

1 Introduction
Il a été montré récemment que les 1-hydroxytryptophanes I jouent un rôle très important dans le système nerveux central, comme intermédiaires de biosynthèse de divers alcaloïdes et acides aminés 〚1〛, bien qu’aucun 1-hydroxyindole n’ait été jusqu’à présent isolé en tant que substance naturelle. Cette caractéristique est due vraisemblablement à la présence du groupement hydroxyle en position 1, qui rend la molécule instable.
La synthèse et la réactivité des dérivés du 1-hydroxyindole ont été particulièrement enrichies par les travaux des équipes de R.M. Acheson 〚2〛 et de M. Somei 〚3〛. Leurs résultats permettent de mieux comprendre la chimie de cette classe de composés. Pour notre part, après avoir synthétisé de nouveaux précurseurs d’alcaloïdes, le 3-acétyl-1-hydroxy-2-méthyl-6-nitroindole et le 3-acétyl-6-amino-1-hydroxy-2-méthylindole 〚4〛, nous étudions leur stabilité et leurs réactivités vis-à-vis des agents alkylants, des réactifs électrophiles carbonylés et des hydrazines. Nous avons envisagé de mettre en jeu les groupements hydroxyle, amino, acétyle et le carbone en position 2.
2 Résultats et discussion
2.1 Réactivité des composés , avec les dérivés halogénés
Très peu de travaux sur l’alkylation du 1-hydroxyindole ont été décrits dans la littérature. Les auteurs ont pu isoler, selon les conditions utilisées, des dérivés O-alkylés 〚5, 6〛, O- et C3-alkylés 〚7〛, O-alkylés, en plus de l’indole. Ce dernier est dû à la perte du groupement hydroxyle lors de la réaction 〚8〛. Pour notre part, nous avons examiné la réaction d’alkylation dans les conditions de la catalyse par transfert de phase solide–liquide des dérivés , avec divers dérivés halogénés. Ainsi, l’iodométhane, le bromoéthane, le bromure de propargyle, les chlorures d’allyle et de benzyle ont été employés. Le carbonate de potassium est utilisé comme base inorganique dans le tétrahydrofurane anhydre et le bromure de tétra-n-butylammonium (BTBA) comme catalyseur.
Ainsi, nous avons constaté que, quel que soit l’halogénure d’alkyle utilisé, l’alkylation du 3-acétyl-1-hydroxy-6-nitroindole conduit, après 24 h sous agitation, à la température ambiante, aux composés O-alkylés avec un bon rendement (54–76%) (Fig. 1).

Alkylation du 3-acétyl-1-hydroxy-6-nitroindole conduisant, après 24 h sous agitation, à la température ambiante, aux composés O-alkylés.
Cependant, dans les mêmes conditions, l’alkylation du 3-acétyl-6-amino-1-hydroxy-2-méthylindole ne nous a pas permis d’isoler de produits identifiables. Cette différence de réactivité avait déjà été soulignée par Somei et al. 〚3〛, qui ont montré que les nitrohydroxy-indoles sont plus stables que leurs homologues aminés.
Les structures des composés isolés ont été établies sur la base des données spectrales RMN 1H, RMN 13C et infrarouge.
Les spectres de RMN 1H des composés 3a–e, enregistrés dans le CDCl3, présentent, outre les signaux correspondant aux protons des deux groupes méthyles et les protons aromatiques de l’hydroxyindole, des signaux relatifs aux protons des groupes O-alkyles. Les différents déplacements chimiques sont rapportés dans le Tableau 1. Les spectres de RMN 13C des composés 3a–e, enregistrés dans le CDCl3, mettent en évidence la O-alkylation, sans ambiguïté, puisque le carbone directement lié à l’oxygène résonne vers les champs faibles (Tableau 2).
