

1 Introduction
Iron is an important element in environment, because it is abundant in the Earth's crust. It is present in soils, essentially as oxides [9], and easily mobilized in natural waters under the form of molecular complexes and colloids. Dissolution of ferric oxides in natural conditions (including acid-base and redox phenomena, microbial mediation, and photochemistry) is of major importance in the cycling of iron [46]. Iron oxides are also present in living organisms (e.g., plants, bacteria, molluscs, birds, animals and humans). Ferrous complexes are the active centre of haemoglobin and ferredoxins, and various biomineralization processes involve ferrous and ferric species for the regulation of iron concentration in organism (ferritin) or, in various occurrences, to produce different oxides, such as goethite, lepidocrocite, magnetite and green rusts. Goethite, lepidocrocite and magnetite are the mains constituents of radulas of limpets and chitons, magnetite is synthesised by magnetotactic bacteria for their guidance and orientation in various aqueous media and in sediments [32]. Of course, the iron oxides are also of high interest in various technological fields, such as metallurgy, coloured pigments, magnetic materials, catalysts... [9].
The structural chemistry of iron oxi(hydroxi)des is very rich, as shown by the large number of structural types for ferrous, ferric and mixed-valent compounds that exist (Fig. 1) [9,58]. Almost all these phases can be formed from solutions giving rise to a puzzling chemistry [9,20,34], and because of the great diversity of physicochemical conditions in the environment (acidity, redox conditions, bacterial activity, temperature, salinity, presence of organic or inorganic ligands...), practically all the iron oxide phases can be found in the natural environment. The high versatility of iron chemistry in aqueous medium comes essentially from two factors: the occurrence of two oxidation states, stable on a large range of acidity, and the high reactivity of iron complexes towards acid-base and condensation phenomena.
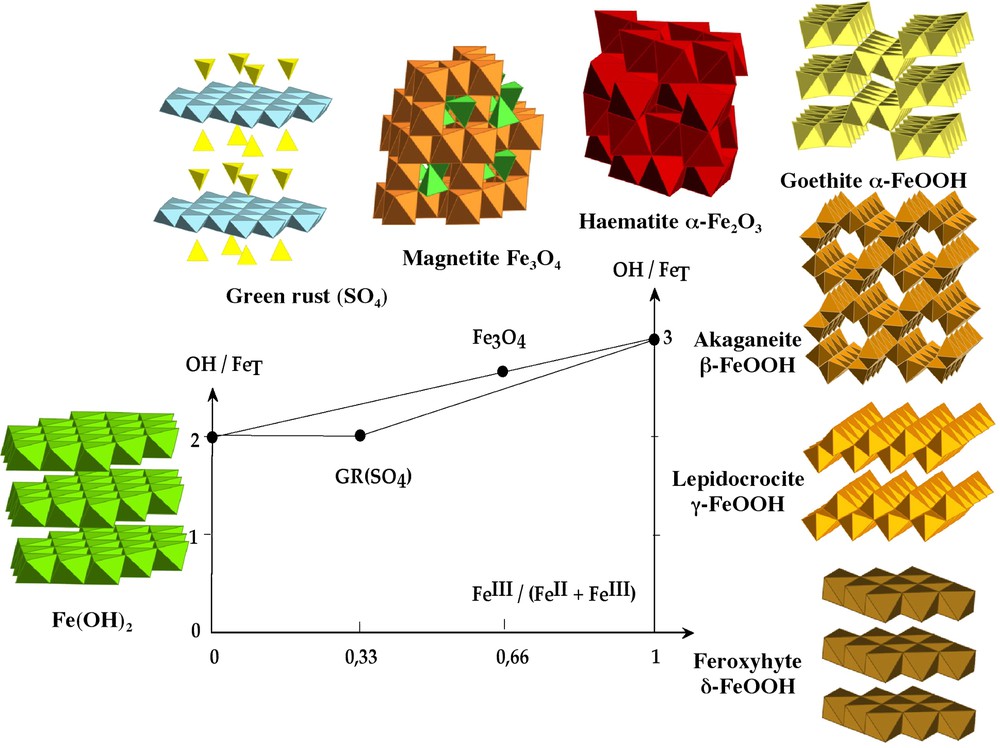
The different phases of iron oxyhydroxides formed as a function of hydroxylation ratio and the composition in the ferrous–ferric system.
