0.1 M NaHCO3 solution, the green rust layer resulting of the immersion at OCP of iron disks is still composed of the hydroxycarbonate, even for NaCl concentration of 4 M [1]. This illustrates the well-known stability of the hydroxycarbonate structure with respect to any other form of GRs and in particular those obtained with monovalent anions [26,27,37]. But the results are different if the corrosion process is accelerated electrochemically. Galvanostatic experiments were performed on iron electrodes dipped in a 0.6 M NaHCO3 + 0.5 M NaCl solution of pH about 8.3 [16]. The corrosion product was a mixture of 35% with 65% FeCO3. In this case, siderite FeCO3 forms, whereas it is not obtained at the OCP. Similarly, the dissolution of E24 steel electrodes anodically polarised in a 0.1 M NaHCO3 + 0.02 M NaCl solution mainly leads to FeCO3 [42]. The difference between the behaviour at OCP and the behaviour under anodic polarisation is likely a consequence of the influence of and concentration ratios. If they are small, Fe(OH)2 can precipitate and is rapidly and totally transformed into . An increase of these ratios should favour FeCO3 at the expense of Fe(OH)2. At the OCP, the reduction of O2 produces two OH− ions when the dissolution of iron produces one Fe2+. This should favour . When an anodic polarisation is applied, the reduction of O2 does not match the production of Fe2+ ions, thus favouring FeCO3.
5 Seawater, and microbially influenced corrosion
Ten- to fifteen-millimetre-thick rust layers formed on steel sheet piles during 25 years proved to be composed of three main layers [40]. The inner one, close to the metal substrate, is essentially made of magnetite. The intermediate one is composed of iron(III) oxyhydroxides. Finally, in the external one, the closest to the interface with seawater, the sulphated form of GRs proved to be the main constituent (Fig. 3). A Raman spectrum of this is displayed (Fig. 4). In agreement with the previous Raman studies of GRs [6,7,21,45], it is composed of two intense peaks at 430 and , that were attributed to FeII–OH and FeIII–OH stretching, respectively [6,7]. The weaker band at was more rarely mentioned [21]. Chemical analyses demonstrated that the GR contained elements Fe, O and S, indicating that it was the hydroxysulphate [40]. However, the molar ratio in seawater is about 19. This illustrates once more the affinity of the GR structure for divalent anions. Note that in seawater the main carbonate species is but is overwhelming, since molar ratio is about 12. This explains why forms even though produces a better stability of the layered structure of GRs [26,27,37].

(a) SEM micrograph showing hexagonal shaped crystals of upon corroded steel sheet left 25 years in seawater and (b) sequence of the rust layers: metal–magnetite–lepidocrocite– [40].
(a) Micrographie par microscopie électronique à balayage montrant des cristaux hexagonaux de rouille verte carbonatée sur une feuille d'acier corrodé abandonnée 25 ans dans l'eau de mer et (b) séquence des couches de rouille : métal–magnétite–lépidocrocite–rouille verte sulfatée [40].

Raman spectrum of the outer part of a marine corrosion rust layer formed on steel.
Spectre Raman de la partie externe d'une couche de rouille formée par corrosion marine sur l'acier.
Additional experiments were performed in the laboratory in order to study the competition between GR(Cl−) and . The oxidation of a precipitate obtained by mixing solutions of FeCl2⋅4 H2O, FeSO4⋅7 H2O and NaOH was studied. The molar ratio was set at 12, whereas two {[Fe2+]/[OH−]} ratios, 0.58 and 1, were considered. The redox potential E and pH versus time curves are displayed in Fig. 5. Those obtained for {[Fe2+]/[OH−]}=0.58 are typical of a two-stage reaction involving the formation of a GR compound as an intermediate product. The first stage elapses from to and corresponds to the formation of a GR from the initial FeII compound, the second one elapses from to and corresponds to the oxidation of the GR into FeOOH phases releasing FeII and anions into solution. Large E and pH variations of around and testify of the disappearance of an initial phase [38,39]. In contrast, the curves obtained for {[Fe2+]/[OH−]}=1 indicate that the reaction involves a supplementary stage, a first one ending at , a second one at and the third one at .