Spectres de RMN 1H des composés 3a–e.
| Produits | CH3 | CH3 | CH3 | CH2O | CH vinylique | CH2 vinylique | CH≡ | CH arom |
| 3a | 2,62 (s, 3H) | 2,81 (s, 3H) | 4,18 (s, 3H) | — | — | — | — | 8,1– 8,34 (m, 3H) |
| 3b | 2,61 (s, 3H) | 2,78 (s, 3H) | 1,51 (t, 3H) J = 7,0 Hz | 4,32 (q, 2H) J = 7,0 Hz | — | — | — | 8,06 (dd, J=8,9 Hz, J = 2,0 Hz,1H) ; 8,12 (d, J= 8,9 Hz, 1H) ; 8,25 (d, J=2,0 Hz, 1H) |
| 3c | 2,58 (s, 3H) | 2,61 (s, 3H) | — | 5,25 (s, 2H) | — | — | — | 7–8,5 (m, 8H) |
| 3d | 2,64 (s, 3H) | 2,81 (s, 3H) | — | 4,75–4,76 (m, 2H) | 6,06–6,22 (m, 1H) | 5,39–5,47 (m, 2H) | — | 8,1 (dd, J = 8,9 Hz, J = 2,0 Hz, 1H) ; 8,15 (d, J = 8,9 Hz, 1H) ; 8,32 (d, J = 2,0 Hz, 1H) |
| 3e | 2,58 (s, 3H) | 2,81 (s, 3H) | — | 5,22 (d, 2H) J = 2,4 Hz | — | — | 3,91 (t, 1H) J = 2,4 Hz | 8,06 (dd, J = 8,9 Hz, J = 2,2 Hz, 1H) ; 8,26 (d, J = 8,9 Hz, 1H) ; 8,44 (d, J = 2,2 Hz, 1H) |
Spectres RMN 13C des composés 3a–e.
| Produits | CH3– | CH3 | CH3 | CH2O | CH vinylique | CH2 vinylique | CH≡ | CH arom | Cq |
| 3a | 11,7 | 31,4 | 66,4 | 105 ;117,9 ; 121,3 | 110,8 ; 127 ; 130,0 ; 143,5 ; 145,3 ; 193,5 | ||||
| 3b | 11,9 | 31,3 | 13,7 | 75,2 | 105,1 ; 117,7 ; 121,14 | 110,8 ; 126,9 ; 130,5 ; 143,3 ; 145,7 ; 193,4 | |||
| 3c | 11,9 | 31,4 | 81,2 | 105,4 ; 117,8 ; 121,2 ; 129,1 ; 130,2 | 110,8 ; 126,9 ; 130,6 ; 132,8 ; 143,3 ; 145,5 ; 193,4 | ||||
| 3d | 12,2 | 31,4 | 79,9 | 123,7 | 129,7 | 105,4 ; 117,8 ; 121,2 | 110,9 ; 126,9 ; 130,7 ; 143,4 ; 145,9 ; 193,4 | ||
| 3e | 12 | 31,0 | 66,7 | 82,3 | 105,6 ; 117,4 ; 121,1 | 110,2 ; 126,4 ; 130,3 ; 142,6 ; 146,5 ; 192,9 |
2.2 Réactivité du composé avec les électrophiles carbonylés
Dans des travaux précédents, nous avons étudié la condensation des aminobenzimidazoles, aminoindazoles et aminoindoles avec l’acétylacétone et les dérivés de la γ-pyrone. Nous avons pu préparer différents systèmes hétérocycliques, dérivés de la quinoxaline, de la benzodiazépine et de la pyridone 〚9–12〛. Aussi nous est-il paru intéressant d’examiner la réactivité de avec l’anhydride acétique, le chlorure de benzoyle, l’acétylacétone et l’acide déhydroacétique (Fig. 2).

Réaction de avec l’anhydride acétique, le chlorure de benzoyle, l’acétylacétone et l’acide déhydroacétique.
Il nous a été possible d’isoler quantitativement les composés d’acylation , de benzoylation et de condensation , . Cependant, en aucun cas, nous n’avons pu obtenir la quinoline ou la pyridone normalement attendues.