Représentation des différentes phases des oxyhydroxydes de fer formés en fonction du taux d'hydroxylation et de la composition dans le système ferreux–ferrique.
We present the main aspects of iron chemistry in aqueous medium involving condensation phenomena and leading to the formation of clusters or nanoparticles. The main factors orienting the crystallization process of ferrous, ferric and mixed-valent solids are examined.
2 Hydroxylation and condensation mechanisms in aqueous solution
Like many transition elements in water, the iron cations form hexacoordinated aquo complexes, [Fe(OH2)6]z+, in which the polarization of coordinated water molecules is strongly dependent on the formal charge (oxidation state) and size of the cation. This makes the ferric aquo complexes strongly more acidic than ferrous complexes; therefore hydroxylation of the cations occurs on very distinct ranges of pH: hydroxylation of Fe(II) cation occurs at room temperature around pH 7–9 while it ranges from pH 1 to pH 4–5 for Fe(III) cation [2].
Hydroxylated complexes are not stable as ‘monomeric’ species in solution; they condense via two basic mechanisms, depending on the nature of the coordination sphere of the cations [20]. In all cases, condensation involves a substitution mechanism induced by the nucleophilic character of hydroxo ligand. For aquohydroxo complexes,
Because of the high lability of coordinated water molecules, olation is more often a very fast reaction. For oxohydroxo complexes,
The hydroxylation rate, h, of the complexes represents their functionality towards condensation and it controls the type and structure of condensed species. h is obviously a function of the pH of the medium. Its also depends on the characteristics of the cation such as size, formal charge and electronegativity. As long as
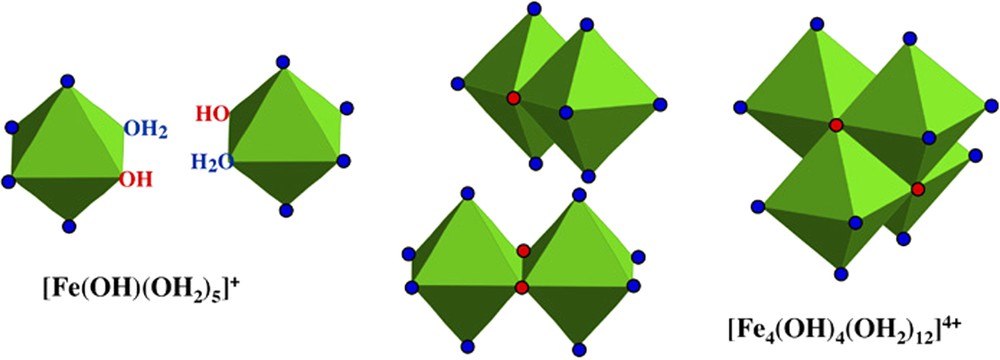
Schematic formation of the ferrous polycation [Fe4(OH)4(OH2)12]4+ from complexes in solution.
Schéma de la formation du polycation ferreux [Fe4(OH)4(OH2)12]4+ à partir de complexes en solution.
3 Formation of solid phases
The precursor of the solid is always a zero-charge aquo hydroxo complex. One may understand the extremely different crystal chemistry of the two cations Fe(II) and Fe(III) not only from the functionality of the precursor, but also from the process involved in its formation, for instance by addition of a base or by thermolysis at temperature in the range 60–100 °C in acidic medium.