(V vs. SHE) and pH vs. time curves obtained during the oxidation of aqueous suspensions of Fe(II)-containing precipitates in the presence of Cl− and ions. . (a) [Fe2+]/[OH−]=0.58; (b) [Fe2+]/[OH−]=1.
Courbes (V par rapport à l'électrode standard à hydrogène, SHE) et pH en fonction du temps, obtenues au cours de l'oxydation de suspensions aqueuses de précipités contenant du FeII en présence d'ions Cl− et . . (a) {[Fe2+]/[OH−]}=0.58 ; (b) {[Fe2+]/[OH−]}=1.
Intermediate compounds were analysed by XRD. Patterns are displayed in Fig. 6. The GR found at for {[Fe2+]/[OH−]}=0.58 is identified as (Fig. 6a). The three main diffraction lines at (1.1 nm), 18.69° (0.55 nm) and 28.22° (0.367 nm) are characteristic of a hydroxysulphate XRD pattern and correspond to (001), (002) and (003) lines of the trigonal structure [46]. Some lines of lepidocrocite γ-FeOOH, end-product of the oxidation process and faint lines of of other indices than are also detected. The abnormal intensity of the lines is due to preferential orientation of micro-crystallites. Thus, in this case, where and {[Fe2+]/[OH−]}=0.58, forms instead of GR(Cl−) even though is in minority, a consequence of the affinity of GRs for divalent anions. The XRD pattern of the product obtained at for {[Fe2+]/[OH−]}=1 is almost identical to the previous one (Fig. 6b). is also obtained here. However, the XRD pattern of the product obtained at is mainly composed of the diffraction lines of GR(Cl−) (Fig. 6c). They are denoted R1. Two of them, at (0.795 nm) and 25.98° (0.398 nm), are extremely intense and correspond to (003) and (006) lines of the conventional hexagonal cell of the rhombohedral structure [38]. Therefore, the first reaction stage corresponds to the formation of GR(Cl−) from the initial precipitate, the second stage to the oxidation of GR(Cl−) into and the last stage to the oxidation of into γ-FeOOH. The diffraction lines of , denoted R2, are seen, together with those of GR(Cl−), as a result of the oxidation of GR(Cl−). Similarly, the two main diffraction lines of γ-FeOOH, denoted L, can be noticed.
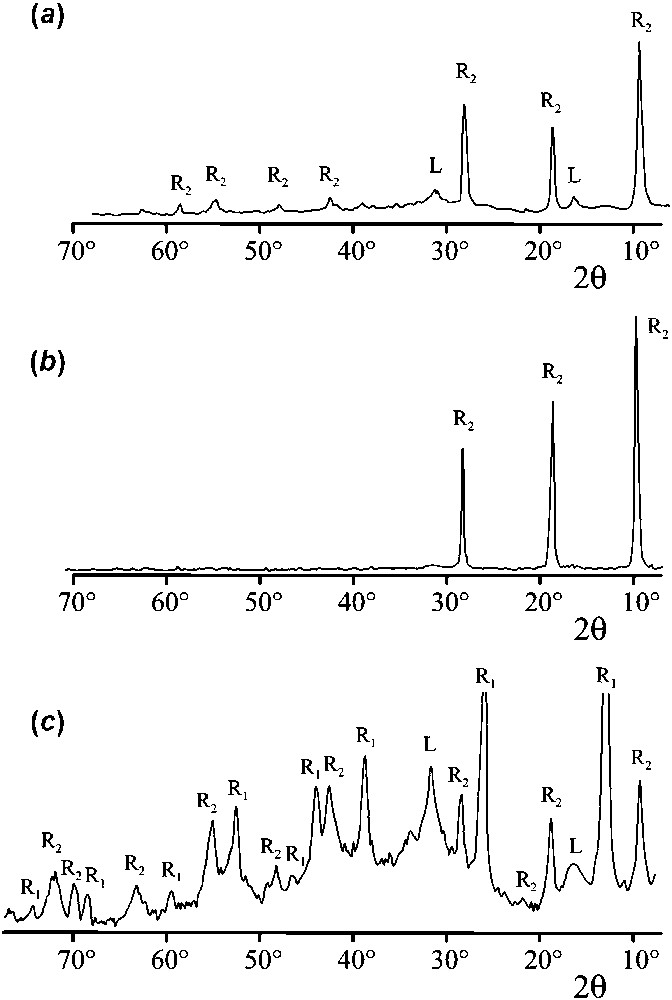
XRD patterns of the intermediate products obtained by oxidation of aqueous suspensions of Fe(II)-containing precipitates in the presence of Cl− and ions. . . (a) Product obtained at for [Fe2+]/ [OH−]=0.58; (b) product obtained at for [Fe2+]/[OH−] =1; (c) product obtained at for [Fe2+]/[OH−]=1. R1 are the lines of GR(Cl−), R2 are the lines of , and L are diffraction lines of lepidocrocite.