Les structures de ces composés ont été établies sans aucune ambiguïté, sur la base de leurs données spectrales RMN 1H, RMN 13C, infrarouge et masse. En effet, on note en particulier, dans les spectres infrarouge des composés , et , enregistrés dans le bromure de potassium, la présence de la bande de vibration νOH vers 3400 cm–1 et dans celui du composé la bande de vibration νC=O à 1767 cm–1. La présence du groupement hydroxyle est également confirmée dans les spectres de masse par l’existence du pic de base à M-16. Ce dernier résultat est une caractéristique de la série des 1-hydroxyindoles 〚3〛. La structure énaminique du composé est mise en évidence par la présence, dans son spectre de RMN 1H, d’un signal à 5,3 ppm, indiquant un proton. Quant à la structure du composé , les spectres de RMN 1H et de RMN 13C attestent la présence des cycles pyranique et indolique par simple comparaison avec les spectres des réactifs de départ.
2.3 Actions des hydrazines sur les composés et
Nous avons également examiné l’action des hydrazines sur les composés et , susceptibles de se comporter comme des cétones α,β éthyléniques dans des réactions de type Michael. Il est à noter que le caractère électrophile de la position 2 est accru par la présence des groupes hydroxyle sur le sommet 1 〚13 〛 et acétyle sur le sommet 3 qui influent par leurs effets –I et –M. Cependant, nous avons constaté que seul le chauffage du composé en présence d’hydrazines dans le n-butanol conduit aux produits de structure pyrazolique attendus 10a–10c (Fig. 3).
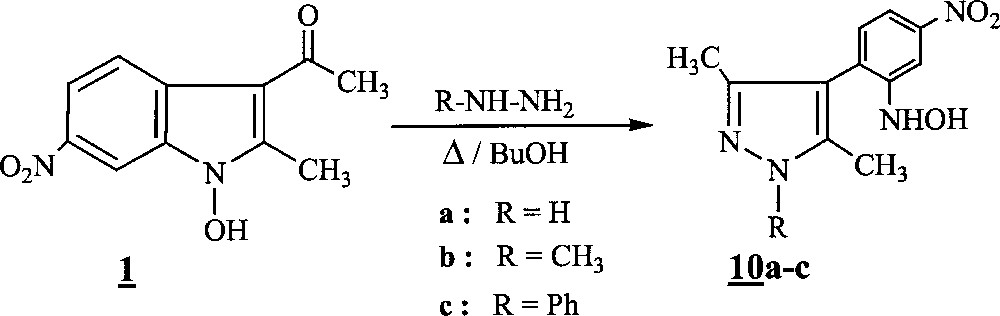
Synthèse des produits de structure pyrazolique 10a–10c à partir du composé , en présence d’hydrazines dans le n-butanol.
Les structures des produits 10a–10c ont été établies sur la base des données spectrales RMN 1H, RMN 13C et infrarouge, corroborées par l’analyse cristallographique réalisée sur le produit 10b (Fig. 4). Ce dernier cristallise dans le système triclinique ; les longueurs des liaisons et les angles correspondants au composé 10b sont rassemblés dans le Tableau 3.
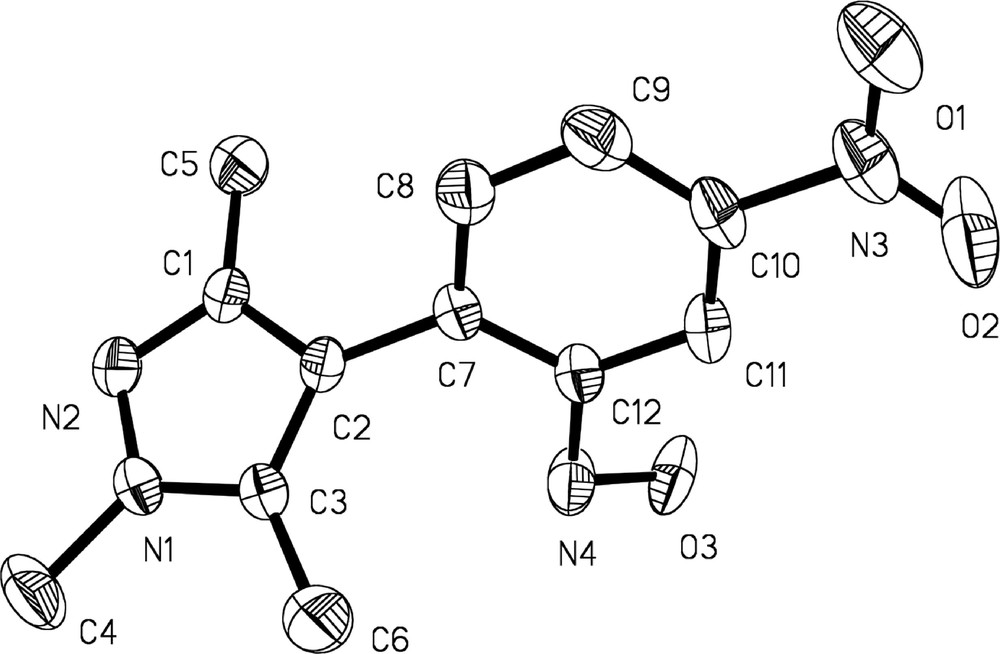
Représentation Ortep du composé 10b.