3.1 Ferrous compounds
Zero-charge ferrous complexes [Fe(OH)2(OH2)4]0 formed at pH > 6–7 under anaerobic conditions lead to the hydroxide Fe(OH)2. Like the hydroxides of many divalent elements, Fe(OH)2 has the brucite structure. It is easy to understand how this structural type is formed (Fig. 3) [20]. As there is no structural relationship between the compact tetramers (Fig. 2) and the hydroxide, the nuclei of the solid phase seems to be planar tetramers [M4(OH)8(OH2)8]0 formed by olation from neutral zero-charge dimers. Because of the bifunctionality of the precursor [Fe(OH)2(OH2)4]0, these planar tetrameric species can further condense and the growth of nuclei, always by olation, must take place rapidly in the plane and leads to the layered brucite-type structure. This example shows very well that the polycations eventually formed during the hydroxylation process are not building blocks for the solid phase. The change in acidity of the medium leads to an important change in the structure of the condensed species during the lowering of their charge. As a rule, there is no structural relationship between the solid and the polycations formed at lower hydroxylation stages.
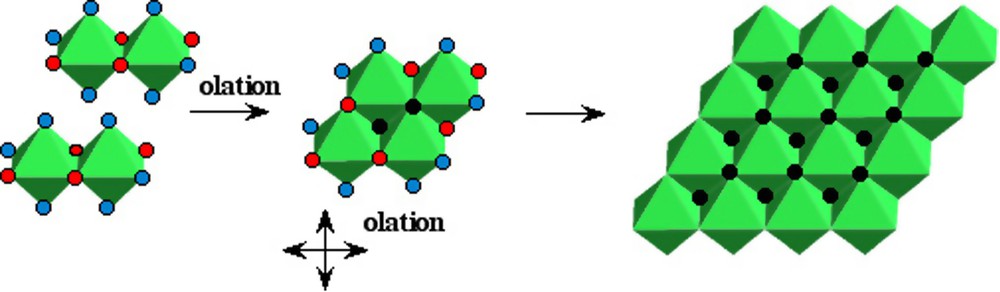
Possible pathway of the formation of ferrous hydroxide Fe(OH)2 from complexes in solution.
Chemin réactionnel possible pour la formation de l'hydroxyde ferreux Fe(OH)2 à partir de complexes en solution.
In the solid state or in aqueous suspension, the ferrous phases are very sensitive to oxidation. Various phases (green rusts, magnetite, goethite, lepidocrocite, and feroxyhyte) can be formed, depending on the conditions, as discussed below.