Clichés de diffraction des rayons X des produits intermédiaires obtenus par oxydation de suspensions aqueuses de précipités contenant du Fe(II) en présence d'ions Cl− et . . . (a) Produit obtenu à pour [Fe2+]/[OH−]=0,58 ; (b) produit obtenu à pour [Fe2+]/[OH−]=1 ; (c) produit obtenu à pour [Fe2+]/[OH−]=1. R1 désigne les raies de GR(Cl−), R2 celles de , et L celles de la lépidocrocite.
proved to have a well defined composition of [15,39,46]. The average oxidation number of iron is then +2.33. In contrast, it was observed that the composition of GR(Cl−) varied continuously [38] as the oxidation advanced. Starting from a compound with formula , chloride and FeIII contents increased, up to an approximate composition of . The average oxidation number of iron varied then from +2.25 to +2.31. We may propose that in the presence of anions, the enrichment in Cl− and FeIII is replaced by a transformation of into and obviously sulphate anions deliver a phase that is more stable than that obtained with chloride anions. The differences observed between the oxidation processes at {[Fe2+]/[OH−]}=1 and 0.58 may result from the nature of the initial precipitate. When {[Fe2+]/[OH−]} is small, close to 0.5, the value corresponding to the stoichiometric conditions of formation of Fe(OH)2, the initial precipitate is indeed Fe(OH)2 or close to Fe(OH)2 [35]. When {[Fe2+]/[OH−]} is larger, Fe(II)-hydroxychlorides may form [35]. At {[FeCl2]/[NaOH]}=2.5, the initial precipitate was identified as β-Fe2(OH)3Cl [36]. This compound gets oxidised the way Fe(OH)2 does into , and we may suppose that, even in the presence of sulphate ions, the oxidation of such an hydroxychloride leads to GR(Cl−). The oxidation of β-Fe2(OH)3Cl into could well be a solid-state reaction.
We demonstrated previously that in marine environments the dissolution of iron in seawater should lead to . Some severe cases of corrosion are due to the concomitant presence of sulphate-reducing bacteria (SRB) and of among the corrosion products [14,30]. The recent paper that described carefully the sequence of rust stratification on steel sheet piles at the anoxic level of sea mud suggests that it is due to bacterial reduction of previously formed lepidocrocite where large hexagonal crystals are clearly identified (Fig. 3) [40]. We thus propose that forms firstly due to the reduction of γ-FeOOH in the outer layer by ubiquitous dissimilatory iron-reducing bacteria (DIRB). Then, the zone enriched in is favourable to the colonisation of the interface by other micro-organisms, the sulphate-reducing bacteria (SRB). SRBs then induce formation of H2S, which is an acidification of the environment, which results in a drastic increase of iron degradation. Microbially influenced corrosion of steels would then be a two-step process involving DIRB that form followed by the reduction of this sulphate reservoir by SRBs.