Principales longueurs de liaisons (A°) et angles (°) du composé 10b.
| C(1)–N(2) | 1.331(4) | C(1)–C(2) | 1.405(4) |
| C(1)–C(5) | 1.482(5) | C(2)–C(3) | 1.390(4) |
| C(2)–C(7) | 1.484(4) | C(3)–N(1) | 1.356(4) |
| C(3)–C(6) | 1.479(5) | C(4)–N(1) | 1.459(4) |
| C(7)–C(8) | 1.395(5) | C(7)–C(12) | 1.404(4) |
| C(8)–C(9) | 1.377(5) | C(9)–C(10) | 1.377(5) |
| C(10)–C(11) | 1.377(5) | C(10)–N(3) | 1.473(4) |
| C(11)–C(12) | 1.390(4) | C(12)–N(4) | 1.409(4) |
| N(1)–N(2) | 1.352(4) | N(3)–O(1) | 1.223(4) |
| N(3)–O(2) | 1.223(4) | N(4)–O(3) | 1.444(4) |
| N(2)–C(1)–C(2) | 110.7(3) | N(2)–C(1)–C(5) | 121.2(3) |
| C(2)–C(1)–C(5) | 128.0(3) | C(3)–C(2)–C(1) | 105.4(3) |
| C(3)–C(2)–C(7) | 126.7(3) | C(1)–C(2)–C(7) | 127.6(3) |
| N(1)–C(3)–C(2) | 106.0(3) | N(1)–C(3)–C(6) | 123.1(3) |
| C(2)–C(3)–C(6) | 130.9(3) | C(8)–C(7) –C(12) | 118.3(3) |
| C(8)–C(7)–C(2) | 119.5(3) | C(12)–C(7)–C(2) | 122.2(3) |
| C(9)–C(B)–C(7) | 122.1(3) | C(10)–C(9)–C(8) | 117.8(3) |
| C(9)–C(10)–C(11) | 122.5(3) | C(9)–C(10)–N(3) | 119.4(3) |
| C(11)–C(10)–N(3) | 118.1(3) | C(10)–C(11)–C(12) | 119.0(3) |
| C(11)–C(12)–C(7) | 120.0(3) | C(11)–C(12)–N(4) | 120.2(3) |
| C(7)–C(12)–N(4) | 119.6(3) | N(2)–N(1)–C(3) | 112.2(3) |
| N(2)–N(1)–C(4) | 120.0(3) | C(3)–N(1)–C(4) | 127.8(3) |
| C(1)–N(2)–N(1) | 105.7(2) | O(1)–N(3)–O(2) | 123.7(3) |
| O(1)–N(3)–C(10) | 117.7(3) | O(2)–N(3)–C(10) | 118.6(3) |
| C(12)–N(4)–O(3) | 111.4(3) |
La formation de ces composés 10a–10c peut s’expliquer selon une réaction d’addition-1,4 de type Michael, mettant en jeu le groupe amino de l’hydrazine utilisée et le carbone en position 2 du 1-hydroxyindole, conduisant à l’intermédiaire 〚A〛. Ce dernier évolue ensuite vers un système tricyclique 〚B〛 instable, résultant de l’attaque nucléophile du carbonyle du groupe acétyle par le deuxième groupe amino hydrazinique. L’élimination d’une molécule d’eau, suivie d’une aromatisation induisant l’ouverture de la liaison N1–C2 indolique, conduit aux systèmes pyrazoliques 10a–c (Fig. 5).