3.2 Ferric compounds
The behaviour of ferric species in solution exemplifies very well the complexity of their structural chemistry. Hydroxylation of ferric ions in solution by addition of base at room temperature (pH > 3) leads quasi instantaneously to 2-line ferrihydrite, a poorly defined highly hydrated phase exhibiting only two broad bands in its X-ray diffraction pattern [18,45]. Due to its poor structural organization, ferrihydrite is thermodynamically unstable. It transforms into different crystalline phases via different pathways depending on the acidity of the medium. At
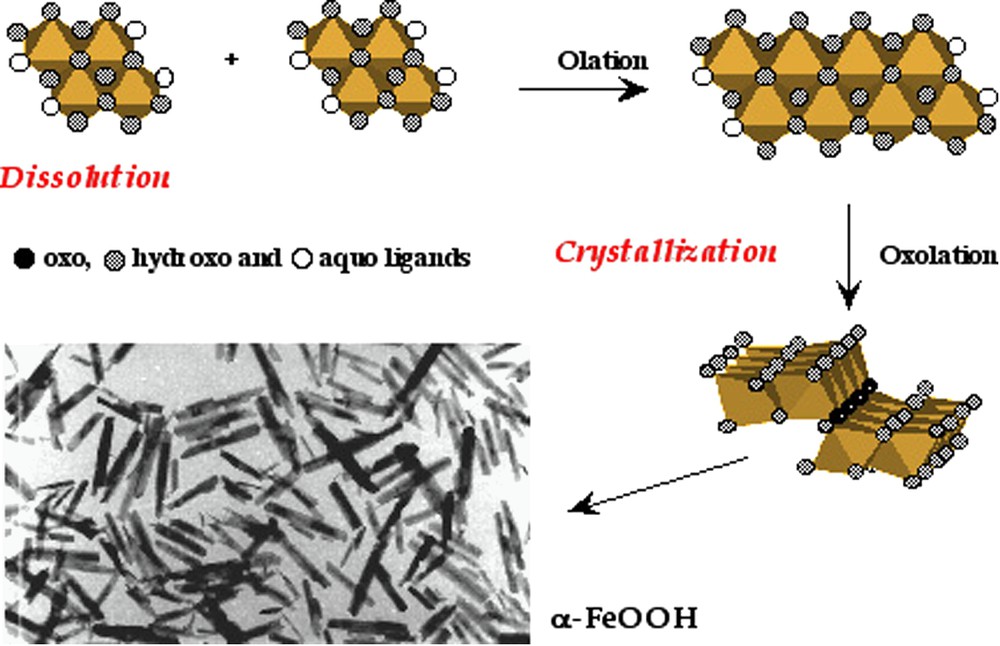
Possible pathway of the formation of goethite α-FeOOH and electron micrograph of particles. From [20].
Chemin réactionnel possible de formation de la goethite α-FeOOH et cliché de microscopie électronique des particules. D'après [20].
Haematite forms by thermolysis of acidic ferric solutions at 90–100 °C (pH ⩽ 3), [9,34]. In these conditions, oxolation is facilitated and leads to oxide [20]. Very small particles of 6-line ferrihydrite form initially at low concentration of chloride (
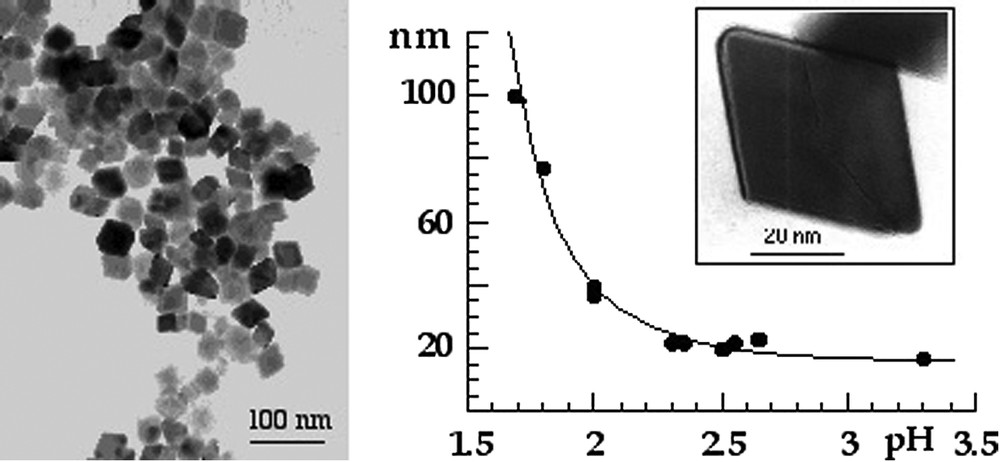
Electron micrographs of haematite particles obtained by thermolysis at 95 °C of ferric nitrate solutions and variation of the particle size as a function of the pH of solutions. From [17].
Clichés de microscopie électronique de particules d'hématite obtenues par thermolyse à 95 °C de solutions de nitrate ferrique et variation de la taille des particules en fonction du pH des solutions. D'après [17].