6 Oxidation of GRs and formation of rust
Other phases can also be obtained from GRs in more specific conditions. Akaganeite forms from GR(Cl−) in solutions containing a large excess of and Cl− [36]. Ferrihydrite was also reported to form from GR(Cl−), when the Fe concentration is very low and the oxidation kinetics fast [41]. Phosphate species modify considerably the oxidation process of aqueous suspensions of . For instance, in a first article, the final product was unambiguously identified as ferrihydrite [3], but more recently it was concluded that this product was a ‘ferric green rust’, that is a FeIII compound characterised by a layered structure similar to that of GRs [23]. The existence of such ‘ferric green rusts’ demonstrates clearly that FeII cations can be oxidised in situ and that the structure of GRs can sustain up to 100% FeIII [17,34,41]. In order to compensate for the increase in positive charge, the oxidation of FeII is accompanied by a deprotonation of the OH− ions of the brucite-like layers. Ferric GRs can be obtained by oxidation of GRs with hydrogen peroxide [4,34,41], electrochemical polarisation of iron [21] or aerial oxidation of dried GR layers [1]. The formation of the ferric was reinvestigated here in the case of steel corrosion.
First, a layer was grown on a steel electrode left at OCP in a 0.1 M NaHCO3 solution, as described here in part 3. The sample was removed from the solution and left in the dry atmosphere of the laboratory. The deep-green corrosion layer turned progressively to brown and was analysed one month later by XRD. Secondly, a 10-ml solution containing approximately 30% H2O2 was added to an aqueous suspension of prepared with the procedure described previously [3]. The suspension turned immediately to brown and the precipitate was filtered, dried to powder and analysed by XRD. Both XRD patterns are presented in Fig. 7.
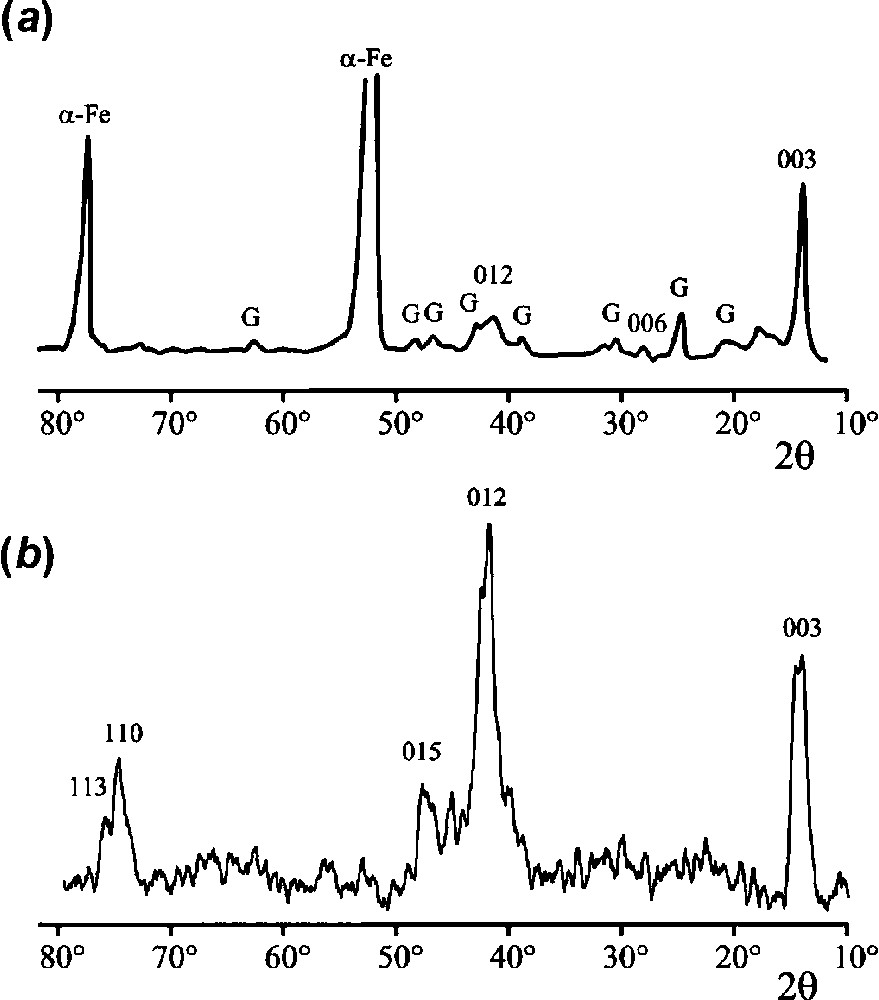
XRD patterns of the ‘ferric ’. The lines denoted by their Miller indices hkl can be ascribed to the ‘ferric green rust’ (see text). . (a) Rust layer obtained by aerial oxidation of a layer formed on steel after 5 days of immersion in a 0.1 M NaHCO3 solution. G are the diffraction lines of goethite and α-Fe those of the substrate. (b) Compound obtained by addition of H2O2 to an aqueous suspension of .
Clichés de diffraction de rayons X de la « rouille verte ferrique ». Les raies désignées par les indices hkl peuvent être attribuées à la rouille verte ferrique (voir le texte). . (a) Couche de rouille obtenue par oxydation à l'air d'une couche de rouille verte carbonatée, formée après 5 jours sur un acier dans une immersion en une solution de NaHCO3 à . G désigne les raies de la goethite et α-Fe celles du substrat. (b) Composé obtenu par adition de H2O2 à une suspension aqueuse de rouille verte carbonatée.