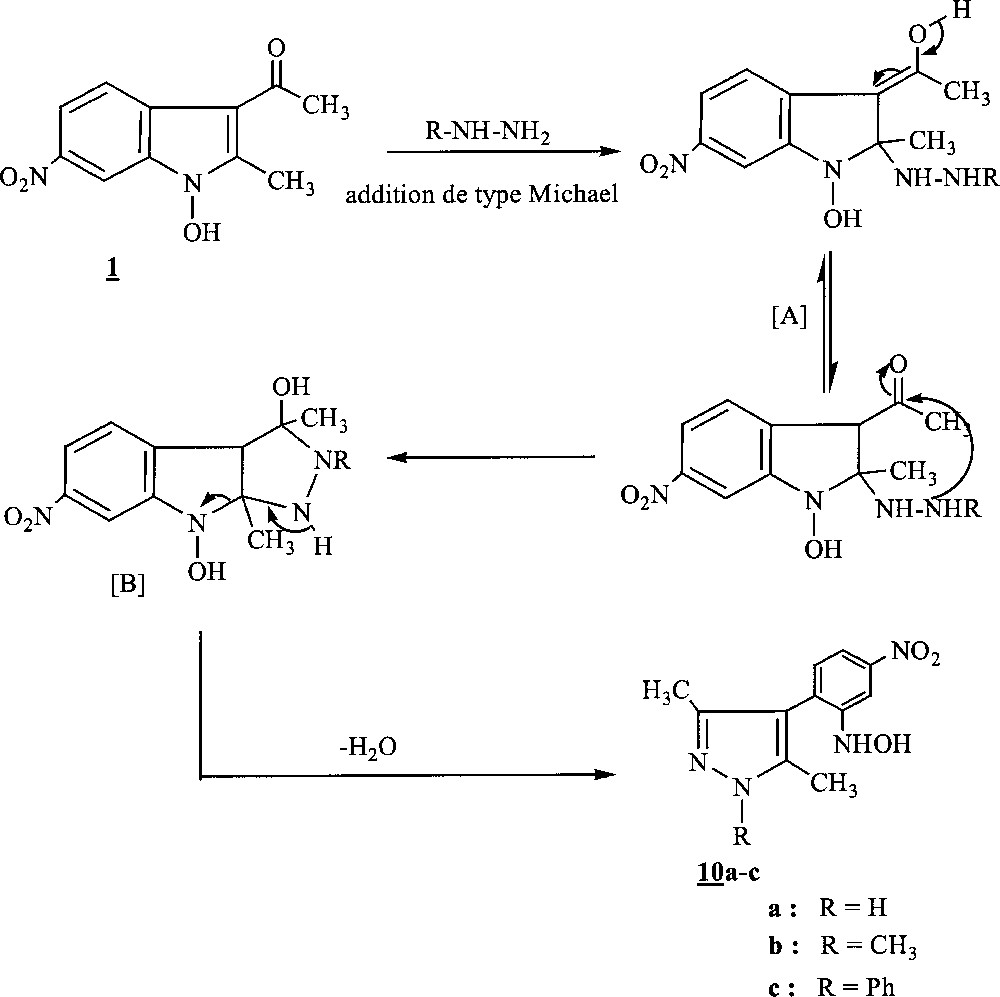
Obtention des systèmes pyrazoliques 10a–c par élimination d’une molécule d’eau, suivie d’une aromatisation induisant l’ouverture de la liaison N1–C2 indolique.
Ce résultat est à rapprocher de celui rapporté par Alberti 〚14〛 lors de l’hydrazinolyse des 3-acétylindoles, car il a pu isoler des 4-(o-aminophényl) pyrazoles.
Ainsi, dans ce travail, nous avons préparé de nouveaux dérivés du 1-hydroxyindole, susceptibles de présenter des propriétés pharmacologiques intéressantes, et vérifié la stabilité relative du dérivé nitré par rapport à son homologue aminé lors des réactions d’alkylation et d’hydrazinolyse ; de même, nous avons montré que le 3-acétyl-1-hydroxyindole constitue un précurseur de choix de nouveaux dérivés pyrazoliques substitués en position 4 par des groupements o-hydroxylaminophényles, difficiles d’accès par d’autres voies de synthèse.