It is worth noting that the unique way to form lepidocrocite, γ-FeOOH, a ferric oxyhydroxide isostructural with boehmite γ-AlOOH, is the oxidation of Fe(OH)2 by air in suspension at pH around 7 [9]. Oxidation occurs at the minimum of solubility of ferric complexes ruling out a dissolution–crystallization process for the structural transformation. It presumably results from solid-state reaction involving local rearrangements of chains of octahedrons forming the corrugated planes of the oxyhydroxide phase. Fragmentation of initial ferrous hydroxide particles forms lath-shaped particles. If oxidation proceeds around pH 7–8 but very fast with H2O2, feroxyhyte, δ-FeOOH, is obtained without any structural rearrangement, even if stacking defects and tetrahedral cations are the more often present [9]. Like lepidocrocite, feroxyhyte is never formed from ferric species. It seems that ferrous ions are oxidized in situ and the hydroxide sheets are simultaneously partially deprotoned, leading to feroxyhyte as the memory of ferrous hydroxide.
3.3 Ferric-ferrous mixed phases: green rusts and magnetite
The presence of ferrous and ferric ions simultaneously in solution orients the condensation process to the formation of specific phases, namely the green rusts, of hydrotalcite structural type, and magnetite or maghemite, of spinel structural type. The formation of these mixed phases depends on many factors such as the iron concentration, pH and especially the composition of the system defined as x=[Fe3+/(Fe2+ + Fe3+)] (Fig. 1). These phases are of high importance because green rusts are reactive intermediate products in aqueous corrosion of iron and are abundantly found in reductomorphic soils. Spinel oxides are ferrimagnetic materials usable for many technological applications. All these phases can have a more or less broad range of composition, x. For green rusts, x is near of 0.33 while x is equal to 0.66 for magnetite. A special feature of these phases lies in the fact that electrons are mobile at room temperature into spinel structure leading to a Fe2.5+ state for octahedral cations, while there is no such electron hopping through the bi-dimensional framework of green rusts.
3.3.1 Green rusts
For
There is a very close structural relation between ferrous hydroxide, green rusts and feroxyhyte. Continuity could be expected in the formation of these phases by hydroxylation of cations in solution, but such continuity does not exist. The reason seems to come from kinetic and electronic reasons. For mixtures with an x ratio higher than 0.33, electron mobility between ferrous and ferric ions drives the spinel crystallization as seen below. Precipitation of ferric ions alone forms goethite and feroxyhyte is obtained only from very fast oxidation of ferrous hydroxide. There is a chromium phase, CrOOH, with a structure very similar to that of feroxyhyte, obtained by thermolysis at 90 °C of acidic solutions. This phase probably results from the chemical inertness of Cr3+ ions towards substitution reactions and, consequently, olation and oxolation reactions are kinetically equivalent [20] so that two-dimensional growth of planes occurs instead chains for goethite (Fig. 4). In fact, ferric ions in solution seem to be too reactive towards condensation and the δ-FeOOH phase cannot be formed directly by precipitation.