The two most intense lines visible in the pattern of the dried rust layer are those of the substrate α-Fe. Lines of goethite α-FeOOH are also observed, together with three other lines that can be attributed to the ferric GR. The main one corresponds to an interplanar distance of 0.735 nm (), that is the distance between interlayers. The occurrence of this peak in the pattern of the ‘ferric GR’ definitively proves that the sequence among hydroxide and anionic layers is conserved in the structure. The decrease of from 0.750 nm in [13] to 0.735 nm in ‘ferric GR’ is attributed to the decrease of the ionic diameter of iron species, from 0.156 nm for FeII to 0.129 nm for FeIII [44]. Two other peaks are visible, at 0.3675 nm () and 0.254 nm ().
The pattern obtained by oxidation of with hydrogen peroxide is composed of five main lines that can all be attributed also to ‘ferric ’. Assuming that the ferric GR keeps the pyroaurite structure of [2], it is computed that the observed diffraction lines are indeed associated with intense lines of the GR structure. The parameters of the conventional hexagonal cell are determined at and . The corresponding Miller index (hkl) was then used to identify the ‘ferric GR’ diffraction lines on the patterns of Fig. 6. However, it is not possible to exclude that ferrihydrite forms together with the ferric GRs if the conditions of corrosion are inhomogeneous as it is surely in actual cases. As a matter of fact, the most poorly ordered form of ferrihydrite, the so-called two-line ferrihydrite, is characterised by an XRD pattern made of two broad lines at and 74°. Intense lines of the ferric GR are also located in these angular regions and may mask those of ferrihydrite.
7 Conclusion
The determining role of GRs in the corrosion processes of ferrous alloys in neutral and alkaline media is now well settled. FeII–III hydroxysalts constitute a major step before the formation of the various components of rust and may control partially the properties of the rust layers formed on steel. Therefore, the positive role of various corrosion inhibitors commonly used to prevent degradation of steels in neutral or alkaline media are partially explained by the influence those species may have upon the formation and/or transformation of GRs. Phosphates, nitrites and nitrates are examples of such inhibitors.