3 Partie expérimentale
3.1 Généralités
Les points de fusion ne sont pas corrigés. Les spectres de RMN 1H et 13C ont été enregistrés sur un appareil Bruker AC 250 à 250 MHz. Les spectres de masse ont été effectués avec un appareil Varian MAT 311A par désorption de champs (DIC, NH3). Les spectres infrarouge ont été réalisés sur un spectromètre Perkin Elmer 1760x.
3.2 Synthèse des 3-acétyl-2-méthyl-6-nitro-1-alkoxyindoles 3a–e
3.2.1 Mode opératoire général
On dissout 0,005 mol de composé 1 et 0,0052 mol d’halogénure d’alkyle (en léger excès) dans 50 ml de THF. On ajoute 0,005 mol de K2CO3 et 0,0005 mol de bromure de tétra-n-butylammonium (BTBA). Le mélange est maintenu sous agitation à température ambiante, pendant 24 h. On filtre et on concentre sous pression réduite. Le produit obtenu est recristallisé dans l’éthanol.
3.2.2 3-acétyl-1-méthoxy-2-méthyl- -6-nitroindole 3a
Rdt : 54% ; P.F. : 193 °C (EtOH) ; IR (KBr) : 1339 et 1502 cm–1 (νNO2) ; 1658 cm–1 (νC=O).
3.2.3 3-acétyl-1-éthoxy-2-méthyl -6-nitroindole 3b
Rdt : 60% ; P.F. : 152 °C (EtOH) ; IR (KBr) : 1341 et 1504 cm–1 (νNO2) ; 1657 cm–1 (νC=O).
3.2.4 3-acétyl-1-benzyloxy-2-méthyl-6-nitroindole 3c
Rdt : 70% ; P.F. : 153 °C (EtOH) ; IR (KBr) : 1340 et 1510 cm–1 (νNO2) ; 1650 cm–1 (νC=O).
3.2.5 3-acétyl-1-allyloxy-2-méthyl-6-nitroindole 3d
Rdt : 76% ; P.F. : 126 °C (EtOH) ; IR (KBr) : 1333 et 1508 cm–1 (νNO2) ; 1648 cm–1 (νC=O).
Masse : 〚M+1〛+ = 275.
3.2.6 3-acétyl-2-méthyl-6-nitro-1-propargyloxyindole 3e
Rdt : 70% ; P.F. : 177 °C (EtOH) ; IR (KBr) : 1343 et 1500 cm–1 (νNO2) ; 1664 cm–1 (νC=O) ; 2118 cm–1 (νC≡C).
3.3 Synthèse du 3-acétyl-1-hydroxy-2-méthyl-6-(acétylamino)indole
Dans un ballon de 250 ml, on introduit 1 g (4,9 mmol) de dans 50 ml d’anhydride acétique et on chauffe au reflux pendant 24 h. On évapore le solvant sous pression réduite. Le produit obtenu est recristallisé dans l’éthanol.
Rdt : 33% ; P.F. : 241 °C (EtOH) ; IR (KBr) : 1634 cm–1 (νC=O) ; 1712 cm–1 (νNCO) ; 3360 cm–1 (νNH) ; 3440 cm–1 (νOH) ; RMN1H (DMSO-d6) δ ppm : 1,94 (s, 3H, CH3) ; 2,38 (s, 3H, CH3) ; 2,51 (s, 3H, CH3) ; 7,04 (dd, J=1,8 Hz, J = 8,6 Hz, 1H) ; 7,81 (d, J = 8,6 Hz, 1H) ; 7,92 (d, J = 1,8, 1H) ; 9,90 (s, 1H, NH) ; 11,5 (s, 1H, OH) ; RMN13C (DMSO-d6) δ ppm : 15,2 (CH3) ; 28,1 (CH3) ; 34,9 (CH3) ; 102,6 ; 118,1 et 124,3 (CHar) ; 112,6 ; 121,8 ; 137,0 ; 138,5 et 144,8 (Car) ; 172,1 et 196,4 (C=O).