3.3.2 Magnetite
Magnetite Fe3O4 is easily obtained by coprecipitation of aqueous Fe3+ and Fe2+ ions with
Nanoparticles of magnetite are very sensitive to oxidation and transform into maghemite, γ-Fe2O3. The high reactivity is obviously due to the high surface-to-volume ratio, but aerial oxidation is not the only way to go to maghemite. Due to the high electron mobility in the bulk, magnetite nanoparticles give rise to an interesting surface chemistry. Different interfacial ionic and/or electron transfers depending on the pH of the suspension can be involved in the transformation. In basic medium, the oxidation of magnetite proceeds by oxygen reduction at the surface of particles (electron transfer only) and coordination of oxide ions, while in acidic medium and anaerobic conditions, surface Fe2+ ions are desorbed as hexaaquo complexes in solution (electron and ion transfer) according to [21]:
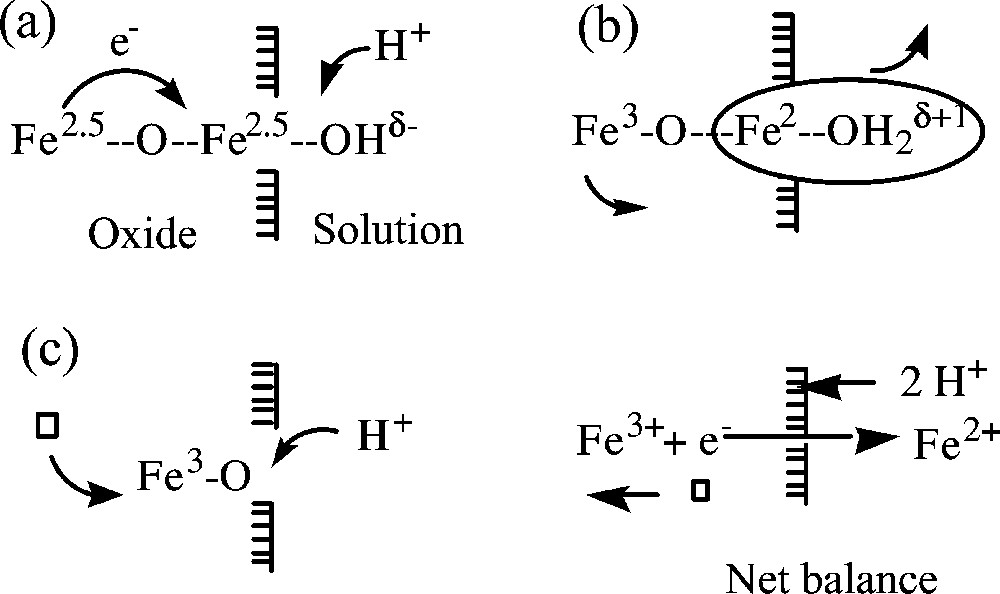
Sketches of the oxidation of magnetite into maghemite in acidic suspension: (a) by protonation of a surface oxygen atom, a mobile electron is localized on a superficial octahedral iron ion; (b) in these acidic conditions (pH around 2), the iron (II) ion formed is released as a soluble hexaaquocation leaving an excess of positive charge inside the particle; (c) the excess of charge is compensated by moving a ferric ion towards the surface in order to create a cationic vacancy in the lattice. The net balance of elimination of Fe(II) ions is the oxidation of material and diffusion of cationic vacancies towards the interior of particles. Masquer
Sketches of the oxidation of magnetite into maghemite in acidic suspension: (a) by protonation of a surface oxygen atom, a mobile electron is localized on a superficial octahedral iron ion; (b) in these acidic conditions (pH around 2), the iron ... Lire la suite
The mobility of electrons on the octahedral sublattice renews the surface ferrous ions allowing the reaction to go to completion. The oxidation in acidic medium (pH ≈ 2) does not lead to noticeable size variation. This reaction is apparently reversible. Adsorption of ferrous ions on maghemite particles by raising the pH proceeds so that the composition of magnetite (
Maghemite particles resulting from oxidation of magnetite can easily form very stable and concentrated aqueous dispersions. In acidic medium and at low ionic strength, maghemite nanoparticles carry a high positive charge density and so are well dispersed in water, forming sols practically free from aggregation [22,26,38]. The formation of such stable sols is a key step to the preparation of magnetic materials such as ferrofluids [1,10,11,13,30]. The dispersion of particles in organic or inorganic matrices enabled the synthesis of various magnetic composite materials well-suited for studies on magnetism of nanoparticles, including measurements on unique particle [56]. Thermal treatment of silica-based composites led to the formation of a less common phase of iron oxide. Silica glass composites were prepared by polymerizing silicic acid or alkoxysilanes inside the aqueous sol of maghemite [7]. Hydrolysis and condensation of silanols take place in situ, forming a gel and a transparent monolithic glass after drying at room temperature (Fig. 6).