3.4 Synthèse du 3-acétyl-1-benzoyloxy-2-méthyl-6-(benzoylamino)indole
Dans un ballon de 250 ml, on introduit 1g (4,9 mmol) du composé et 1,37 g (9,8 mmol) de chlorure de benzoyle dans 40 ml de pyridine à 0 °C. Le mélange est ensuite laissé sous agitation à la température ambiante pendant 24 h. Le précipité obtenu est essoré, puis recristallisé dans l’éthanol.
Rdt : 60% ; P.F. : 209 °C (EtOH) ; IR (KBr) : 1619 cm–1 (νC=O) ; 1666 cm–1 (νNCO) ; 1767 cm–1 (νCO (ester)) RMN1H (DMSO-d6) δ ppm : 2,59 (s, 3H, CH3) ; 2,63 (s, 3H, CH3) ; 7,48–8,31 (m, 13H, Har) ; 10,35 (s, 1H, NH) ; RMN13C (DMSO-d6) δ ppm : 11,0 (CH3) ; 31(CH3) ; 99,1 ; 116,3 ; 118,2 ; 127,5 ; 128,2 ; 129,5 ; 130,2 et 131,4 ; 135,7 (CHar) ; 110,4 ; 121,0 ; 124,5 ; 132,0 ; 134,7 ; 135,4 ; et 140,7 (Car) ; 163,7 ; 165,4 et 193,2 (C=O). Masse : 〚M+1〛+ = 413.
3.5 Synthèse du 4-N-(3-acétyl-1-hydroxy-2-méthylindole-6-yl)amino-pent-3-en-2-one
Une solution de 0,5 g (2,45 mmol) du produit dans 3 ml (0,029 mol) d’acétylacétone est chauffée à 60 °C pendant 5 min. Après refroidissement, le précipité obtenu est essoré, puis recristallisé dans l’éthanol.
Rdt : 70% ; P.F. : 228 °C (EtOH) ; IR (KBr) : 1593,6 cm–1 (νC=O) ; 1630,7 cm–1 (νC=O) ; 3446,8 cm–1 (νOH) ; RMN1H (DMSO-d6) δ ppm : 2,01 (s, 3H, CH3) ; 2,03 (s, 3H, CH3) ; 2,52 (s, 3H, CH3) ; 2,85 (s, 3H, CH3) ; 5,26 (s, 1H, CH vinylique) ; 7,04 (dd, J = 8,5 Hz, J = 2 Hz, 1H) ; 7,25 (d, J = 8,5 Hz, 1H) ; 8,05 (d, J = 2 Hz, 1H) ; 11,8 (s, 1H, NH) ; 12,6 (s, 1H, OH) ; RMN 13C (DMSO-d6) δ ppm : 12,1 (CH3) ; 20,2 (CH3) ; 29,7 (CH3) ; 31,7 (CH3) ; 98,0 ; 104,9 ; 120,1 et 122,1 (CHar + C3) ; 109,6 ; 120,6 ; 133,9 ; 134,0 ; 142,7 et 161,2 (Car + C4) ; 193,3 et 195,6 (C=O). Masse : 〚M〛+ = 286.
3.6 Synthèse du 3-〚(3-acétyl-1-hydroxy-2-méthylindole-6-yl)éthanimidoyl〛-2-hydroxy-6-méthyl-4H-4-pyranone
Une solution de 0,5 g (2,45 mmol) du composé et de 0,41 g (2,45 mmol) d’acide déhydroacétique dans 40 ml d’éthanol est agitée à température ambiante pendant 24 h. Le précipité obtenu est essoré, puis recristallisé dans l’éthanol.