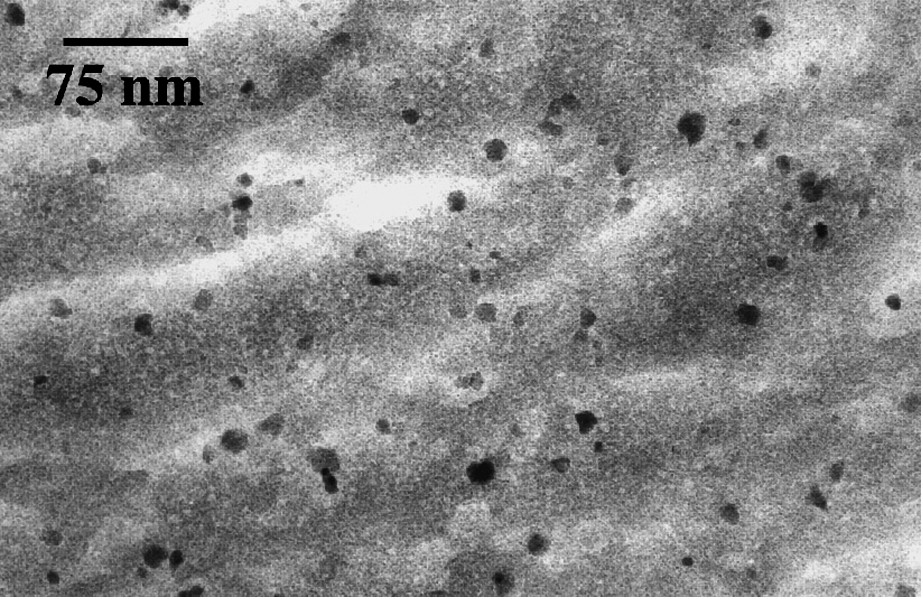
Transmission electron microscopy image of γ-Fe2O3–18wt% silica composite.
Cliché de microscopie électronique en transmission du composite silice–γ-Fe2O3 18% en masse.
The silica matrix acts as an anti-sintering agent that stabilizes the maghemite particles against the thermal transformation into haematite. In composites with sufficiently low particle concentration, no transformation occurs as long as the matrix prevents the migration and coalescence of particles, that is, up to around 1000 °C, the temperature at which the glass softens or starts crystallizing. By heating at 1200 °C, the matrix partly crystallizes into cristobalite and the major iron oxide phase is the rarely observed polymorph ɛ-Fe2O3 [55]. The particles are spherical in shape (Fig. 7) and correspond to a few tens of maghemite nanoparticles in volume. ɛ-Fe2O3 isotype with kappa alumina, is ferrimagnetic with a high coercivity [19]. It is probably piezoelectric.

TEM micrograph of silica composite heated at 1200 °C and crystal structure of ɛ-Fe2O3.
Cliché de microscopie électronique en transmission du composite silicique chauffé à 1200 °C et structure de ɛ-Fe2O3.
At 1400 °C, the crystallization of the matrix is complete and iron oxide is transformed into haematite in the form of crystals larger than those of ɛ-Fe2O3 by at least one order of magnitude. All observations indicate that ɛ-Fe2O3 forms as a result of the sintering of a limited number of γ-Fe2O3 nanoparticles. With powdered uncoated particles, the

Crystal structure of β-Fe2O3.
Structure cristalline de β-Fe2O3.
4 Conclusions
This overview emphasizes the high versatility of aqueous iron chemistry. It shows how to control the crystalline structure and offers many possibilities for tailoring materials for a wide range of utilizations. The careful control of the size and degree of dispersion of the particles in composite materials may reveal unexpected phenomena.
Acknowledgements
The authors wish to thank their collaborators P. Belleville, P. Prené, L. Vayssières, and J. Hernandez. Prof. J.-M. Génin (LCPME, ‘Université Henri-Poincaré, Nancy, France) is gratefully acknowledged for fruitful discussions.