Rdt : 84% ; P.F. : 279 °C (EtOH) ; IR (KBr) : 1670 cm–1 (νC=O) ; 1700 cm–1 (νNCO) ; 3400 cm–1 (νOH) ; RMN 1H (DMSO-d6) δ ppm : 2,14 (s, 3H, CH3) ; 2,54 (s, 3H, CH3) ; 2,55 (s, 3H, CH3) ; 2,71 (s, 3H, CH3) ; 5,85 (d, J = 0,7 Hz, 1H) ; 7,15 (dd, J = 8,6 Hz, J = 1,8 Hz, 1H) ; 7,47 (d, J = 1,8 Hz, 1H) ; 8,16 (d, J = 8,6 Hz, 1H) ; 11,92 (s, 1H, OH) ; 15,74 (s, 1H, OH) ; RMN13C (DMSO-d6) δ ppm : 11,2 (CH3) ; 19,2 (CH3) ; 19,9 (CH3) ; 30,8 (CH3) ; 105,7 ; 106,6 ; 119,4 et 121,2 (CHar) ; 96,4 ; 108,8 ; 121,4 ; 130,3 ; 132,7 ; 142,7 ; 163,1 et 175,0 (Car) ; 183,8 et 192,5 (C=O).
3.7 Synthèses de 10a, 10b et 10c
3.7.1 Synthèse du 3,5-diméthyl-4-(2-hydroxylamino-4-nitro-phényl)pyrazole 10a
Dans un ballon de 250 ml contenant 60 ml de butanol, on place 0,0042 mol du produit avec 0,016 mole d’hydrazine ; le mélange est porté à reflux pendant 1 h. Après évaporation du solvant sous pression réduite, le produit obtenu est lavé à l’éther éthylique.
Rdt : 42% ; P.F. : 192 °C (EtOH) ; IR (KBr) : 1346 cm–1 et 1525 cm–1 (νNO2) ; 3279 cm–1 (νNH) ; 3437 cm–1 (νOH) ; RMN1H (DMSO-d6) δ ppm : 2,3 (s, 6H, 2CH3) ; 7,30–8,20 (m, 3H, Har) ; 9,01 (s, 2H, NHOH). Masse : 〚M+1〛+ = 249.
3.7.2 Synthèse du 1,3,5-triméthyl-4-(2-hydroxylamino-4-nitro-phényl)pyrazole 10b
Le mode opératoire est le même que pour le produit 10a, avec 0,016 mol de méthylhydrazine ; le mélange est porté à reflux pendant 24 h.
Rdt : 23% ; P.F. : 189 °C (EtOH) ; IR (KBr) : 1339 cm–1 et 1522 cm–1 (νNO2) ; 1619 cm–1 (νC=N) ; 3319 cm–1 (νNH) ; 3416 cm–1 (νOH) ; RMN1H (DMSO-d6) δ ppm : 1,97 (s, 3H, CH3) ; 2,04 (s, 3H, CH3) ; 3,68 (s, 3H, CH3) ; 7,05–7,95 (m, 3H, Har) ; 8,81 (s, 2H, NHOH) ; RMN 13C (DMSO-d6) δ ppm : 10,1 (CH3) ; 12,1 (CH3) ; 35,7 (CH3) ; 106,3 ; 113,2 et 131,6 (CHar) ; 112,6 ; 125,2 ; 137,4 ; 143,9 ; 147,3 ; 150,8 (Car).
3.7.3 Synthèse du 3,5-diméthyl-4-(2-hydroxylamino-4-nitro-phényl)-1-phénylpyrazole 10c
Même mode opératoire que pour le produit 10b, avec 0,016 mol de phénylhydrazine.
Rdt : 29% ; P.F. : 203 °C (EtOH) ; IR (KBr) : 1330 cm–1 et 1501 cm–1 (νNO2) ; 1599 cm–1 (νC=N) ; 3359 cm–1 (νNH) ; 3475 cm–1 (νOH) ; RMN 1H (DMSO-d6) δ ppm : 2,39 (s, 3H, CH3) ; 2,66 (s, 3H, CH3) ; 7,20–8,31 (m, 8H, Har) ; 9,11 (s, 2H, NHOH) ; RMN 13C (DMSO-d6) δ ppm : 11,1 (CH3) ; 16,8 (CH3) ; 104,6 ; 112,4 ; 114,6 ; 118,3 ; 119,9 et 128,9 (CHar) ; 109,9 ; 125,3 ; 131,0 ; 138,7 ; 139,4 ; 141,3 et 146,3 (Car).
Remerciements
Le présent travail a été réalisé dans le cadre du contrat PARS (Chimie 015) et du pôle de compétence Pharchim.